
Abstract
New cyanobenzofurans derivatives 2–12 were synthesised, and their antiproliferative activity was examined compared to doxorubicin and Afatinib (IC50 = 4.17–8.87 and 5.5–11.2 µM, respectively). Compounds 2 and 8 exhibited broad-spectrum activity against HePG2 (IC50 = 16.08–23.67 µM), HCT-116 (IC50 = 8.81–13.85 µM), and MCF-7 (IC50 = 8.36–17.28 µM) cell lines. Compounds 2, 3, 8, 10, and 11 were tested as EGFR-TK inhibitors to demonstrate their possible anti-tumour mechanism compared to gefitinib (IC50 = 0.90 µM). Compounds 2, 3, 10, and 11 displayed significant EGFR TK inhibitory activity with IC50 of 0.81–1.12 µM. Compounds 3 and 11 induced apoptosis at the Pre-G phase and cell cycle arrest at the G2/M phase. They also increased the level of caspase-3 by 5.7- and 7.3-fold, respectively. The molecular docking analysis of compounds 2, 3, 10, and 11 indicated that they could bind to the active site of EGFR TK.
1. Introduction
Cancer remains among the leading causes of death worldwide owing to the resistance of cancer cells to the existing anti-tumour agentsCitation1–3. Treatment of cancer is a significant challenge for medicinal chemists due to the pressing need for novel and effective anti-cancer drugsCitation4–10. Moreover, receptor tyrosine kinases (RTK) play crucial roles in activating signal transduction pathways in the cell, resulting in cell division, differentiation, and activation of regulatory mechanismsCitation11,Citation12. Epidermal growth factor receptor tyrosine kinase (EGFR-TK) is a tyrosine kinase receptor of the ErbB family. It regulates numerous biological processes, including cell motility, adhesion, regulation, angiogenesis, apoptosis, and metastasisCitation13–19. Notably, overexpression of these receptors is found in various cancer cells (e.g. colon, ovarian, prostate, and breast cancer cells)Citation17–20. Hence, simultaneous inhibition of EGFR is expected to provide superior efficacy to single receptor targeting, making EGFR a critical target for the design and development of anti-tumour agentsCitation15,Citation21–26. In recent years, afatinib (I), and gefitinib (II), have been reported as effective EGFR inhibitors for the treatment of several cancer types ()Citation27–32. It has been found that compounds are possessing a nitrile moiety exhibit exciting and diverse biological activitiesCitation33,Citation34. For instance, neratinib (IV), pelitinib (V), and bosutinib (VI) are tyrosine kinase inhibitors incorporating nitrile groups. They have been shown to be effective in the treatment of breast cancer, solid tumours, and chronic myelogenous leukaemia, respectively ()Citation35–37. Intriguingly, the benzofuran core is one of the essential oxygen-containing scaffolds. Valuable therapeutic agents can be obtained by integrating suitable pharmacophores on the benzofuran moietyCitation38,Citation39. Recent studies indicated that benzofuran derivatives possessed anti-tumour activityCitation39–41. In addition, benzofuran-containing compounds have been demonstrated to be kinase inhibitors. For instance, compound VII exhibited good inhibitory activity against c-Src, while chalcone-benzofuran XIII was a strong inhibitor of vascular endothelial growth factor receptor 2 (VEGR-2) ()Citation42,Citation43. Recently, compounds incorporating a benzofuran core, such as derivatives IX, X, and XI, displayed significant EGFR TK inhibitory activity compared to the reference drug erlotinib. Compounds IX, X, and XI could effectively induce apoptosis ()Citation26,Citation44,Citation45. Furthermore, hybridisation of benzofuran with 4-aminoquinoline afforded compound XII, which showed inhibitory activity against EGFR ()Citation46.
Figure 1. The reported anti-tumour agents with inhibitory activity against EGFR.
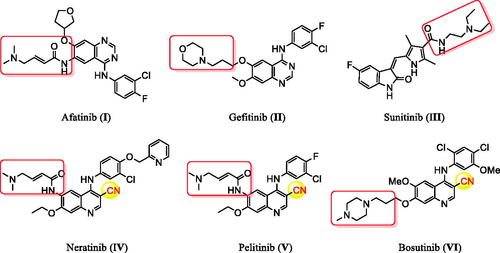
Figure 2. Previously reported benzofuran derivatives VII–XII with anti-tumour and kinase inhibition activities as well as compounds 2–12 designed herein.
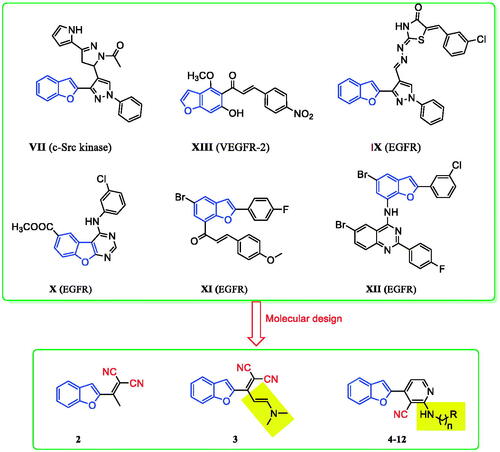
Considering the above results, in this work, a series of benzofuran scaffolds 2–12 () was synthesised based on bioisosteric modifications of compounds shown in and . In the prepared derivatives, the benzofuran core was linked with alkylnitrile or nicotinonitrile moieties. The anti-cancer activity of the designed compounds was analysed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The most active compounds were also evaluated against the target EGFR TK. Moreover, the induction of apoptosis and the effects of the most active derivatives on the caspase-3 level were assessed using a flow cytometry technique. The cell cycle activity was also detected for the most potent compounds to determine the possible cell cycle stage at which the new derivatives could suppress the growth of cancer cells. Lastly, molecular modelling was conducted to explore the plausible binding modes of the most promising derivatives in the binding site of EGFR.
