
Abstract
Metachromatic leukodystrophy (MLD) is a rare genetic disease characterised by a dysfunction of the enzyme arylsulphatase A leading to the lysosomal accumulation of cerebroside sulphate (sulphatide) causing subsequent demyelination in patients. The enzyme galactosylceramide (cerebroside) sulphotransferase (CST) catalyses the transfer of a sulphate group from 3′-phosphoadenosine-5'-phosphosulphate (PAPS) to cerebrosides producing sulphatides. Substrate reduction therapy for arylsulphatase A by inhibition of CST was proposed as a promising therapeutic approach. To identify competitive CST inhibitors, we synthesised and investigated analogues of the substrate galactosylceramide with variations at the anomeric position, the acyl substituent and the carbohydrate moiety, and investigated their structure–activity relationships. While most of the compounds behaved as substrates, α-galactosylceramide 16 was identified as the first competitive CST inhibitor. Compound 16 can serve as a new lead structure for the development of drugs for the treatment of this devastating disease, MLD, for which small molecule therapeutics are currently not available.
1. Introduction
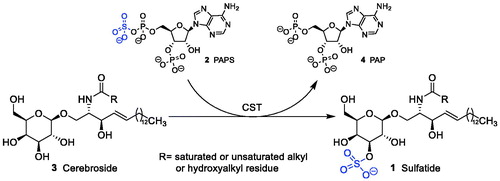
Metachromatic leukodystrophy (MLD) is a rare genetic disease characterised by a dysfunction of the enzyme arylsulphatase ACitation1. This defect leads to the lysosomal accumulation of cerebroside sulphate (sulphatide, 1) in various cells such as tubular kidney cells, bile duct epithelia, some neurons, oligodendrocytes and Schwann cells. In particular accumulation in the latter two results in progressive demyelination finally causing lethal symptoms in patients. Recently haematopoetic stem cell-based gene therapy has been shown to be effective in patients in early preclinical states of disease onlyCitation2. Thus, there is an urgent need to develop alternative strategies to treat MLD. One of these strategies is substrate reduction therapy in which galactosylceramide (cerebroside) sulphotransferase (CST; EC 2.8.2.11), the enzyme which synthesises sulphatide, is inhibited. This would diminish the load of accumulated sulphatide in the patient. Such a strategy has been shown to be effective in Gaucher disease, another lysosomal sphingolipid storage disorderCitation3. Inhibition of galactosylceramide sulphotransferase has been proposed as a promising new therapeutic strategy for the treatment of MLDCitation1,Citation4. CST catalyses the transfer of a sulphate group from the coenzyme 3′-phosphoadenosine-5′-phosphosulphate (PAPS, 2) to galactosylceramide (3) yielding galactosylceramide sulphate (2) and adenosine-3′,5′-bisphosphate (PAP, 4) ()Citation5–7.
Figure 1. Sulphatide synthesis by CST: galactosylceramide (cerebroside) is converted to sulphatide by CST in the presence of PAPS as sulphate donor.
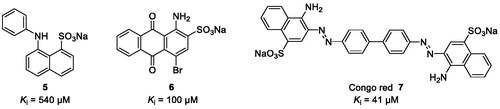
We, therefore, aim at developing CST inhibitors to reduce the biosynthesis of sulphatide to prevent sulphatide aggregation in the central and peripheral nervous systemCitation7. Sulphation is a widely observed biological reaction conserved from bacterium to human that plays a key role in various biological processesCitation8,Citation9. Deficiencies due to the lack of the ubiquitous sulphate donor PAPS are lethal in humansCitation8–10. A large group of enzymes called sulphotransferases catalyses the transfer reaction of the sulphuryl group of PAPS to the acceptor group of numerous biochemical and xenobiotic substratesCitation11. Structure-based sequence alignments based on X-ray crystal structures indicate that the PAPS-binding site is conservedCitation8,Citation9,Citation12. Therefore, competitive inhibitors for the CST substrate galactosylceramide are expected to have less side effects compared to inhibitors competing with the co-substrate PAPS. However, so far only few weakly potent, non-selective CST inhibitors have been described ()Citation13.
Figure 2. Chemical structures of aromatic dyes known as CST inhibitors competing with the co-substrate PAPS.
A reaction mechanism between the enzyme, the co-substrate PAPS, and the substrate galactosylceramide has been proposed based on crystal structuresCitation14–16. A transition state mimetic might efficiently inhibit the CST-catalysed reactionCitation17. In the present study, we synthesised and investigated a series of substrate analogues with the aim to study their structure–activity relationships (SARs) as substrates and to identify competitive inhibitors.
2. Materials and methods
2.1. Commercial compounds
Psychosine was purchased from Sigma (Steinheim, Germany). Galactosylceramide and glucosylceramide were obtained from Matreya LLC (Pleasant Gap, PA); according to the supplier, cerebroside consists of a mixture of saturated or unsaturated fatty acid residues (C16:0, C18:0, C20:0, C22:0, C23:0, C24:0–C27:0, C24:1–C27:1) or hydroxyacyl residues (C18:0(2–OH), C20:0(2–OH), C22:0(2–OH), C23:0(2–OH), C24:0(2–OH), C24:1(2–OH), C25:0(2–OH)) and glucosylceramide are a mixture of glucosylceramide with saturated or unsaturated fatty acid residues (C16:0, C18:0, C20:0, C22:0, C23:0, C24:0, C24:1). 3-Phosphoadenosine-5-phosphosulphate (PAPS) was purchased from Bellbrook Labs (No. 2059) in high purity. Other commercial sources of PAPS typically contain significant amounts of PAP and are, therefore, not suitable for the assayCitation18. α-Galactosylceramide (KRN7000) was purchased from Avanti Polar Lipids, (Alabaster, AL). β-KRN7000 was synthesised and provided by the laboratory of S. van Calenbergh.
