
Abstract
This paper presents the production and kinetic and inhibitory characterisation of β-carbonic anhydrase from the opportunistic bacterium Staphylococcus aureus (SauBCA). From the eight different carbonic anhydrase (CA) families known to date, humans have only the α-form, whereas many clinically relevant pathogens have β- and/or γ-form(s). Based on this discovery, β- and γ-CAs have been introduced as promising new anti-infective targets. The results of this study revealed that recombinant SauBCA possesses significant CO2 hydration activity with a kcat of 1.46 × 105 s−1 and a kcat/KM of 2.56 × 107 s− 1M−1. Its enzymatic function was inhibited by various sulphonamides in the nanomolar − micromolar range, and the Ki of acetazolamide was 628 nM. The best inhibitor was the clinically used sulfamide agent famotidine (Ki of 71 nM). The least efficient inhibitors were zonisamide and dorzolamide. Our work encourages further investigations of SauBCA in an attempt to discover novel drugs against staphylococcal infections.
Introduction
Staphylococcal species represent a significant part of our microbiota, e.g. on our skin and in our mouth and gutCitation1–3. Most of these species are harmless, commensal bacteria that do not cause inflammation (e.g. S. epidermidis)Citation3. However, one particular strain of staphylococci has been at the centre of attention since our first encounter with it over a century agoCitation4. Despite huge improvements in health care, Staphylococcus aureus has caused increasing morbidity and, in some cases, mortalityCitation4,Citation5. Due to the emergence of multi-drug-resistant strains (collectively termed MRSA, i.e. methicillin-resistant S. aureus), treatment has remained particularly challengingCitation4. The first nosocomial MRSA emerged not too long after Ian Fleming discovered penicillinCitation6,Citation7, and more strains have emerged, causing it to be one of the most prominent causative agents of surgical-site infections and invasive bacterial diseasesCitation8–10. In the past 20 years, S. aureus infections have increased dramatically, causing an increase in MRSA strains displaying resistance to penicillin-derived β-lactam antibioticsCitation11,Citation12. The antibiotic resistance of MRSA is based on the single gene mecA, which encodes a penicillin inactivating enzyme on the surface of the bacteriaCitation13,Citation14. In addition, the virulence of staphylococci is based on their ability to attach to foreign bodies by specialised adhesins and form a biofilm to shield itself from antibioticsCitation15. MRSA is particularly resilient in hospital settings, where patients with diseases such as type I diabetes, immunodeficiencies or ongoing haemodialysis are at the greatest risk of acquiring the infectionCitation12,Citation16,Citation17. Most cases occur through hospital personnel who are infected by their own reservoir or by infected patientsCitation1,Citation18. The infection requires physical contact and is activated when the skin or mucosal barrier is breached, allowing the bacteria to enter adjoining tissues or the bloodstreamCitation4. Infections vary from mild skin lesions to severe cases of sepsis, endocarditis, osteomyelitis and pneumoniaCitation4. Not all individuals show signs of S. aureus infection; hence, it can easily be passed on unnoticed via the skin in addition to from surgical instrumentation and other inanimate surfacesCitation15,Citation19.
Novel innovative therapies are not only needed but also quickly becoming a necessity as the number of antibiotic-resistant pathogen strains increases. Scientists worldwide are thus exploring novel strategies for the prevention of these most threatening pathogenic infections. Among the promising biomolecular targets are carbonic anhydrases (CAs), a group of metalloenzymes found in all lifeforms. These vitally important enzymes catalyse a reversible reaction in which CO2 is converted to bicarbonate ions and protons. This simple reaction is responsible for numerous vital cellular functions, such acid-base homeostasis, CO2 transportation, and photosynthesisCitation20–22. Among the eight evolutionarily divergent but functionally convergent CA gene families (α, β, γ, δ, ζ, η, θ, and ι), humans have only the α-formsCitation23,Citation24. Interestingly, numerous pathogens have been identified with only β- and/or γ-CA genes in their genome. This has been regarded as a promising starting point for discovering novel, specific anti-infectives against pathogens. The fundamental differences discovered between the active sites of different CA families support the idea of specifically targeting β- and/or γ-CAs with minimal effects on human α-CAs. Numerous studies have been conducted demonstrating the efficiency of sulphonamides and anions as CA inhibitors (CAIs)Citation25–31. Detailed characterisation of druggable CAs is considered a prerequisite for the design of more specific and efficient CAIs. To date, several crystal structures of pathogenic β-CAs have been solved and can consequently be exploited for more efficient CAI designCitation32–39. Such structure-based design allows for in vivo targeted therapeutics against pathogenic diseases without the disadvantage of affecting the human or other mammalian CAs.