2. Results and discussion
2.1. Chemistry
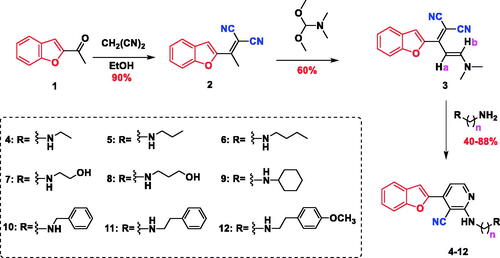
The synthetic pathway adopted to prepare the novel series of benzofuran-incorporating nitrile derivatives is shown in Scheme 1. Knoevenagel condensation of 2-acetyl benzofuran (1) in an ethanolic solution of malononitrile afforded 2-(1-(benzofuran-2-yl)ethylidene)malononitrile (2), which reacted with N,N-dimethylformamide dimethylacetal (DMFDMA) to give (E)-2-(1-(benzofuran-2-yl)-3-(dimethylamino)allylidene)malononitrile (3). The subsequent condensation reaction with different primary amines yielded 4-(benzofuran-2-yl)-2-(substituted)-nicotinonitriles 4–12. The structure of compound 2 was confirmed by infra-red (IR) spectroscopy, which showed absorption bands at 2222 cm−1 (CN) and 1572 cm−1 (C=C). Additionally, the disappearance of a band at 1680 cm−1 was detected (C=O). The 13C nuclear magnetic resonance (NMR) spectrum revealed the disappearance of carbon signals at 186 ppm (C=O) as well as the appearance of two peaks at 113.24 and 113.60 ppm, which were attributed to two nitrile groups. Moreover, the presence of the methyl group of the ethylidene moiety (CH3–C=C) was confirmed by the singlet peaks at 2.63 and 19.38 ppm in the 1H NMR and 13C NMR spectra, respectively. The structure of compound 2 was verified by elemental analysis and mass spectrometry, which showed a molecular ion peak (M+) at m/z 208. The NMR spectrum of compound 3 was characterised by the absence of the methyl signal of the ethylidene moiety (CH3–C=C) at 2.63 and 19.38 ppm. In addition, two singlet peaks corresponding to the dimethylamino group (N(CH3)2) were observed at 3.09 and 3.23 ppm in the 1H NMR spectrum as well as at 37.86 and 45.77 ppm in the 13C NMR spectrum. The structures of compounds 4–12 were confirmed by IR, 1H NMR, 13C NMR, and mass spectrometry data. The IR spectra showed absorption signals at 3343–3370 and 2209–2219 cm−1 due to the presence of (NH) and (CN) groups, respectively. Moreover, the peaks corresponding to the dimethylamino group (N(CH3)2) at 3.09 and 3.23 ppm in the 1H NMR spectra as well as at 37.86 and 45.77 ppm in the 13C NMR spectra of compounds 4–12 disappeared. However, the signals ascribed to the (NH) group were detected at 6.66–7.18 ppm. The presence of the aliphatic residue was confirmed by the peaks at 0.90–4.56 and 13.49–59.28 ppm in the 1H NMR and 13C NMR spectra, respectively.
Scheme 1. Synthetic route for the preparation of cyanobenzofuran derivatives 2–12.
2.2. Biological screening
2.2.1. In vitro antiproliferative activity and structure activity relationship (SAR)
The antiproliferative activity of the newly synthesised compounds 2–12 against five human cancer cell lines, including hepatocellular carcinoma (HePG2), colorectal carcinoma (HCT-116), human breast adenocarcinoma (MCF-7), human prostate carcinoma (PC3), cervical carcinoma (HeLa), and normal cell (WI38) was evaluated by an MTT assay employing a previously described procedureCitation47,Citation48. Doxorubicin (DOX) and Afatinib were used as positive control. The antiproliferative activity of the tested compounds is summarised in and . DOX and Afatinib exhibited IC50 values of (4.50 and 5.53 µM), (5.23 and 11.23 µM), (4.17 and 7.23 µM), (8.87 and 7.6 3 µM), and (5.57 and 6.3 µM) against HePG2, HCT-116, and MCF-7, PC3, and HeLa cells, respectively. Compound 2, which possessed two nitrile groups, showed strong antiproliferative activity, with IC50 values of 8.81 and 8.36 µM against HCT-116, and MCF-7 cell lines, respectively. However, 2 showed moderate activity against HePG2 with an IC50 value of 16.08 µM, and weak activity against PC3 and HeLa cell lines, with IC50 values of 26.82 and 39.03 µM, respectively. Enaminonitrile 3 displayed strong antiproliferative activity against the HCT-116 cell line (IC50 of 10.84 µM) and weak activity against the other tested cell lines. The benzofuran–nicotinonitrile derivatives 4–12 bearing secondary amine side chains exhibited varying antiproliferative activity. Derivative 4 with an ethylamine fragment showed strong antiproliferative activity against the PC3 and good activity against HeLa cell lines, with IC50 values of 14.27 and 21.10 µM, respectively. Nonetheless, compound 4 only weak activities were observed against HePG2 and MCF-7 cell lines (IC50 values of 58.75 and 55.04 µM, respectively). Notably, elongation of the ethyl chain (e.g. compound 4, IC50 = 14.27–58.75 µM) to a propyl (e.g. derivative 5) or a butyl moiety (e.g. compound 6) led to decreased antiproliferative activity against HePG2, MCF-7, PC3, and HeLa cell lines, with IC50 values in the range of 75.19–100.0 µM. In contrast, derivative 6 showed better activity against the HCT-116 cell line than 4 and 5 with IC50 values of 56.36, 72.4, and 94.14 µM, respectively. The introduction of a hydroxyl group in compound 4 gave derivative 7. It increased the antiproliferative activity against HePG2, HCT-116, and MCF-7 cell lines, with IC50 values of (58.75, 72.40, and 55.04 µM) (53.43, 38.64, and 40.83 µM), respectively. Conversely, the PC3 and HeLa cell lines were less sensitive to compound 7 than derivative 4 with IC50 (70.10 and 73.29 µM) and (14.27 and 21.10 µM). Intriguingly, introducing a hydroxyl group in compound 5 afforded derivative 8 and led to increased antiproliferative activity against all tested cell lines (IC50 range of 75.19 to >100.0 µM and 13.85 to 58.76 µM, respectively).
Figure 3. Structure activity relationship of benzofuran–nicotinonitrile derivatives as anti-cancer agents.
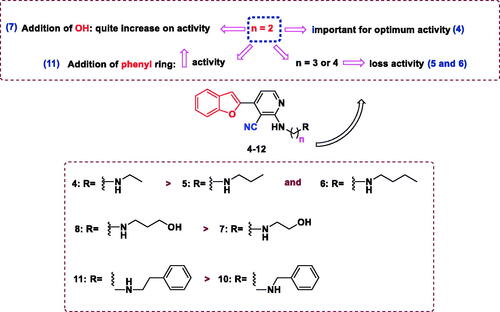
Table 1. In vitro antiproliferative activities (IC50, µM)a,b of the synthesised compounds 2–12.
Similarly, compared with derivative 7, compound 8 exhibited a drastic increase in the antiproliferative activity against all evaluated cell lines (IC50 range of 38.64–73.29 µM and 13.85–58.76 µM, respectively). Replacement of the ethyl moiety in compound 4 with a cyclohexyl fragment (compound 9) increased the antiproliferative activity against HePG2, HCT-116, and MCF-7 cell lines with IC50 values of (58.75, 72.40, and 55.04 µM) and 44.70, 29.52 and 35.17 µM), respectively. Furthermore, PC3 and HeLa cell lines were more susceptible to derivative 4 than compound 9 IC50 values of (14.27 and 21.0 µM) and 51.06–73.59 µM), respectively. Moreover, benzylamine derivative 10 comparatively inhibited the growth of MCF-7 cells (IC50 value of 19.69 µM) and showed weak activity against the other tested cell lines. It is noteworthy that the replacement of the benzyl moiety in compound 10 with a phenylethyl fragment gave compound 11 and resulted in good antiproliferative activity against HePG2, MCF-7, and PC3 cell lines, with IC50 values of (34.32, 19.69, and 30.90 µM) and (20.43, 14.55 and 18.75 µM), respectively. Introduction of a 4-methoxyl moiety in compound 11 afforded derivative 12, which displayed weak antiproliferative activity. The cytotoxic activity of the new compounds was also examined against normal W138 fibroblast cell to study the safety of the newly synthesised compounds, using (MTT) colorimetric assay (). The tested compounds did not display cytotoxicity towards W138 cells (IC50 values of 59.49–204.00 µM) compared to doxorubicin (IC50 values of 55.29 µM).