2.2. Chemistry
Precoated Macherey-Nagel SIL G/UV254 plates were used for TLC and spots were examined under UV light at 254 nm and further visualised by sulphuric acid-anisaldehyde spray or by spraying with a solution of (NH4)6Mo7O24·4H2O (25 g/L) and (NH4)4Ce(SO4)4·2H2O (10 g/L) in H2SO4 (10%) followed by charring. Column chromatography was performed on Biosolve silica gel (32–63 µm, 60 Å). NMR spectra were obtained with a Varian Mercury 300 Spectrometer. Chemical shifts are given in ppm (δ) relative to the residual solvent signals, in the case of CDCl3: δ = 7.26 ppm for 1H and δ = 77.4 ppm for 13 C and in the case of pyridine-d5: δ = 8.74, 7.58 and 7.22 ppm for 1H and δ = 149.9, 135.5 and 123.5 ppm for 13C. Exact mass measurements were performed on a Waters LCT Premier XE TOF equipped with an electrospray ionisation interface and coupled to a Waters Alliance HPLC system. Samples were infused in a CH3CN/HCO2H (1000:1) mixture at 10 ml/min.
2.2.1. General procedure for Staudinger reduction and acylation reaction for compounds 24 and 25
To a solution of azide 23 (400 mg, 0.42 mmol, 1 eq.) in 16 ml of tetrahydrofuran (THF) at room temperature, a 1 M solution of PMe3 in THF (6.3 ml, 6.3 mmol, 15 eq.) was added dropwise. After stirring for 3 h, 2 ml of H2O were added and the reaction mixture was allowed to stir overnight at room temperature. Then the solvent was removed under reduced pressure and the residue was co-evaporated with toluene to afford the crude amine. A mixture of the crude amine, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 131 mg, 0.84 mmol, 2 eq.) and the appropriate fatty acid (0.63 mmol, 1.5 eq.) in 10 ml of CH2Cl2 was stirred for 24 h at room temperature. The reaction mixture was diluted with CH2Cl2, washed with H2O (2 × 10 ml) and brine (1 × 10 ml), dried over Na2SO4, filtered, and evaporated to dryness. Purification by column chromatography using 10% ethyl acetate in hexane gave the desired amides in the indicated yield.
2.2.1.1. [(1S,2S,3R)-1-[[[2,3-Bis-O-(phenylmethyl)-4,6-O-[(S)-phenylmethylene]-α-d-galactopyranosyl]oxy]methyl]-2,3-bis(phenylmethoxy)heptadecyl]-N-pentanamide (24)
Yield: 74%. 1H NMR (300 MHz, CDCl3): δ 0.87 (t, J = 7.2 Hz, terminal CH3), 0.88 (t, J = 6.7 Hz, terminal CH3), 1.18 − 1.35 (m, 26 H, CH2), 1.37–1.53 (m, 2 H, CH2), 1.55–1.69 (m, 2 H, CH2), 1.85–1.92 (m, 2 H, CH2), 3.51–3.57 (m, 1 H, H-4), 3.58 (br s, 1 H, H-4”), 3.74–3.82 (m, 2 H, Hb-1 and H-3), 3.88–3.99 (m, 3 H, Hb-6”, Ha-1 and H-3”), 4.05–4.15 (m, 2 H, H-2” and Ha-6”), 4.18 (d, J = 3.3 Hz, 1 H, H-5”), 4.24–4.34 (m, 1 H, H-2), 4.46–4.57 (m, 3 H, CH2Ph), 4.60–4.66 (m, 1 H, CH2Ph), 4.69–4.80 (m, 3 H, CH2Ph), 4.85 (d, J = 11.6 Hz, 1 H, CH2Ph), 4.96 (d, J = 3.5 Hz, 1 H, H-1”), 5.46 (s, 1 H, H-8”), 5.76 (d, J = 8.2 Hz, 1 H, NHCO), 7.19–7.42 (m, 23 H, arom H), 7.48–7.55 (m, 2 H, arom H). 13 C NMR (75 MHz, CDCl3) δ 13.82, 14.11, 22.37, 22.67, 25.82, 27.71, 29.36, 29.68, 29.71, 29.79, 30.27, 31.91, 36.41, 50.35, 62.91, 68.22, 69.41, 71.72, 71.90, 73.28, 73.79, 74.37, 75.69, 76.16, 77.20, 79.44, 79.92, 99.68, 101.00, 126.31, 127.54, 127.57, 127.60, 127.66, 127.71, 127.82, 127.88, 128.08, 128.31, 128.35, 128.43, 128.82, 137.82, 138.40, 138.52, 138.56, 138.66, 172.85.