In the present study, we produced and isolated a novel β-CA from S. aureus (SauBCA) as a recombinant protein and tested its kinetic properties and inhibition profile against several known sulphonamides. Our results demonstrate that SauBCA represents a druggable enzyme target that should be further tested both in vitro and in vivo using different classes of potential CA inhibitors.
Materials and methods
Protein production
The SauBCA gene, obtained from the Universal Protein Resource Database (UniProt, protein entry EZX15767Citation40), was cloned into the expression vector pBVboostFGCitation41 by GeneArt (Thermo Fisher Scientific, Germany). The synthesised insert was composed of Gateway-compatible recombination sites (attL1 and attL2), Shine-Dalgarno and Kozak sequences, N-terminal 6× His-tag with surrounding spacer regions (MSTT and ATAIPTTCitation42), SauBCA, and a thrombin cleavage site (LVPRGS)Citation43. Chemically competent E. coli (OneShot® BL21 Star™ (DE3) cells, #C601003, Thermo Fisher Scientific) were transformed according to the Thermo Fisher Scientific OneShot® BL21(DE3) competent cells manual (part no. 28–0182). The culture medium used was Luria-Bertani (LB) supplemented with 10 mg/mL gentamicin (1:1000, v/v). The cells were grown in a fermenter at 28 °C for 12 h and subsequently induced by adding 1 mM isopropyl β-D-1-isopropyl-thiogalactopyranoside (IPTG). After another 12 h of culture at 25 °C, the cells were harvested by centrifugation at 4000 g for 40 min at 4 °C and mechanically disrupted with an EmulsiFlex-C3 homogeniser (AVESTIN, Canada). Subsequently, the cells were centrifuged at 13000 g for 20 min at 4 °C, and the supernatant was mixed with Protino® Nickel-nitrilotriacetic acid (Ni2+-NTA) agarose affinity chromatography resin (Macherey-Nagel GmbH Co., Germany) and 50 mM Na2HPO4, 0.5 M NaCl and 50 mM imidazole binding buffer (BB; pH 8.0). The incubation took place for 2 h at room temperature (RT) with gentle agitation, followed by overnight incubation at 4 °C without agitation. The resin was loaded into a chromatography column with an EMD Millipore™ vacuum filtering flask (Merck, #XX1004705) and washed generously with BB. The protein was eluted from the column with 50 mM Na2HPO4, 0.5 M NaCl and 350 mM imidazole (pH 7.0). After elution, the eluted fractions were analysed with reducing sodium dodecyl sulphate gel electrophoresis (SDS-PAGE) using a 12% (w/v) polyacrylamide gel and visualised with PageBlue protein staining solution (Thermo Fisher Scientific, #24620). The polypeptide bands on SDS-PAGE were used to identify the protein by means of tandem mass spectrometry (MS/MS, Meilahti Clinical Proteomics Core Facility, University of Helsinki, Finland). A 6× His-tag was enzymatically cleaved using thrombin (#RECOMT, Sigma-Aldrich) according to the Thrombin CleanClive™ kit manual instructions (Sigma-Aldrich). The tag was separated from the core protein with Ni2+-NTA affinity chromatography as mentioned above. Prior to further characterisation, the buffer was exchanged for 50 mM Tris-Cl (pH 7.5).