2.2.2. EGFR TK inhibition assay
The most active derivatives, that is, 2, 3, 8, 10, and 11, were subjected to the EGFR TK inhibition assayCitation7,Citation10,Citation23–25. The results revealed that several of the tested compounds were promising EGFR TK inhibitors (). It was evident that compounds 2, 3, 10, and 11 exhibited strong inhibitory activities against EGFR (IC50 values of 1.09, 0.93, 1.12, and 0.81, respectively). Notably, this activity was comparable to that of the reference drug gefitinib (IC50 value of 0.90 µM). It was observed that all of the tested derivatives showed 50% inhibition against EGFR of less than 1.2 µM, except compound 8, which was found to be the least effective EGFR inhibitor (IC50 = 4.24 µM). It was determined that compounds incorporating a phenethyl moiety, such as derivative 11, displayed higher inhibitory activity against EGFR than the corresponding compounds containing a benzyl fragment (e.g. 10) or a propanol group (e.g. 8).
Table 2. IC50 values of compounds 2, 3, 8, 10, and 11 against EGFR.
2.2.3. Caspase-3 assay and induction of apoptosis
Caspases are critical mediators of programmed cell death, that is, apoptosisCitation49. Caspase-3 is important in processes involving dissociation of the cell and the formation of the apoptotic element; therefore, it is regarded as one of the best biochemical hallmarks of apoptosisCitation49. This, to examine the apoptotic activity of compounds 3 and 11, the level of caspase-3 was measured after treating the HCT-116 and MCF-7 cells with 3 and 11, respectively (). The concentration of active caspase-3 was measured using the ELISA techniqueCitation49. In addition, the fluorescence density produced by the tested compounds is illustrated in . Interestingly, compound 3 significantly induced apoptosis in HCT-116 cells after 24 h of treatment. The level of caspase-3 increased 5.7-fold compared to the control. Moreover, a considerable 7.3-fold increase in the caspase-3 level was detected following the treatment of the MCF-7 cells with compound 11. The bioluminescent intensities of caspase-3 indicated the apoptotic activity of compounds 3 and 11.
Figure 4. Caspase-3 enzyme assay for compounds 3 and 11.
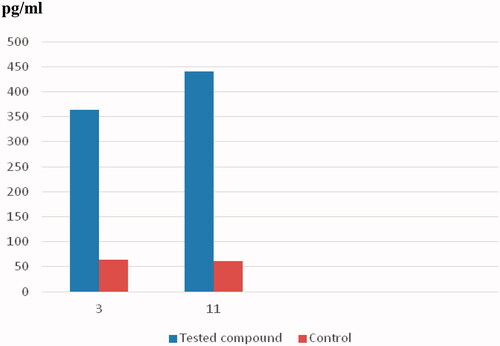
Table 3. Effects of compounds 3 and 11 on the levels of human caspase-3.
2.2.4. Cell cycle arrest analysis and detection of apoptosis
The cell cycle is a sequence of growth and development steps that lead to DNA replication and cell division. It consists of four distinct phases: the G1 phase, S phase (synthesis), G2 phase, and M phaseCitation23,Citation24,Citation50–52. Apoptosis, that is, programmed cell death, is considered an important target of the most anti-tumour agents, resulting in G2/M arrestCitation23,Citation24,Citation50–52. Our promising derivatives 3 and 11 were subjected to cell cycle analysis and an apoptotic assay to investigate their roles in the cell cycle progression of HCT-116 and MCF-7 cells, respectively. To better characterise the mode of cell death induced by the tested compounds, following treatment of the HCT-116 and MCF-7 cells with compounds 3 and 11 at a concentration of 10 µM for 24 h, respectively, the cells were stained with propidium iodide (PI). The DNA contents were measured by flow cytometry ( and ; ). Compared with the control, which was treated with DMSO, following treatment of HCT-116 and MCF-7 cells with compounds 3 and 11, the cell proportion at the S phase decreased to 20.91% and 21.36%, respectively. In addition, compounds 3 and 11 increased the cell proportion at the G2/M phase to 12.62% and 15.28%, respectively, compared to the control cells (4.19% and 3.66%, correspondingly). These results indicated that the cells were arrested at the G2/M phase. Furthermore, the pre-G1 population was detected following treatment with compounds 3 and 11 (13.06% and 16.25% compared to 0.39% and 0.55% in the control cells, respectively). Moreover, annexin-5/PI stainingCitation23,Citation24,Citation52 was performed for compounds 3 and 11 using HCT-116 and MCF-7 cells. A comparison was made to the control (DMSO) and reference (gefitinib). The gefitinib results showed an early apoptosis of 7.22% (HCT-116) and 4.28% (MCF-7), whereas the values for late apoptosis were 7.47% (HCT-116) and 7.63% (MCF-7). The results demonstrated in , and suggest an increase in the early apoptosis from 0.16% (control sample in DMSO) to 5.95% for compound 3. In contrast, derivative 11 showed an increase in the early apoptosis to 6.89%. Compounds 3 and 11 increased the late apoptosis from 0.11% (DMSO) to 5.79% and 8.13%, respectively. It was also evident that 3 and 11 preferentially activated the apoptotic pathway rather than the necrotic pathway. This induced action was the result of the cell cycle arrest at the G2/M phase.
Figure 5. Determination of apoptosis in the HCT-116 cell line and analysis of the cell cycle arrest using flow cytometry. (A) Effect of compound 3 on the cell cycle distribution of HCT-116. (B) Apoptosis effect on the human HCT-116 cell line induced by compound 3.
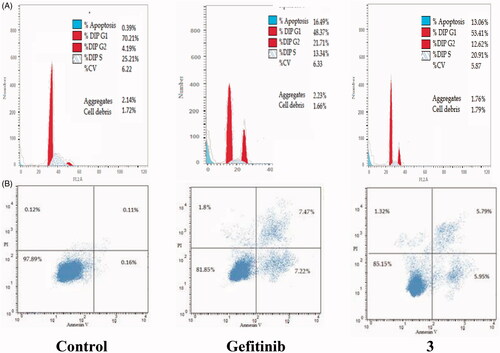
Figure 6. Determination of apoptosis in the MCF-7 cell line and analysis of the cell cycle arrest using flow cytometry. (A) Effect of compound 11 on the cell cycle distribution of MCF-7. (B) Apoptosis effect on the human MCF-7 cell line induced by compound 11.
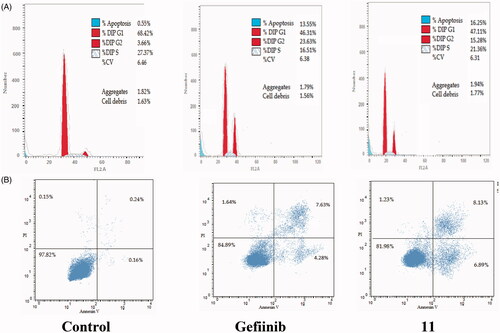
Figure 7. Cell cycle arrest analysis of compounds 3 and 11 in comparison with gefitinib.