2.2.1.2. [(1S,2S,3R)-1-[[[2,3-Bis-O-(phenylmethyl)-4,6-O-[(S)-phenylmethylene]-α-d-galactopyranosyl]oxy]methyl]-2,3-bis(phenylmethoxy)heptadecyl]-N-non-adecanamide (25)
Yield 84%. 1H NMR (300 MHz, CDCl3): δ 0.86–0.92 (m, 6 H, 2 × terminal CH3) 1.16–1.37 (m, 54 H, CH2), 1.37–1.55 (m, 2 H, CH2), 1.55–1.72 (m, 2 H, CH2), 1.81–1.97 (m, 2 H, CH2), 3.50–3.57 (m, 1 H, H-4), 3.59 (br s, 1 H, H-4”), 3.74–3.82 (m, 2 H, Hb-1 and H-3), 3.88–3.98 (m, 3 H, Hb-6”, Ha-1 and H-3”), 4.04–4.15 (m, 2 H, H-2” and Ha-6”), 4.18 (d, J = 3.1 Hz, 1 H, H-5”), 4.25–4.35 (m, 1 H, H-2), 4.47–4.63 (m, 3 H, CH2Ph), 4.65–4.80 (m, 4 H, CH2Ph), 4.86 (d, J = 11.7 Hz, 1 H, CH2Ph), 4.95 (d, J = 3.4 Hz, 1 H, H-1”), 5.46 (s, 1 H, H-8”), 5.77 (d, J = 8.2 Hz, 1 H, NHCO), 7.22–7.43 (m, 23 H, arom H), 7.48–7.55 (m, 2 H, arom H). 13C NMR (75 MHz, CDCl3) δ 14.11, 22.67, 25.71, 25.83, 29.36, 29.45, 29.71, 29.80, 30.26, 31.91, 36.73, 50.33, 62.93, 68.19, 69.41, 71.72, 71.90, 73.29, 73.81, 74.36, 75.69, 76.15, 77.20, 79.49, 79.85, 99.64, 101.00, 126.31, 127.56, 127.68, 127.82, 127.88, 128.08, 128.29, 128.32, 128.35, 128.43, 128.84, 137.82, 138.40, 138.53, 138.64, 172.89.
2.2.2. General procedure for the debenzylation reaction to prepare 12 and 17
A solution of the protected glycoside (0.06 mmol) in CHCl3 (3 ml) and EtOH (9 ml) was hydrogenolysed under atmospheric pressure in the presence of palladium black (35 mg). Upon reaction completion, the mixture was filtered through celite. The filter cake was rinsed with CHCl3 and EtOH and the filtrate was evaporated to dryness. After purification by column chromatography (10% →; 18% MeOH in CH2Cl2), the final compounds were obtained as white powders in the indicated yield.
2.2.2.1. [(1S,2S,3R)-1-[(α-d-Galactopyranosyloxy)methyl]-2,3-dihydroxyheptadecyl]-N-pentanamide (12)
Yield: 70%. 1H NMR (300 MHz, pyridine-d5): δ 0.79 (t, J = 7.4 Hz, 3H, terminal CH3), 0.87 (t, J = 6.7 Hz, 3H, terminal CH3), 1.16–1.51 (m, 24 H, CH2), 1.57–1.79 (m, 3 H, CH2), 1.80–1.99 (m, 2 H, CH2), 2.21–2.35 (m, 1 H, CH2), 2.39 (t, J = 7.9 Hz, 2H, CH2), 4.27–4.33 (m, 2 H), 4.34–4.46 (m, 4 H), 4.51 (t, J = 6.0 Hz, 1 H), 4.56 (d, J = 2.7 Hz, 1 H), 4.62–4.71 (m, 2 H), 5.21–5.31 (m, 1 H, H-2), 5.57 (d, J = 3.8 Hz, 1 H, H-1”), 6.39 (br s, 6 H, OH), 8.43 (d, J = 8.5 Hz, 1 H, NH). 13 C NMR (75 MHz, pyridine-d5) δ 14.55, 14.84, 23.25, 23.48, 27.06, 28.93, 30.15, 30.46, 30.54, 30.69, 30.89, 32.67, 34.84, 36.99, 52.06, 63.16, 69.02, 70.85, 71.49, 72.13, 73.03, 73.54, 77.14, 102.01, 173.90. HRMS (ESI) m/z: calculated for C29H58NO9 [M + H]+ 564.4106; found 564.4094.
2.2.2.2. [(1S,2S,3R)-1-[(α-d-Galactopyranosyloxy)methyl]-2,3-dihydroxyheptadecyl]-N-non-adecanamide (17)
Yield 42%. 1H NMR (300 MHz, pyridine-d5): δ 0.88 (t, J = 6.4 Hz, 6H, 2 × terminal CH3), 1.11–1.50 (m, 52 H, CH2), 1.60–1.75 (m, 1 H, CH2), 1.76–1.99 (m, 4 H, CH2), 2.23–2.37 (m, 1 H, CH2), 2.46 (t, J = 7.5 Hz, 2 H, CH2), 4.30–4.36 (m, 2 H), 4.38–4.48 (m, 4 H), 4.53 (t, J = 6.1 Hz, 1 H), 4.56 (d, J = 3.1 Hz, 1 H), 4.63–4.72 (m, 2 H), 5.23–5.33 (m, 1 H, H-2), 5.59 (d, J = 3.8 Hz, 1 H, H-1”), 6.24 (br s, 6 H, OH), 8.48 (d, J = 8.8 Hz, 1 H, NH). 13 C NMR (75 MHz, pyridine-d5) δ 14.68, 23.34, 26.79, 26.91, 30.02, 30.16, 30.21, 30.27, 30.33, 30.40, 30.42, 30.55, 30.76, 32.53, 34.75, 37.20, 51.85, 63.06, 69.08, 70.71, 71.39, 72.01, 72.89, 73.45, 77.13, 101.94, 173.62. HRMS (ESI) m/z: calculated for C43H86NO9 [M + H]+ 760.6297; found 760.6267.