Kinetics and inhibition studies
An Applied Photophysics stopped-flow instrument was used for assaying CA-catalysed CO2 hydration activityCitation44. Phenol red (at a concentration of 0.2 mM) was used as the pH indicator at the absorbance maximum of 557 nm with 20 mM TRIS (pH 8.4) as the buffer with 20 mM Na2SO4 (for maintaining constant ionic strength) and following the initial rates of the CA-catalysed CO2 hydration reaction for over the period of 10 − 100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and sulphonamide inhibition constants. Six traces of the initial 5 − 10% of the reaction were used to determine the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. A stock solution of inhibitor (0.1 mM) was prepared in distilled-deionized water, and dilutions of up to 0.01 nM were prepared thereafter with distilled-deionized water. Inhibitor (I) and enzyme (E) solutions were preincubated together for 15 min at RT prior to the assay to allow formation of the E − I complex. Inhibition constants were obtained by using the Cheng-Prusoff equation and nonlinear least squares methods (with PRISM 3) and are presented as the means from at least three different determinations. Kinetic constants were obtained using Lineweaver-Burke plots as reported earlierCitation45–47.
Results and discussion
Protein production
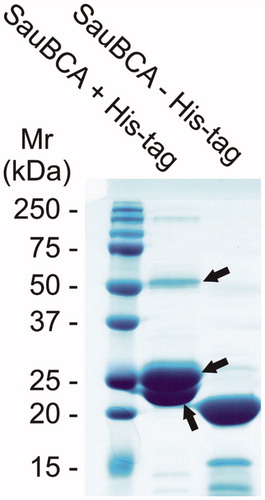
A single β-CA enzyme of S. aureus was identified from the UniProt databaseCitation40 and named SauBCA. The enzyme was successfully expressed in E. coli and purified by affinity chromatography. SDS-PAGE was used to monitor the purification process and successful cleavage of the His-tag. The tag was cleaved prior to further characterisation experiments. The gel from SDS-PAGE was stained with PageBlue and is shown in . The relative molecular mass of the major band seen on the gel after His-tag removal corresponded to approximately 21 kDa. The theoretical molecular mass of SauBCA was calculated to be 21.1 kDa, suggesting that the purification procedure yielded the correct protein. This was further confirmed by MS/MS analysis, where three major polypeptides were analysed and identified as SauBCA. The relatively strong ∼52 kDa polypeptide seemed to represent a dimeric form of the protein.
Figure 1. SDS-PAGE of the purified SauBCA with and without the His-tag. The polypeptides marked with arrows were subjected to MS/MS analysis and identified as SauBCA.
Kinetics
The obtained kinetic parameters for SauBCA are shown in together with human α-CA isoforms and other representative β-CAs for comparison. Kinetic analysis of SauBCA revealed that it is a moderately efficient enzyme with a kcat of 1.46 × 105 s−1 and kcat/KM of 2.56 × 107 s−1M−1. SauBCA showed kinetic properties fairly similar to several human CA isozymes, such as CA I, CA VA, CA VI, CA XII, CA XIII, and CA XIV. Notably, the kcat value of SauBCA was identical to that of hCA XIII, whereas it was the most different from the clinically relevant isozymes hCA II and hCA IX, as well as to the low activity enzyme hCA III. Similar kinetics were observed with the β-CAs from Burkholderia pseudomallei and Flaveria bidentis. As seen in , the binding affinities of SauBCA and Burkholderia pseudomallei β-CA to acetazolamide (AAZ) are rather similar, whereas the inhibition constant for Flaveria bidentis β-CA is very different. This finding suggests major structural differences in the active sites of these β-CA enzymes.
Table 1. Kinetic data of SauBCA and the inhibition results for the standard sulphonamide inhibitor acetazolamide (AAZ). All human isozymesCitation22,Citation48 and two other β-CAsCitation26,Citation27,Citation49 with corresponding properties are shown for comparison. The kcat and kcat/KM values are rounded to one decimal place.