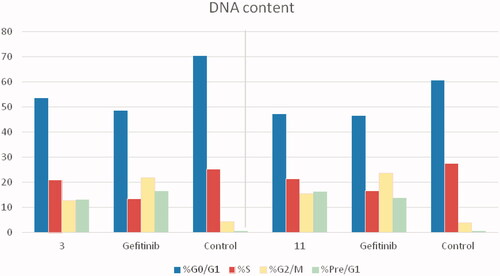
Figure 8. Percentage of apoptosis for compounds 3 and 11 in comparison with gefitinib and control cells.
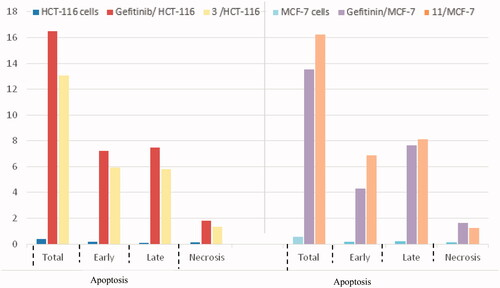
Table 4. Analysis of the effects of compounds 3 and 11 on the cell cycle progression in HCT-116 and MCF-7 cells, respectively, using flow cytometry.
Table 5. Apoptotic activity of compounds 3 and 11 in HCT-116 and MCF-7 cell lines, respectively.
2.2.5. Molecular modelling study
Molecular modelling is a tool used to inspect bioactive molecules within a putative binding site of a particular enzyme or receptorCitation53–57. It can also be employed for studying the molecular structure and structural activity relationship of different moleculesCitation6,Citation58,Citation59. In this study, the MOE 2008.10 software obtained from the Chemical Computing Group Inc. (Montreal, QC, Canada) was used for the docking protocol. The docked compounds and the co-bound inhibitor were docked into the putative binding site of the protein to generate an appropriate binding orientation. Molecular docking of the most active compounds 2, 3, 10, and 11 was conducted to explore their binding modes and interactions with the constitutive amino acids in the active site of EGFR. Molecular operating environment (MOE) software version 2008.10 was used for the analysis (). The crystal structure of the EGFR TK receptor in complex with erlotinib was obtained from the RCSB protein data bank (PDB ID: 1M17) and was utilised to establish the starting docking model of EGFR TKCitation60. The quinazoline core of the erlotinib inhibitor exhibited a hydrogen bond with Met769Citation60. Erlotinib was subjected to one run of docking into the binding site to verify and validate the docking process. Docking of 2, 3, 10, and 11 revealed that all compounds fit into the enzyme active site almost at the same position as erlotinib (). One of the nitrile groups of compound 2 (S = −8.53 kcal/mol) is bound to the active site of EGFR TK through hydrogen bonding with the vital amino acid Met769. The second nitrile moiety interacted with the amino acid residues Thr766 and Cys751 through water-mediated hydrogen bonding.
Figure 9. 2D binding modes and residues involved in the recognition of the most active compounds docked and minimised in the EGFR binding pocket: Compound 2 (upper left panel), 3 (upper right panel), 10 (lower left panel), and 11 (lower right panel).
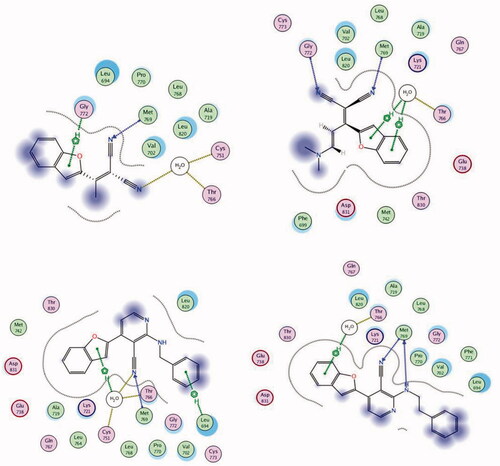
Moreover, the benzofuran core is connected to Gly772 by cation–H interactions. Derivative 3 (S = −9.38 kcal/mol) interacted with residues Met769 and Gly772 via hydrogen bonding using its two nitrile moieties. The benzofuran ring of 3 bound to Thr766 by water-mediated arene–H interactions. The nitrile moiety in compound 10 (S = −9.61 kcal/mol) connected to Met769 by hydrogen bonding, while the benzofuran ring interacted with amino acid residues Thr766 and Cys751 via water-mediated cation–π bonding. In addition, the benzyl fragment is bound to residue Leu694 by cation–π interactions. The best docking score was achieved for compound 11 (−10.38 kcal/mol). 11 bound to the active site of EGFR TK through two hydrogen bonds to the critical amino acid Met769 using the N atom of the nitrile moiety and the N atom of phenethylamine. Lastly, the benzofuran core bound to Thr766 by water-mediated arene–H interactions ().
3. Conclusion
To develop potent anti-tumour agents, a series of cyanobenzofuran hybrids were designed and synthesised in this work. The in vitro antiproliferative activity of the prepared compounds was evaluated for HePG2, HCT-116, MCF-7, PC3, and HeLa cancer cell lines. The biological results revealed that compounds 2, 3, 10, and 11 exhibited a broad spectrum antiproliferative activity against selected cell lines. Moreover, the most active derivatives were further evaluated for their inhibitory activity against EGFR kinase. Compounds 3 and 11 displayed significant EGFR TK inhibitory activity (IC50 0.93 and 0.81 µM, respectively) even compared to the reference drug gefitinib (IC50 0.9 µM). Compounds 2 and 10 also showed good EGFR TK inhibitory activity (IC50 1.09 and 1.12 µM, respectively). The apoptosis assay and cell cycle analysis results demonstrated that derivatives 3 and 11 induced apoptosis of cancer cells and arrested the cell cycle at the G2/M phase. In addition, 3 and 11 led to an increase in caspase-3 by 5.7- and 7.3-fold, respectively. These outcomes indicated that the potent pro-apoptotic activity of compounds 3 and 11 was a result of the induction of the intrinsic apoptotic pathway rather than the necrotic pathway. Compared to erlotinib, the molecular docking analysis of the most active compounds 2, 3, 10, and 11 showed good fitting and suitable interactions with the key amino residues in the binding site of the EGFR kinase. The presence of the cyano group in the compounds enabled hydrogen bonding interactions with the Met769 amino acid. Additionally, the benzofuran moiety exhibited van der Waals interactions with the EGFR binding site. Based on these findings, it can be concluded that derivatives 3 and 11 are promising scaffolds for further modification and optimisation to obtain potent and selective anti-tumour agents with EGFR inhibitory activity.