2.2.3. Synthesis of tert-butyl-N-[(1S,2S,3R)-1-[[[2,3-bis-O-(phenylmethyl)-4,6-O-[(S)-phenylmethylene]-α-d-galactopyranosyl]oxy]methyl]-2,3-bis(phenylmethoxy)heptadecyl]carbamate (26)
To a solution of azide 23 (800 mg, 0.84 mmol) in THF (30 ml) at room temperature, a 1 M solution of PMe3 in THF (12.6 ml, 12.6 mmol) was added dropwise. After stirring for 3 h at room temperature, H2O (4 ml) was added and the reaction mixture was allowed to stir overnight at room temperature. Then the solvent was removed under reduced pressure and additional co-evaporation with toluene to afford the crude amine. The latter was dissolved in CH2Cl2 (13 ml) and Et3N (3.3 ml) followed by the addition of Boc2O (1.1 g, 5.03 mmol). The reaction mixture was stirred overnight at room temperature, evaporated under reduced pressure and purified by column chromatography (0%→20% EtOAc in hexanes) to yield 26 (703 mg, 81%) as a colourless oil. 1H NMR (300 MHz, CDCl3): δ 0.90 (t, J = 6.6 Hz, terminal CH3), 1.21–1.34 (m, 20 H, CH2), 1.43 (br s, 9 H, tBu), 1.47–1.73 (m, 6 H, CH2), 3.52–3.58 (m, 1 H, H-4), 3.59 (br s, 1 H, H-5”), 3.72–3.81 (m, 2 H, Hb-1 and H-3), 3.82–3.96 (m, 3 H, Hb-6”, Ha-1 and H-2), 3.96–4.09 (m, 2 H, H-2” and H-3”), 4.09–4.13 (m, 1 H, Ha-6”), 4.15–4.21 (m, 1 H, H-4”), 4.42–4.56 (m, 2 H, CH2Ph), 4.56–4.68 (m, 2 H, CH2Ph), 4.73–4.88 (m, 5 H, CH2Ph and NHCO), 4.95 (d, J = 3.1 Hz, 1 H, H-1”), 5.47 (s, 1 H, H-8”), 7.18–7.44 (m, 23 H, arom H), 7.50–7.55 (m, 2 H, arom H). 13 C NMR (75 MHz, CDCl3) δ 14.11, 22.69, 25.83, 28.01, 28.40, 29.36, 29.65, 29.71, 31.92, 51.68, 62.82, 68.48, 69.41, 71.83, 73.61, 74.51, 75.60, 76.10, 77.20, 79.21, 79.43, 79.75, 99.42, 101.01, 126.31, 127.54, 127.57, 127.79, 127.88, 128.08, 128.26, 128.29, 128.32, 128.35, 128.82, 137.83, 138.47, 138.55, 138.64, 138.73, 155.34. HRMS (ESI) m/z: calculated for C64H86NO10 [M + H]+ 1028.6246; found 1028.6260.
2.2.4. Synthesis of tert-butyl-N-[(1S,2S,3R)-1-[(α-d-galactopyranosyloxy)methyl]-2,3-dihydroxyheptadecyl]carbamate (27)
A solution of 26 (703 mg, 0.68 mmol) in CHCl3 (6 ml) and EtOH (18 ml) was hydrogenolysed under atmospheric pressure in the presence of palladium black (50 mg). Upon reaction completion, the mixture was filtered through celite. The filter cake was rinsed with CHCl3 and EtOH and the filtrate was evaporated to dryness. After purification by column chromatography (10% →18% MeOH in DCM), compound 27 (242 mg, 61%) was obtained as a pale yellowish solid. 1H NMR (300 MHz, pyridine-d5): δ 0.88 (t, J = 6.4 Hz, 3H, terminal CH3), 1.15–1.34 (m, 21 H, CH2), 1.37–1.46 (m, 1 H, CH2), 1.51 (s, 9 H, tBu), 1.60–1.75 (m, 1 H, CH2), 1.80–1.98 (m, 2 H, CH2), 2.22–2.35 (m, 1 H, CH2), 4.24–4.35 (m, 3 H, Ha-1, H-3”, H-4), 4.38–4.53 (m, 4 H, CH2-6”, H-4” and H-3”), 4.57 (d, J = 3.0 Hz, 1 H, H-5”), 4.61–4.74 (m, 2 H, H-2” and Hb-1), 4.91–5.01 (m, 1 H, H-2), 5.56 (d, J = 3.8 Hz, 1 H, H-1”), 6.41 (br s, 6 H, OH), 7.46 (d, J = 9.1 Hz, 1 H, NH). 13 C NMR (75 MHz, pyridine-d5) δ 14.86, 23.50, 27.03, 29.13, 30.17, 30.48, 30.54, 30.58, 30.68, 30.90, 32.68, 34.87, 53.09, 63.10, 68.94, 70.82, 71.47, 72.16, 72.94, 73.41, 77.24, 79.06, 101.81, 157.14. HRMS (ESI) m/z: calculated for C34H63N2O10 [M + pyridine + H]+ 659.4477; found 659.4462.