Inhibition studies
A set of clinically used sulphonamide drugs and sulphonamide analogues were investigated against SauBCA. The obtained inhibition constants can be seen in , along with the results from human α-CA isoform II for comparison. The molecular structures of sulphonamides 1–24 and of the clinically used agents tested in this study are shown in .
Figure 2. The molecular structures of the sulphonamide analogues used in this study (1–24) as well as selected clinically used agents.
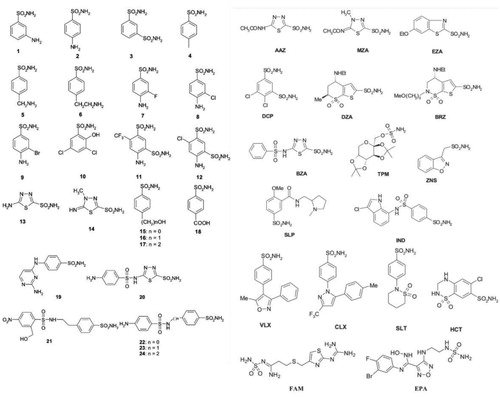
Table 2. Inhibition data for SauBCA and hCA II with sulphonamide analogues 1–24 and selected clinically used agents.
SauBCA was successfully inhibited by selected sulphonamide analogues 1–24 and standard sulphonamide inhibitors AAZ-epacadostat (EPA) in the nanomolar range. In general, all studied compounds resulted in strong to medium Ki values spanning between 71 and 4551 nM. The most efficient inhibition was obtained with sulphonamide analogues 9, 11, 12, 21, 22, 24 and famotidine (FAM). Their inhibition affinities were < 100 nM, which indicates strong inhibition against the SauBCA catalytic function. The least efficient inhibitors were the following sulphonamides: zonisamide (ZNS), dorzolamide (DZA), 19, celecoxib (CLX), methazolamide (MZA), sulthiame (SLT), and brinzolamide (BRZ). Despite showing the lowest efficiency in this study, their inhibition can still be regarded as medium/medium-weak. AAZ ranked 29th out of 41 studied sulphonamides with a Ki of 628 nM. In general, the clinically used sulphonamides showed inferior inhibition compared to designed sulphonamide analogues 1–24, except for FAM, which was in fact the most effective inhibitor. However, FAM (together with EPA) are the only sulfamide derivatives in the series, and FAM has a quite diverse scaffold compared to the other clinically used drugs investigated here. It has a large aliphatic fragment present in the structure, which is absent in the other compounds investigated in this study. Future work entails the design of more potent inhibitors against SauBCA.
Conclusion
In the current study, we successfully produced recombinant SauBCA and investigated its kinetics and inhibition profile with sulphonamides. The data showed that SauBCA possesses significant catalytic activity with a kcat of 1.46 × 105 s−1 and kcat/KM of 2.56 × 107 s−1 M−1. The successful inhibition of SauBCA with selected sulphonamides in the nanomolar range warrants further investigation of potent SauBCA inhibitors for developing future anti-infective agents against staphylococcal infections. One of the most effective inhibitors detected in the study was famotidine, a clinically used antiulcer drug, with a Ki of 71 nM.
Author contributions
All authors designed and conceived the experiments. LJU, MK, SB and AA performed the experiments. All authors analysed the data. LJU wrote the first draft of the manuscript. All authors have given approval to the final version of the manuscript.
Disclosure statement
The authors do not declare any conflicts of interest.
Additional information
Funding
References
- Casewell MW, Hill RL. The carrier state: methicillin-resistant staphylococcus aureus. J Antimicrob Chemother 1986;18( Suppl A):1–12.
- Noble WC, Valkenburg HA, Wolters CH. Carriage of staphylococcus aureus in random samples of a normal population. J Hyg (Lond) 1967;65:567–73.
- Microbiota of the human body. Advances in experimental medicine and biology. Switzerland, Cham: Springer.