4. Experimental
4.1. Chemistry
All melting points (°C) were recorded using a Fisher–John melting point apparatus and were uncorrected. The IR spectra were determined for KBr discs on a Thermo Fischer Scientific Nicolet IS10 spectrometer (wavenumber in cm−1) at the Faculty of Pharmacy, Mansoura University, Egypt. The 1H NMR spectra were obtained in DMSO-d6 or CDCl3 employing a Jeol 500 MHz spectrometer at the Faculty of Science, Mansoura University, Egypt. The 13C NMR spectra were obtained in DMSO-d6 using a Jeol 500 MHz spectrometer at the Faculty of Science, Mansoura University, Egypt. Electron ionisation mass spectrometry (EI MS) was performed on a Hewlett Packard 5988 spectrometer at the Al-Azhar University, Cairo, Egypt. Microanalyses (C, H, N) were conducted at the Microanalytical Unit, Cairo University, and the results were within ±0.4% of the theoretical values. The antiproliferative screening of all newly synthesised compounds, enzyme activity inhibition assay, caspase-3 assay, apoptosis induction analysis, and cell cycle analysis was conducted at the Holding Company for Biological Products and Vaccines (VACSERA), Cairo, Egypt.
4.1.1. 2-(1-Benzofuran-2-yl)ethylidene)malononitrile (2)
A solution of 2-acetylbenzofuran (1) (0.32 g, 2.0 mmol) and CH2(CN)2 (malononitrile) (0.132 g, 2.0 mmol) in EtOH (15 ml) was refluxed for 24 h. Yellow crystals were formed after cooling and evaporating ethanol under reduced pressure. The crystals were collected by filtration and recrystallized from EtOH to afford the titled compound crystalline yellow needles.
Yield = 90%, m.p. = 140–142 °C, crystalline yellow needles; IR (KBr, cm−1): 3018 (=C–H), 2222 (CN), 1610 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 2.63 (s, 3H, CH3), 7.42 (dd, J = 8.9, 8.9 Hz, 1H, ArH), 7.60 (dd, J = 9.3, 9.3 Hz, 1H, ArH), 7.70 (d, J = 10.5 Hz, 1H, ArH), 7.87 (d, J = 9.6 Hz, 1H, ArH), 8.08 (s, 1H, ArH); 13C NMR (125 MHz, DMSO-d6): δ = 19.38, 78.23, 111.56, 113.24, 113.60, 117.39, 123.23, 124.28, 126.83, 129.83, 149.91, 155.11, 156.94; MS (EI) m/z (C13H8N2O): 209.08 (M + 1, 60.5%), 208 (M+, 39.53%), 18.05 (20.28%), 167.08 (13.06%), 153.08 (7.61%), 143.04 (4.71%), 118.06 (6.95%), 94.07 (15.20%), 67.08 (6.83%), 44.03 (25.09%); Elemental Analysis for C13H8N2O, Calcd.: C, 74.8; H, 3.87; N, 13.45; Found: C, 74.98; H, 3.88; N, 13.46.
4.1.2. (E)-2-(1-(benzofuran-2-yl)-3-(dimethylamino)allylidene)malononitrile (3)
DMFDMA (2.69 g, 3 ml, 22.57 mmol) was added to compound 2 (0.1 g, 0.48 mmol) and stirred at room temperature overnight in a solvent free environment. The mixture was then washed with diethyl ether and filtered to give crystalline yellow needles. Yield= 60%, m.p. = 161–163 °C, crystalline yellow needles, IR (KBr, cm−1): 3060 (=C–H), 2197 (CN), 1610 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 3.09 (s, 3H, CH3), 3.23 (s, 3H, CH3), 5.70 (d, J = 12.0 Hz, 1H, CH), 7.35 (t, J = 7.4 Hz, 1H, ArH), 7.43 (s, 1H, ArH), 7.46 (m, 1H, ArH), 7.68 (d, J = 8.7 Hz, 1H, ArH), 7.72 (d, J = 12.1 Hz, 1H, CH), 7.78 (d, J = 7.7 Hz, 1H, ArH); 13C NMR (125 MHz, DMSO-d6): δ = 37.86, 45.77, 95.72, 111.81, 112.18, 122.28, 123.69, 126.46, 127.42, 148.44, 154.83, 156.05, 157.59; Elemental Analysis for C16H13N3O, Calcd.: C, 72.8; H, 4.98; N, 15.96; Found: C, 72.98; H, 4.8; N, 15.97.
4.1.3. General procedure for the preparation of 4-(benzofuran-2-yl)-2-(substituted amino)nicotinonitriles 4–12
A mixture of compound 3 (0.2 g, 0.7 mmol) and an appropriate primary amine (2 ml) was refluxed for one h and stirred at room temperature overnight. The reaction mixture was diluted with CH2Cl2 (20 ml) and washed with brine (5 ml). The aqueous layer was extracted with CH2Cl2 (20 ml × 3), and the organic extracts were dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The obtained residue was purified by column chromatography (SiO2:CH2Cl2/MeOH = 30:1) to give the titled products 4–12 as yellow solids.
4.1.3.1. 4-(Benzofuran-2-yl)-2-(ethylamino)nicotinonitrile (4)
Yield = 40%, m.p. = 155–158 °C, yellow powder, IR (KBr, cm−1): 3350 (NH), 2215 (CN); 1H NMR (500 MHz, DMSO-d6): δ = 1.15 (t, J = 7.1 Hz, 3H, CH3), 3.46 (q, J = 5.6 Hz, 2H, CH2), 7.15 (d, J = 5.3 Hz, 1H, NH), 7.25 (t, J = 5.4 Hz, 1H, ArH), 7.35 (t, J = 7.8 Hz, 1H, ArH), 7.47 (dd, J = 11.4, 11.4 Hz, 1H, ArH), 7.69 (d, J = 8.2 Hz, 1H, ArH), 7.83 (d, J = 7.8 Hz, 1H, ArH), 7.86 (s, 1H, ArH), 8.36 (d, J = 5.3 Hz, 1H, ArH); Elemental Analysis for C16H13N3O, Calcd.: C, 72.8; H, 4.98; N, 15.96; Found: C, 72.97; H, 4.8; N, 15.95.
4.1.3.2. 4-(Benzofuran-2-yl)-2-(propylamino)nicotinonitrile (5)
Yield = 88%, m.p. = 118–120 °C, yellow powder, IR (KBr, cm−1): 3361 (NH), 2212 (CN), 1583 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 0.90 (t, J = 7.4 Hz, 3H, CH3), 1.59 (sextet, J = 7.4 Hz, 2H, CH2), 3.39 (td, J = 7.1, 7.5 Hz, 2H, CH2), 7.15 (d, J = 5.2 Hz, 1H, NH), 7.26 (t, J = 5.6 Hz, 1H, ArH), 7.36 (t, J = 7.4 Hz, 1H, ArH), 7.47 (m, 1H, ArH), 7.70 (d, J = 8.7 Hz, 1H, ArH), 7.83 (d, J = 7.7 Hz, 1H, ArH), 7.87 (s, 1H, ArH), 8.37 (d, J = 5.2 Hz, 1H, ArH); Elemental Analysis for C17H15N3O, Calcd.: C, 73.63; H, 5.45; N, 15.15; Found: C, 73.64; H, 5.46; N, 15.17.