2.2.5. Synthesis of [(1S,2S,3R)-1-[(α-d-galactopyranosyloxy)methyl]-2,3-dihydroxyheptadecyl]-N-tetracos-15-enamide (22)
The Boc-protected glycophytosphingosine 27 (150 mg, 0.26 mmol) was dissolved in CH2Cl2 (20 ml) and 1 M HCl in 90% aqueous AcOH solution (100 μL) was added at room temperature. After 35 min TLC showed incomplete conversion of the starting material and another amount of the HCl solution (100 μL) was added. This process was repeated till TLC (CH2Cl2/MeOH: 8/2) showed full conversion of the starting material. This required a total addition of 800 μL of the HCl solution. The solvents were removed under reduced pressure and the residue was co-evaporated with MeOH (3 × 5 ml). The resulting crude amine (used without further purification) was dissolved in a biphasic mixture of THF (2.5 ml) and saturated aqueous NaOAc (2.5 ml). In a separate flask, nervonic acid (114 mg, 0.31 mmol) was refluxed for 2 h in oxalyl chloride (4 ml) and the crude formed acyl chloride, obtained after evaporation of the solvent by a stream of nitrogen and subsequent drying on high-vacuum, was dissolved in THF (2.5 ml) and added dropwise to the biphasic mixture. The reaction mixture was stirred for 2 h at room temperature and TLC showed complete conversion of the starting material. Next, the aqueous layer was extracted with THF (3 × 15 ml) and the combined organic layer was dried over Na2SO4, filtered and evaporated. The crude residue was purified by column chromatography (10% → 16% MeOH in CH2Cl2) furnishing final compound 22 (79 mg, 37%) as a pale solid. 1H NMR (300 MHz, pyridine-d5): δ 0.86 (dt, J = 6.7, 5.4 Hz, 3H, terminal CH3), 1.15–1.49 (m, 54 H, CH2), 1.55–1.74 (m, 1 H, CH2), 1.74–1.98 (m, 4 H, CH2), 2.07–2.17 (m, 4 H, CH2), 2.21–2.35 (m, 1 H, CH2), 2.43 (t, J = 7.4 Hz, 2 H, CH2), 4.21–4.37 (m, 2 H, H-3, H-4), 4.38–4.48 (m, 4 H, Ha-1, H-3” and CH2-6”), 4.53 (t, J = 6.3 Hz, 1 H, H-5”), 4.57 (d, J = 2.9 Hz, 1 H, H-4”), 4.63–4.73 (m, 2 H, H-2” and Hb-1), 4.96 (br s, 1 H, OH), 5.24–5.33 (m, 1 H, H-2), 5.49–5.55 (m, 2H, CH = CH), 5.59 (d, J = 3.7 Hz, 1 H, H-1”), 6.10 (br s, 1 H, OH), 6.21–6.79 (m, 3 H, 3 x OH), 6.96 (br s, 1 H, OH), 8.48 (d, J = 8.8 Hz, 1 H, NH). 13 C NMR (75 MHz, pyridine-d5) δ 14.77, 23.41, 26.89, 26.99, 28.02, 30.03, 30.11, 30.26, 30.31, 30.38, 30.41, 30.49, 30.60, 30.64, 30.86, 32.58, 32.61, 34.81, 37.28, 51.97, 63.14, 69.13, 70.79, 71.48, 72.09, 72.97, 73.52, 77.17, 102.01, 130.73, 173.77. HRMS (ESI) m/z: calculated for C48H94NO9 [M + H]+ 828.6923; found 828.6942.
2.3. Biological evaluation
The CST reaction was carried out in a total volume of 50 µl containing 3′-phosphoadenosine-5′-phosphosulphate and galactosylceramide (concentrations of 3′-phosphoadenosine-5′-phosphosulphate and galactosylceramides varied according to assay type) in reaction buffer (10 mM HEPES, 16 mM MgCl2, 0.2% (v/v) Triton X-100, pH 7.1). All lipids and Triton X-100 were dissolved in chloroform/methanol (1:1), pipetted into reaction vials, and the chloroform/methanol mixture was removed by drying before adding reaction buffer. Reactions were initiated by the addition of 938 ng of human galactosylceramide sulphotransferase (CST), and then incubated at 37 °C for 30 min. All enzymatic reactions were stopped by heating for 10 min at 60 °C.
Analytical experiments were carried out by using a P/ACE MDQ capillary electrophoresis (CE) system (Beckman Instruments, Fullerton, CA) equipped with a DAD detection system. The capillary temperature was kept constant at 15 °C. The electrophoretic separations were carried out by using fused-silica capillary of 60 cm total length (50 cm effective length) × 75.5 µm (id) × 363.7 µm (od) obtained from Optronis GmbH. The following conditions were applied: λmax = 260 nm, voltage = −15 kV, running buffer 75 mM phosphate buffer, 0.002% polybrene, pH 5.6 (adjusted by phosphoric acid), electrokinetic injection (−10 kV, 30 s). The capillary was washed with 0.2 M NaOH for 2 min, and running buffer for 2 min before each injection. Data collection and corrected peak area analysis were performed with the 32 Karat software obtained from Beckman Coulter (Fullerton, CA). Further data analysis was carried out with Graph Pad Prism 4 (Graph Pad Software, Inc. San Diego, CA) and Excel. The human CST enzyme was obtained by heterologous expression in CHO cells in analogy to a described procedureCitation19. The CE assay method has previously been reportedCitation18.