- Lowy FD. Staphylococcus aureus infections. N Engl J Med 1998;339:520–32.
- Keynan Y, Rubinstein E. Staphylococcus aureus bacteremia, risk factors, complications, and management. Crit Care Clin 2013;29:547–62.
- Chatterjee SS, Otto M. Improved understanding of factors driving methicillin-resistant staphylococcus aureus epidemic waves. Clin Epidemiol 2013;5:(205–17.
- Deurenberg RH, Stobberingh EE. The evolution of staphylococcus aureus. Infect Genet Evol 2008;8:747–63.
- Petti CA, Sanders LL, Trivette SL, et al. Postoperative bacteremia secondary to surgical site infection. Clin Infect Dis 2002;34:305–8.
- Klein E, Smith DL, Laxminarayan R. Hospitalizations and deaths caused by methicillin-resistant staphylococcus aureus, united states, 1999–2005. Emerging Infect Dis 2007;13:1840–6.
- Kallen AJ, Mu Y, Bulens S, et al. Health care-associated invasive mrsa infections, 2005–2008. JAMA 2010;304:641–8.
- Fuda CC, Fisher JF, Mobashery S. Beta-lactam resistance in staphylococcus aureus: the adaptive resistance of a plastic genome. Cell Mol Life Sci 2005;62:2617–33.
- Tong SYC, Davis JS, Eichenberger E, et al. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 2015;28:603–61.
- Berger-Bachi B. Genetic basis of methicillin resistance in staphylococcus aureus. Cell Mol Life Sci 1999;56:764–70.
- Ubukata K, Nonoguchi R, Matsuhashi M, Konno M. Expression and inducibility in staphylococcus aureus of the meca gene, which encodes a methicillin-resistant s. Aureus-specific penicillin-binding protein. J Bacteriol 1989;171:2882–5.
- Guyot A, Layer G. MRSA - 'bug-bear' of a surgical practice: reducing the incidence of MRSA surgical site infections. Ann R Coll Surg Engl 2006;88:222–3.
- Jacobson MA, Gellermann H, Chambers H. Staphylococcus aureus bacteremia and recurrent staphylococcal infection in patients with acquired immunodeficiency syndrome and aids-related complex. Am J Med 1988;85:172–6.
- Tuazon CU, Perez A, Kishaba T, Sheagren JN. Staphylococcus aureus among insulin-injecting diabetic patients. An increased carrier rate. Jama 1975;231:1272
- Sherertz RJ, Reagan DR, Hampton KD, et al. A cloud adult: The staphylococcus aureus-virus interaction revisited. Ann Intern Med 1996;124:539–47.
- Kramer A, Schwebke I, Kampf G. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect Dis 2006;6:(130
- Supuran CT, De Simone G, Carbonic anhydrases as biocatalysts: from theory to medical and industrial applications. In Carbonic anhydrases as biocatalysts: from theory to medical and industrial applications; 2015.
- Claudiu TS. Carbonic anhydrases as drug targets - an overview. Curr Top Med Chem 2007;7:825–33.
- Supuran CT. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Kikutani S, Nakajima K, Nagasato C, et al. Thylakoid luminal θ-carbonic anhydrase critical for growth and photosynthesis in the marine diatom Phaeodactylum tricornutum. Proc Natl Acad Sci USA 2016;113:9828–33.
- Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J 2019;13:2094–106.
- Syrjanen L, Kuuslahti M, Tolvanen M, et al. The β-carbonic anhydrase from the malaria mosquito Anopheles gambiae is highly inhibited by sulfonamides. Bioorg Med Chem 2015;23:2303–9.
- Vullo D, Del Prete S, Di Fonzo P, et al. Comparison of the sulfonamide inhibition profiles of the beta- and gamma-carbonic anhydrases from the pathogenic bacterium Burkholderia pseudomallei. Molecules 2017;22:421.
- Supuran CT. Bortezomib inhibits bacterial and fungal β-carbonic anhydrases. Bioorg Med Chem 2016;24:4406–9.