4.1.3.3. 4-(Benzofuran-2-yl)-2-(butylamino)nicotinonitrile (6)
Yield = 60%, m.p. = 112–114 °C, yellow powder, IR (KBr, cm−1): 3354 (NH), 2865 (C–H), 2215 (CN), 1588 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 0.90 (t, J = 7.4 Hz, 3H, CH3), 1.32 (sextet, J = 7.5 Hz, 1H, ArH), 1.55 (tt, J = 7.5 Hz, 2H, CH2), 3.42 (d, J = 6.3 Hz, 2H, CH2), 7.13 (d, J = 5.3 Hz, 1H, NH), 7.22 (s, 1H, ArH), 7.35 (d, J = 7.8 Hz, 1H, ArH, 7.54 (m, 1H, ArH), 7.69 (d, J = 8.8 Hz, 1H, ArH), 7.82 (d, J = 7.8 Hz, 1H, ArH), 7.85 (d, J = 0.7 Hz, 1H, ArH), 8.35 (d, J = 5.2 Hz, 1H, ArH); 13C NMR (125 MHz, DMSO-d6): δ = 13.49, 19.29, 30.63, 40.33, 83.8, 107.77, 108.59, 111.18, 116.42, 122.15, 123.52, 126.49, 127.43, 140.12, 149.75, 152.57, 153.91, 159.23; Elemental Analysis for C18H17N3O, Calcd.: C, 74.20; H, 5.88; N, 14.42; Found: C, 74.21; H, 5.89; N, 14.40.
4.1.3.4. 4-(Benzofuran-2-yl)-2-(2-hydroxyethylamino)nicotinonitrile (7)
Yield= 70%, m.p. = 164–166 °C, yellow powder, IR (KBr, cm−1): 3000 (OH), 3344 (NH), 2938, 2219 (CN), 1591 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 3.51 (td, J = 7.5, 5.1 Hz, 2H, CH2), 3.56 (td, J = 7.5, 5.2 Hz, 2H, CH2), 4.79 (t, J = 5.2 Hz, 1H, OH), 7.04 (t, J = 5.1 Hz, 1H, NH), 7.17 (d, J = 5.3 Hz, 1H, ArH), 7.35 (t, J = 7.4 Hz, 1H, ArH), 7.47 (m, 1H, ArH), 7.70 (d, J = 8.8 Hz, 1H, ArH), 7.83 (d, J = 7.7 Hz, 1H, ArH), 7.87 (d, J = 0.8 Hz, 1H, ArH), 8.36 (d, J = 5.2 Hz, 1H, ArH); 13C NMR (125 MHz, DMSO-d6): δ = 43.52, 59.28, 84.58, 108.42, 109.02, 111.55, 116.72, 122.53, 123.98, 126.89, 127.76, 140.43, 150.02, 152.84, 154.28, 159.59; Elemental Analysis for C16H13N3O2, Calcd.: C, 68.81; H, 4.69; N, 15.05; Found: C, 68.82; H, 4.67; N, 15.04.
4.1.3.5. 4-(Benzofuran-2-yl)-2-(3-hydroxypropylamino)nicotinonitrile (8)
Yield = 88%, m.p. = 124–126 °C, yellow powder, IR (KBr, cm−1): 3100 (OH), 3343 (NH), 2218 (CN), 1594 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 1.73 (p, J = 6.5 Hz, 2H, CH2), 3.50 (m, 4H, 2CH2), 5.75 (s, 1H, OH), 7.15 (d, J = 5.3 Hz, J 1H, NH), 7.24 (t, J = 5.4 Hz, 1H, ArH), 7.34 (dd, J = 11.4, 11.4 Hz, 1H, ArH), 7.46 (m, 1H, ArH), 7.69 (d, J = 7.9 Hz, 1H, ArH), 7.82 (d, = 7.7 Hz, 1H, ArH), 7.86 (s, 1H, ArH), 8.36 (d, J = 5.3 Hz, 1H, ArH); Elemental Analysis for C17H15N3O2, Calcd.: C, 69.61; H, 5.15; N, 14.33; Found: C, 69.63; H, 5.14; N, 14.32.
4.1.3.6. 4-(Benzofuran-2-yl)-2-(cyclohexylamino)nicotinonitrile (9)
Yield = 50%, m.p. = 186–188 °C, yellow powder, IR (KBr, cm−1): 3355 (NH), 2210 (CN), 1579 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 1.14 (m, 1H, CH), 1.31 (m, 2H, CH2), 1.42 (m, 2H, CH2), 1.61 (d, J = 12.7 Hz, 1H, CH), 1.73 (d, J = 13.1 Hz, 2H, CH2), 1.87 (d, J = 9.8 Hz, 2H, CH2), 4.00 (m, 1H, CH), 6.66 (d, J = 7.9 Hz, 1H, NH), 7.15 (d, J = 5.2 Hz, 1H, ArH), 7.35 (t, J = 7.4 Hz, 1H, ArH), 7.46 (dd, J = 11.7, 4.3 Hz, 1H, ArH), 7.69 (d, J = 8.2 Hz, 7.83 (d, J = 7.7 Hz, 1H, ArH), 7.86 (s, 1H, ArH), 8.36 (d, J = 5.3 Hz, 1H, ArH); 13C NMR (125 MHz, DMSO-d6): δ = 24.8, 25.26, 31.93, 49.79, 84.42, 108.33, 108.98, 111.52, 117.61, 122.50, 123.86, 126.84, 127.76, 140.64, 150.04, 152.87, 154.25, 158.75; Elemental Analysis for C20H19N3O, Calcd.: C, 75.69; H, 6.03; N, 13.24; Found: C, 75.68; H, 6.01; N, 13.25.
4.1.3.7. 4-(Benzofuran-2-yl)-2-(benzylamino)nicotinonitrile (10)
Yield = 43%, m.p. = 180–182 °C, yellow powder. IR (KBr, cm−1): 3029 (=C–H), 3369 (NH), 2211 (CN), 1580 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 4.56 (d, J = 6.0 Hz, 2H, CH2), 7.17 (d, J = 5.2 Hz, 1H, NH), 7.21 (dd, J = 10.0, 10.0 Hz, 1H, ArH), 7.30 (s, 1H, ArH), 7.31 (d, J = 7.4 Hz, 1H, ArH), 7.33 (s, 2H. ArH), 7.35 (d, J = 2.6 Hz, 1H, ArH), 7.47 (s, 1H, ArH), 7.69 (d, J = 7.8 Hz, 1H, ArH), 7.83 (d, J = 7.7 Hz, 1H, ArH), 7.87 (d, J = 6.0 Hz, 1H, ArH), 7.88 (s, 1H, ArH), 8.32 (d, J = 5.2 Hz, 1H, ArH); 13C NMR (125 MHz, DMSO-d6): δ = 44.03, 84.62, 108.69, 109.06, 111.53, 116.65, 122.52, 123.88, 126.65, 126.87, 127.08, 127.75, 128.18, 139.94, 140.53, 150.01, 152.83, 154.29, 159.41; Elemental Analysis for C21H15N3O, Calcd.: C, 77.52; H, 4.65; N, 12.91; Found: C, 77.53; H, 4.64; N, 12.93.