2.3.1. Determination of kinetic parameters for CST
For the determination of kinetic parameters (Km and Vmax), eight different substrate concentrations were chosen. Negative controls were performed in the presence of heat-inactivated enzyme (10 min, 60 °C). Each analysis was repeated three times in independent experiments.
2.3.2. Investigation of CST inhibitors
For CST inhibitor characterisation, full concentration–inhibition curves were determined by testing a suitable range of inhibitor concentrations, to determine IC50 values. Ki values were calculated according to the Cheng–Prusoff equationCitation20. The substrate concentration was 100 µM of galactosylceramide (3), and 466 µM of KRN7000 (18), respectively, and the concentration of the cofactor PAPS was 30 μM. Substrate conversion was strictly controlled to be below 15%. Negative controls were performed in the presence of heat-inactivated enzyme (10 min, 60 °C). Each analysis was repeated three times in independent experiments.
3. Results and discussion
3.1. Chemistry
A series of analogues of the natural CST substrate galactocerebroside with variations in the galactose moiety (α- and β-glycosides, substitution of the sugar moiety), in the hydroxylated alkyl chain, and in the fatty acid residue was designed and synthesised. β-KRN7000Citation21 and α-glycosides 11, 13–16 and 18–21 were prepared as previously described.Citation22–27 The synthetic route to obtain α-galactosylceramides 12, 17 and 22 is depicted in Scheme 1.
Scheme 1. Reagents and conditions: (a) (i) PMe3, THF, then H2O; (ii) appropriate RCOOH, EDC, CH2Cl2; (b) H2, Pd black; (c) (i) PMe3, THF, then H2O; (ii) (Boc)2O, Et3N, CH2Cl2; (d) (i) HCl, AcOH, H2O; (ii) nervonic acid, oxalyl chloride, reflux; the acyl chloride is added to crude amine in THF/aq. NaOAc.
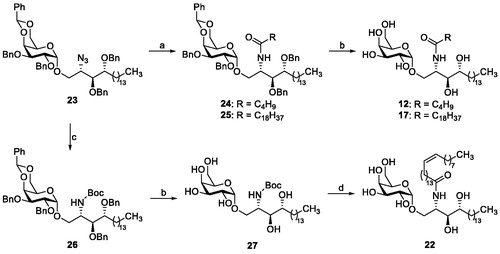
The required azidoglycoside 23 was obtained through Lewis acid-catalysed glycosidation reaction as previously reportedCitation28. Staudinger reduction and subsequent EDC-mediated acylation with the appropriate fatty acid furnished 24 and 25. Final catalytic hydrogenolysis yielded α-galactosylceramides 12 and 17. To avoid saturation of the cis-double bond of 15-tetracosanoic acid, the amino group generated after Staudinger reduction was Boc-protected prior to the removal of the benzyl groups to afford intermediate 27. Subsequent removal of the Boc moiety with HCl in acetic acid gave the corresponding psychosine derivative, which was subjected to Schotten–Baumann acylation with 15-tetracosanoyl chloride to afford glycolipid 22.
3.2. Biological evaluation
The synthesised compounds were studied using a previously developed capillary electrophoresis-based assay, in which the conversion of the co-substrate PAPS, acting as a sulphate donor, to adenosine-3′,5′-bisphosphate (PAP) was measuredCitation18. In addition to the natural substrate galactosylceramide (3), which contains a β-galactosyl residue and is present in nerve tissues as a constituent of myelin, the corresponding β-glucosyl derivative glucocerebroside (9) was investigated, which is mainly found in liver and spleen. As another naturally occurring sphingolipid psychosine (galactosyl-β-sphingosine, 8) was investigated, which is a cytotoxic derivative of galactosylceramide (3) that lacks its fatty acid residue. Moreover, 13 glycolipids, four of which (12, 17, 20 and 22) are new compounds, not previously described in literature, were investigated as artificial substrate analogues and/or competitive inhibitors of CST (). Enzyme kinetic parameters (Michaelis–Menten constant and maximal velocity) were determined for all (artificial) substrates for which significant conversion of >20% as compared to the natural substrate (set at 100%) was observed.
Table 1. Investigation of analogues of galactocerebroside as substrates of CSTa.
3.3. Structure–activity relationships
3.3.1. Substrates
The natural substrates of CST in nerve tissues are β-galactosylceramides (3) which are converted into 3-O-sulphogalactosylcerebroside that constitutes about 4% of total myelin lipids. They are sphingolipids consisting of (i) a sphingosine residue, (ii) a β-galactose, and (iii) a fatty acid residue attached via an amide linkage. Natural galactosylceramide (3) may contain different saturated or unsaturated (hydroxy)fatty acid residues with chain lengths mostly between C18 and C27, frequently C24. Galactosylceramide displayed a Km value of 60.3 µM determined in our recently developed capillary electrophoresis-based assay and was the best substrate of all compounds investigated in the present studyCitation19. The corresponding β-glucosylceramide (9) were shown to be much weaker substrates with only 19% conversion compared to that of 3 (set at 100%) determined under the same conditions. Another natural sphingolipid that had previously been shown to be a substrate of CST is psychosine (8) which is lacking the fatty acid residue of galactosylceramides (3). Psychosine was still efficiently sulphurylated by the enzyme showing an only moderately reduced Km value of 103 µM in the same assayCitation19. A synthetic galactosylceramide analogue, β-KRN7000 (10), in which the double bond of the sphingosine core structure is hydrated and, therefore, contains an additional hydroxy group, led to reduced conversion (42% compared to 100% for galactosylceramide) and an increased Km value of 550 µM (compared to 60 µM for galactosylceramide) determined under the same conditions. Interestingly, the α-galactoside anomer of compound 10, KRN7000 (18) showed about the same conversion rate and Km value indicating that for this series of more polar, hydrated sphingosine-derived synthetic lipids, the enzyme did not discriminate between α- and β-glycosidic configuration. Thus, we investigated further α-galactosyl-lipids (11–17, 19–22) derived from KRN7000 (18) mainly with modification of the fatty acid residue. KRN7000 (18) had previously been found to display immunostimulatory and antitumor activity in several in vivo models, and was advanced to clinical trialsCitation29–31.