- Innocenti A, Hall RA, Schlicker C, et al. Carbonic anhydrase inhibitors. Inhibition of the beta-class enzymes from the fungal pathogens candida albicans and cryptococcus neoformans with aliphatic and aromatic carboxylates. Bioorg Med Chem 2009;17:2654–7.
- Nishimori I, Minakuchi T, Kohsaki T, et al. Carbonic anhydrase inhibitors: the beta-carbonic anhydrase from Helicobacter pylori is a new target for sulfonamide and sulfamate inhibitors. Bioorg Med Chem Lett 2007;17:3585–94.
- Carta F, Maresca A, Covarrubias AS, et al. Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active beta-carbonic anhydrase from mycobacterium tuberculosis, rv3588c. Bioorg Med Chem Lett 2009;19:6649–54.
- Del Prete S, Vullo D, De Luca V, et al. Sulfonamide inhibition studies of the β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem 2016;24:1115–20.
- Murray AB, Aggarwal M, Pinard M, et al. Structural mapping of anion inhibitors to β-Carbonic Anhydrase psCA3 from Pseudomonas aeruginosa. ChemMedChem 2018;13:2024–9.
- Ferraroni M, Del Prete S, Vullo D, et al. Crystal structure and kinetic studies of a tetrameric type ii β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Crystallogr D Biol Crystallogr 2015;71:2449–56.
- Dostal J, Brynda J, Blaha J, et al. Crystal structure of carbonic anhydrase cance103p from the pathogenic yeast candida albicans. BMC Struct Biol 2018;18:14
- Schlicker C, Hall RA, Vullo D, et al. Structure and inhibition of the co2-sensing carbonic anhydrase can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol 2009;385:1207–20.
- Cronk JD, Rowlett RS, Zhang KY, et al. Identification of a novel noncatalytic bicarbonate binding site in eubacterial beta-carbonic anhydrase. Biochemistry 2006;45:4351–61.
- Huang S, Xue Y, Sauer-Eriksson E, et al. Crystal structure of carbonic anhydrase from Neisseria gonorrhoeae and its complex with the inhibitor acetazolamide. J Mol Biol 1998;283:301–10.
- Suarez Covarrubias A, Larsson AM, Hogbom M, et al. Structure and function of carbonic anhydrases from mycobacterium tuberculosis. J Biol Chem 2005;280:18782–9.
- Pinard MA, Lotlikar SR, Boone CD, et al. Structure and inhibition studies of a type ii beta-carbonic anhydrase psca3 from Pseudomonas aeruginosa. Bioorg Med Chem 2015;23:4831–8.
- Uniprot UniProt Consortium01.03.2017].
- Laitinen OH, Airenne KJ, Hytonen VP, et al. A multipurpose vector system for the screening of libraries in bacteria, insect and mammalian cells and expression in vivo. Nucleic Acids Res 2005;33:e42.
- Piao S, Xu Y, Ha NC. Crystallization and preliminary x-ray crystallographic analysis of MacA from Actinobacillus actinomycetemcomitans. Acta Crystallogr Sect F Struct Biol Cryst Commun 2008;64:391–3.
- Hilvo M, Baranauskiene L, Salzano AM, et al. Biochemical characterization of ca ix, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799–809.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Del Prete S, Bua S, Supuran CT, Capasso C. Escherichia coli γ-carbonic anhydrase: characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. J Enz Inhib Med Chem 2020;35:1545–54.
- Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev 2020.
- De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- Hilvo M, Innocenti A, Monti SM, et al. Recent advances in research on the most novel carbonic anhydrases, ca xiii and xv. Curr Pharm Des 2008;14:672–8.
- Monti SM, De Simone G, Dathan NA, et al. Kinetic and anion inhibition studies of a β-carbonic anhydrase (FbiCA 1) from the C4 plant Flaveria bidentis. Bioorg Med Chem Lett 2013;23:1626–30.