4.1.3.8. 4-(Benzofuran-2-yl)-2-(phenethylamino)nicotinonitrile (11)
Yield = 56%, m.p. = 157–159 °C, yellow powder, IR (KBr, cm−1): 3022 (=C–H), 3370 (NH), 2209 (CN), 1588 (C=C); 1H NMR (500 MHz, DMSO-d6): δ = 2.90 (t, J = 6.2 Hz, 2H, CH2Ph), 3.66 (td, J = 6.2, 5.3 Hz, 2H, CH2N), 7.18 (d, J = 5.3 Hz, NH), 7.21 (t, J = 7.2 Hz, 1H, ArH), 7.25 (s, 1H, ArH), 7.26 (s, 1H, ArH), 7.30 (s, 1H, ArH), 7.31 (d, J = 3.1 Hz, 1H, ArH), 7.33 (s, 1H, ArH), 7.36 (t, J = 7.6 Hz, 1H, ArH), 7.47 (m, 1H, ArH), 7.70 (d, J = 8.4 Hz, 1H, ArH), 7.83 (d, J = 7.8 Hz, 1H, ArH), 7.88 (s, 1H, ArH), 8.40 (d, J = 5.2 Hz, 1H, ArH); Elemental Analysis for C22H17N3O, Calcd.: C, 77.86; H, 5.05; N, 12.38; Found: C, 77.87; H, 5.03; N, 12.36.
4.1.3.9. 4-(Benzofuran-2-yl)-2-(4-methoxyphenethylamino)nicotinonitrile (12)
Yield = 66%, m.p. = 156–158 °C, yellow powder, IR (KBr, cm−1): 3013 (=C–H), 3352 (NH), 2217 (CN), 158 (C=C); 1H-NMR (500 MHz, DMSO-d6): δ = 2.82 (m, 2H, CH2), 3.60 (m, 2H, CH2), 3.71 (s, 3H, CH3), 6,85 (s, 1H, NH), 6.87 (s, 1H, ArH), 7.15 (s, 1H, ArH), 7.16 (d, J = 2.8 Hz, 1H, ArH), 7.17 (s, 1H, ArH), 7.25 (s, 1H, ArH), 7.35(s, 1H, ArH), 7.46 (s, 1H, ArH), 7.69 (d, J = 8.4 Hz, 1H, ArH), 7.82 (d, J = 7.9 Hz, 1H, ArH), 7.86 (s, 1H, ArH), 8.39 (d, J = 5.3 Hz, 1H, ArH); 13C NMR (125 MHz, DMSO-d6): δ = 34.0, 42.83, 54.98, 84.50, 108.34, 108.8, 111.54, 113.79, 116.68, 122.51, 123.89, 126.87, 129.63, 131.40, 140.45, 150.05, 152.97, 154.27, 157.66, 159.46; Elemental Analysis for C23H19N3O2, Calcd.: C, 74.78; H, 5.18; N, 11.37; Found: C, 74.77; H, 5.17; N, 11.36.
4.2. Biological evaluation
4.2.1. Antiproliferative screening
The in vitro antiproliferative activity of all synthesised compounds was evaluated by an MTT assay according to the reported methodCitation47,Citation48.
4.2.2. Epidermal growth factor inhibition assay
EGFR enzyme assay was conducted as described in our previous reportsCitation23–25.
4.2.3. Caspase-3 assay
Sandwich enzyme-linked immunosorbent assay (ELISA) was used to determine the level of active human caspase-3 as previously reported and according to the manufacturer’s instructionsCitation49.
4.2.4. Cell cycle analysis and induction of apoptosis
4.2.4.1. Flow cytometry analysis of the cell cycle distribution
Cell cycle analysis was performed according to our previous report using the HCT-116 and MCF-7 cell lines stained with the DNA fluorochrome PI and analysed by FACSCalibur flow cytometerCitation50–52.
4.4.2.2. Analysis of cellular apoptosis
Apoptosis induction was performed using the HCT-116 and MCF-7 cell lines and well-established Annexin 5-FITC/PI detection kit similar to the report procedureCitation50–52. The cell line samples were analysed using FACSCalibur flow cytometer.
4.3. Docking study
The molecular modelling calculations and docking studies were performed using the MOECitation61 software version 2008.10 (Chemical Computing Group Inc., Montreal, Quebec, Canada). The X-ray crystallographic structure of EGFR with erlotinib was obtained from the RCSB protein data bank (PDB ID: 1m17).
Supplemental Material
Download PDF (942.9 KB)Acknowledgements
The authors thank the Deanship of Scientific Research and RSSU at King Saud University for their technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Varmus H. The new era in cancer research. Science 2006;312:1162–5.
- Li Q, Xu W. Novel anticancer targets and drug discovery in post genomic age. Curr Med Chem Anti-Cancer Agents 2005;5:53–63.
- C, Avendaño JC, Menendez Medicinal chemistry of anticancer drugs. Amsterdam, The Netherlands: Elsevier; 2015.
- Abdel-Aziz AA, El-Azab AS, Alanazi AM, et al. Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzyme Inhib Med Chem 2016;31:796–809.
- El-Azab AS, Alanazi AM, Abdel-Aziz NI, et al. Synthesis, molecular modeling study, preliminary antibacterial, and antitumor evaluation of N-substituted naphthalimides and their structural analogues. Med Chem Res 2013;22:2360–75.
- Alanazi AM, Abdel-Aziz AA, Shawer TZ, et al. Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: in silico studies. J Enzyme Inhib Med Chem 2016;31:721–35.
- El-Sayed MA, El-Husseiny WM, Abdel-Aziz NI, et al. Synthesis and biological evaluation of 2-styrylquinolines as antitumour agents and EGFR kinase inhibitors: molecular docking study. J Enzyme Inhib Med Chem 2018;33:199–209.
- Alanazi AM, El-Azab AS, Al-Swaidan IA, et al. Synthesis, single-crystal, in vitro antitumor evaluation and molecular docking of 3-substitued 5, 5-diphenylimidazolidine-2, 4-dione derivatives. Med Chem Res 2013;22:6129–42.
- Alanazi AM, Al-Suwaidan IA, Alaa A-M, et al. Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents. Med Chem Res 2013;22:5566–77.
- El-Husseiny WM, El-Sayed MA, Abdel-Aziz NI, et al. Synthesis, antitumour and antioxidant activities of novel α,β-unsaturated ketones and related heterocyclic analogues: EGFR inhibition and molecular modelling study. J Enzyme Inhib Med Chem 2018;33:507–18.
- Kolibaba KS, Druker BJ. Protein tyrosine kinases and cancer. Biochim Biophys Acta 1997;1333:F217–48.
- Black JD, Brattain MG, Krishnamurthi SA, et al. ErbB family targeting. Curr Op Investig Drugs 2003;4:1451–4.
- Ji Y, Sun Q, Zhang J, Hu H. MiR-615 inhibits cell proliferation, migration and invasion by targeting EGFR in human glioblastoma. Biochem Biophys Res Communic 2018;499:719–26.
- Antonello A, Tarozzi A, Morroni F, et al. Multitarget-directed drug design strategy: a novel molecule designed to block epidermal growth factor receptor (EGFR) and to exert proapoptotic effects. J Med Chem 2006;49:6642–5.
- Yewale C, Baradia D, Vhora I, et al. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials 2013;34:8690–707.