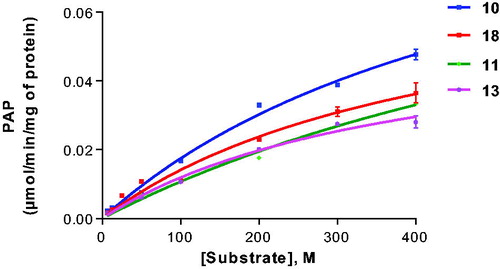
Using the artificial CST substrate KRN7000 (18) as a lead structure, we modified the fatty acid amide moiety. Compound 21, which lacks the fatty acid residue, was not well accepted as a substrate in this series (19% conversion). We subsequently investigated in a systematic manner the introduction of saturated fatty acid residues with increasing chain length. Compound 11 having a short fatty acid residue, namely acetyl, was tolerated as a substrate (40% conversion) confirming that a long fatty acid residue is not required for interaction with the enzyme and acceptance as a substrate. Probing the optimal length of the fatty acid residue led to the following result: C2 (acetyl, 11), C5 (12), C8 (13), C11 (14) and C19 (17) were about similarly good substrates (). Medium chain fatty acid residues appeared to be less well tolerated than some shorter chain analogs. In particular, palmitic acid (C16, 16) showed a sulphurylation rate below 10%. Interestingly, further increase of the chain length in compound 18 (C26) led to the best substrate in this series of α-galactosides with 51% conversion. Aromatic substitution in fatty acid amide analogue 19 was tolerated (compare 19 with 13 and 14), while the C24-fatty acid containing a cis-double bond was not a very good substrate. A bulky aromatic substitution on the sugar moiety in 20 strongly reduced the conversion rate (compare 20 with 18). Typical Michaelis–Menten curves are shown in for selected substrates (10, 11, 13 and 18).
Figure 3. Enzyme kinetics of galactosylceramide sulphotransferase for selected substrates. For Km and Vmax values see .
3.3.2. Inhibitors
Substrate analogues that were not or only poorly converted by CST with a conversion rate below 20% as compared to galactosylceramide might still be able to bind to the enzyme and thereby act as competitive inhibitors. Therefore, we investigated compounds 9, 16, and 10–22 for inhibition of CST activity using two different substrates, the natural galactosylceramide (3) and the synthetic α-galactoside KRN7000 (18). Initial inhibitor screening was performed at 100 µM concentration. Only derivative 16 displayed measurable enzyme inhibition at this concentration ().
Table 2. CST inhibitory activity of galactocerebroside analogues
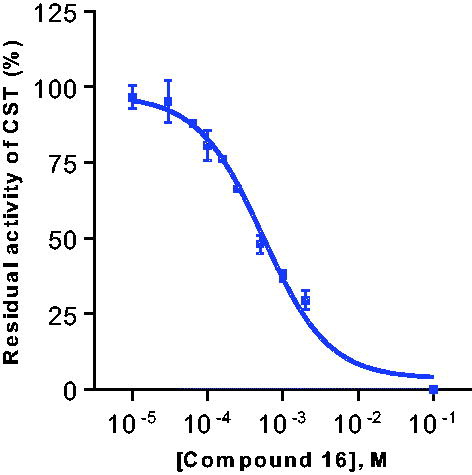
Subsequently, concentration-dependent inhibition was determined against both substrates, and nearly identical Ki values were determined against both substrates, 127 µM and 159 µM, respectively (see and ). To our knowledge, this is the first reported substrate-competitive inhibitor of CST.
Figure 4. Concentration–inhibition curve of the galactosylceramide sulphotransferase (CST) inhibitor 16. The curve had to be extrapolated due to limited solubility of 16. A Ki value of 127 ± 12 µM versus galactosylceramide as a substrate was determined.
4. Conclusions
In conclusion, we synthesised and investigated selected analogues of the natural CST substrate galactosylceramide (3), which were related to the artificial substrate KRN7000 (18). Our aim was to study structure–activity relationships for substrates of the enzyme, and to identify competitive inhibitors. We obtained detailed SAR information for the CST galactosylceramide binding site by analysing artificial substrates. Most importantly, a novel, competitive CST inhibitor (16) was identified. Compound 16 can serve as a lead structure for optimisation to obtain potent competitive CST inhibitors, which are urgently needed for the treatment of MLD. Our future goal is to develop CST inhibitors for substrate reduction therapy to help MLD patients to survive this devastating genetic disease.
Disclosure statement
The authors declare no conflict of interest, financial or otherwise.
Additional information
Funding
References
- Köhler W, Curiel J, Vanderver A. Adulthood leukodystrophies. Nat Rev Neurol 2018;14:94–105.
- Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016;388:476–87.
- Coutinho MF, Santos JI, Alves S. Less is more: substrate reduction therapy for lysosomal storage disorders. Int J Mol Sci 2016;17:1065.
- Gieselmann V, Krägeloh-Mann I. Metachromatic leukodystrophy – an update. Neuropediatrics 2010;41:1–6.
- Honke K. Biological functions of sulfoglycolipids and the EMARS method for identification of co-clustered molecules in the membrane microdomains. J Biochem 2018;163:253–63.
- Honke K. Biosynthesis and biological function of sulfoglycolipids. Proc Jpn Acad, Ser B, Phys Biol Sci 2013;89:129–38.
- Eckhardt M. The role and metabolism of sulfatide in the nervous system. Mol Neurobiol 2008;37:93–103.
- Yoshinari K, Petrotchenko EV, Pedersen LC, Negishi M. Crystal structure-based studies of cytosolic sulfotransferase. J Biochem Mol Toxicol 2001;15:67–75.
- Negishi M, Pedersen LG, Petrotchenko E, et al. Structure and function of sulfotransferases. Arch Biochem Biophys 2001;390:149–57.
- Superti-Furga A. A defect in the metabolic activation of sulfate in a patient with achondrogenesis type IB. Am J Hum Genet 1994;55:1137–45.
- Chen Y, Liu Y, Sullards MC, Merrill AH Jr. An introduction to sphingolipid metabolism and analysis by new technologies. Neuromol Med 2010;12:306–19.
- Rath VL, Verdugo D, Hemmerich S. Sulfotransferase structural biology and inhibitor discovery. Drug Discov Today 2004;9:1003–11.
- Zaruba M, Hilt D, Tennekoon G. Inhibition of rat brain galactocerebroside sulfotransferase by triazine aromatic dyes: interaction with the 3'-phosphoadenosine 5'-phosphosulfate binding site. Biochem Biophys Res Commun 1985;129:522–9.
- Pedersen LC, Petrotchenko E, Shevtsov S, Negishi M. Crystal structure of the human estrogen sulfotransferase-PAPS complex: evidence for catalytic role of Ser137 in the sulfuryl transfer reaction. J Biol Chem 2002;277:17928–32.
- Edavettal SC, Lee KA, Negishi M, et al. Crystal structure and mutational analysis of heparan sulfate 3-O-sulfotransferase isoform 1. J Biol Chem 2004;279:25789–97.
- Pedersen LC, Darden TA, Negishi M. Crystal structure of beta 1,3-glucuronyltransferase I in complex with active donor substrate UDP-GlcUA. J Biol Chem 2002;277:21869–73.
- Copeland RA. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. Hoboken: John Wiley & Sons; 2005.
- Li W, Zech I, Gieselmann V, Müller CE. A capillary electrophoresis method with dynamic pH junction stacking for the monitoring of cerebroside sulfotransferase. J Chromatogr A 2015;1407:222–7.
- Eckhardt M, Fewou SN, Ackermann I, Gieselmann V. N-glycosylation is required for full enzymic activity of the murine galactosylceramide sulphotransferase. Biochem J 2002;368:317–24.
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099–108. PMID 4202581.
- Ding N, Zhang W, Lv G, Li Y. Synthesis and biological evaluation of antifungal activities of novel 1,2-trans glycosphingolipids. Arch Pharm (Weinheim) 2011;344:786–93.
- Goff RD, Gao Y, Mattner J, et al. Effects of lipid chain lengths in alpha-galactosylceramides on cytokine release by natural killer T cells. J Am Chem Soc 2004;126:13602–3.
- Li Q, Ndonye RM, Illarionov PA, et al. Rapid identification of immunostimulatory alpha-galactosylceramides using synthetic combinatorial libraries. J Comb Chem 2007;9:1084–93.
- Lim C, Kim JH, Baek DJ, et al. Design and evaluation of ω-hydroxy fatty acids containing α-GalCer analogues for CD1d-mediated NKT cell activation. ACS Med Chem Lett 2014;5:331–5.
- Xia C, Yao Q, Schümann J, et al. Synthesis and biological evaluation of alpha-galactosylceramide (KRN7000) and isoglobotrihexosylceramide (iGb3). Bioorg Med Chem Lett 2006;16:2195–9.
- Fujio M, Wu D, Garcia-Navarro R, et al. Structure-based discovery of glycolipids for CD1d-mediated NKT cell activation: tuning the adjuvant versus immunosuppression activity. J Am Chem Soc 2006;128:9022–3.
- Guillaume J, Pauwels N, Aspeslagh S, et al. Synthesis of C-5″ and C-6″-modified α-GalCer analogues as iNKT-cell agonists. Bioorg Med Chem 2015;23:3175–82.
- Figueroa-Pérez S, Schmidt RR. Total synthesis of alpha-galactosyl cerebroside. Carbohydr Res 2000;328:95–102.
- Schneiders FL, Scheper RJ, von Blomberg BM, et al. Clinical experience with α-galactosylceramide (KRN7000) in patients with advanced cancer and chronic hepatitis B/C infection. Clin Immunol 2011;140:130–41.
- Yamasaki K, Horiguchi S, Kurosaki M, et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin Immunol 2011;138:255–65.
- Washah H, Agoni C, Olotu FA, et al. Tweaking α-galactoceramides: probing the dynamical mechanisms of improved recognition for invariant natural killer T-cell receptor in cancer immunotherapeutics. Curr Pharm Biotechnol 2020; In Press.DOI:10.2174/1389201020666191118103342