- Qu X, Yang L, Shi Q, et al. Lidocaine inhibits proliferation and induces apoptosis in colorectal cancer cells by upregulating mir-520a-3p and targeting EGFR. Pathol-Res Pract 2018;214:1974–9.
- Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Therap 1999;82:241–50.
- Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev. Cancer 2013;13:663–73.
- Gullick W. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Brit Med Bull 1991;47:87–98.
- Cai J, Sun M, Ge X, Sun Y. EGFR tyrosine kinase inhibitors differentially affect autophagy in head and neck squamous cell carcinoma. Biochem Biophys Res Commun 2017;486:1027–33.
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Critical Rev in Oncol/Hematol 1995;19:183–232.
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28–39.
- Alkahtani HM, Alanazi MM, Aleanizy FS, et al. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: molecular docking studies. Saudi Pharm J 2019;27:682–93.
- El-Azab AS, Alaa A-M, AlSaif NA, et al. Antitumor activity, multitarget mechanisms, and molecular docking studies of quinazoline derivatives based on a benzenesulfonamide scaffold: cell cycle analysis. Bioorganic Chem 2020;104:104345.
- Alkahtani HM, Abdalla AN, Obaidullah AJ, et al. Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: dual inhibitors of EGFR/HER2. Bioorg Chem 2020;95:103461.
- Abbas H-AS, Abd El-Karim SS. Design, synthesis and anticervical cancer activity of new benzofuran-pyrazol-hydrazono- thiazolidin-4-one hybrids as potential EGFR inhibitors and apoptosis inducing agents. Bioorg Chem 2019;89:103035.
- Barker AJ, Gibson KH, Grundy W, et al. Studies leading to the identification of ZD1839 (Iressa™): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett 2001;11:1911–4.
- Yang Z, Hackshaw A, Feng Q, et al. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: a meta-analysis. Int J Cancer 2017;140:2805–19.
- Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol 2008;26:2442–9.
- Christensen J. A preclinical review of sunitinib, a multitargeted receptor tyrosine kinase inhibitor with anti-angiogenic and antitumour activities. Annals Oncol 2007;18:x3–10.
- Madhusudan S, Ganesan TS. Tyrosine kinase inhibitors in cancer therapy. Clin BioChem 2004;37:618–35.
- Takimoto CH, Awada A. Safety and anti-tumor activity of sorafenib (Nexavar) in combination with other anti-cancer agents: a review of clinical trials. Cancer Chemother Pharmacol 2008;61:535–48.
- Kouznetsov VV, Galvis CEP. Strecker reaction and α-amino nitriles: recent advances in their chemistry, synthesis, and biological properties. Tetrahedron 2018;74:773–810.
- Fleming FF, Yao L, Ravikumar PC, et al. Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J Med Chem 2010;53:7902–17.
- Dhillon S. Neratinib in early-stage breast cancer: a profile of its use in the EU. Clin Drug Investig 2019;39:221–9.
- Barbuti AM, Zhang G-N, Gupta P, et al., EGFR and HER2 inhibitors as sensitizing agents for cancer chemotherapy. In: Protein kinase inhibitors as sensitizing agents for chemotherapy. Amsterdam, The Netherlands: Elsevier; 2019: 1–11.
- Isfort S, Brümmendorf TH. Bosutinib in chronic myeloid leukemia: patient selection and perspectives. J Blood Med 2018;9:43–50.
- Nevagi RJ, Dighe SN, Dighe SN. Biological and medicinal significance of benzofuran. Euro J Med Chem 2015;97:561–81.
- Khanam H, Shamsuzzaman . Bioactive benzofuran derivatives: a review. Euro J Med Chem 2015;97:483–504.
- Radadiya A, Shah A. Bioactive benzofuran derivatives: an insight on lead developments, radioligands and advances of the last decade. Euro J Med Chem 2015;97:356–76.
- Mao Z-W, Zheng X, Lin Y-P, et al. Design, synthesis and anticancer activity of novel hybrid compounds between benzofuran and N-aryl piperazine. Bioorg Med Chem Lett 2016;26:3421–4.
- Abd El-Karim SS, Anwar MM, Mohamed NA, et al. Design, synthesis, biological evaluation and molecular docking studies of novel benzofuran-pyrazole derivatives as anticancer agents. Bioorg Chem 2015;63:1–12.
- Abdelhafez OM, Amin KM, Ali HI, et al. Design, synthesis and anticancer activity of benzofuran derivatives targeting VEGFR-2 tyrosine kinase. RSC Advances 2014;4:11569–79.
- Mphahlele MJ, Maluleka MM, Parbhoo N, Malindisa ST. Synthesis, evaluation for cytotoxicity and molecular docking studies of benzo[c]furan-chalcones for potential to inhibit tubulin polymerization and/or EGFR-tyrosine kinase phosphorylation. Int J Mol Sci 2018;19:2552.
- Sheng J, Liu Z, Yan M, et al. Biomass-involved synthesis of N-substituted benzofuro[2,3-d]pyrimidine-4-amines and biological evaluation as novel EGFR tyrosine kinase inhibitors. Org Biomol Chem 2017;15:4971–7.
- Mphahlele MJ, Maluleka MM, Aro A, et al. Benzofuran-appended 4-aminoquinazoline hybrids as epidermal growth factor receptor tyrosine kinase inhibitors: synthesis, biological evaluation and molecular docking studies. J Enzyme Inhib Med Chem 2018;33:1516–28.
- Vega-Avila E, Pugsley MK. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc Western Pharmacol Soc 2011;54:10–4.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth 1983;65:55–63.
- Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differentiation 1999;6:99–104.
- Othman DI, Selim KB, Magda A-A, et al. Design, synthesis and anticancer evaluation of new substituted thiophene-quinoline derivatives. Bioorg Med Chem 2019;27:115026.
- Fares S, Selim KB, El-Sayed M, Goda FE. Synthesis, biological evaluation and molecular modeling of novel benzofuran-N-heterocyclic hybrids as anticancer agents. Journal of American Science 2017;13(11):65–76.
- Turky A, Bayoumi AH, Ghiaty A, et al. Design, synthesis, and antitumor activity of novel compounds based on 1,2,4-triazolophthalazine scaffold: apoptosis-inductive and PCAF-inhibitory effects. Bioorg Chem 2020;101:104019.
- Alaa A-M, El-Azab AS, Abou-Zeid LA, et al. Synthesis, anti-inflammatory, analgesic and COX-1/2 inhibition activities of anilides based on 5,5-diphenylimidazolidine-2,4-dione scaffold: molecular docking studies. Eur J Med Chem 2016;115:121–31.
- Abdel-Aziz AA, Asiri YA, Al-Agamy MH. Design, synthesis and antibacterial activity of fluoroquinolones containing bulky arenesulfonyl fragment: 2D-QSAR and docking study. European J Med Chem 2011;46:5487–97.
- El-Azab AS, Mary YS, Panicker CY, et al. DFT and experimental (FT-IR and FT-Raman) investigation of vibrational spectroscopy and molecular docking studies of 2-(4-oxo-3-phenethyl-3, 4-dihydroquinazolin-2-ylthio)-N-(3, 4, 5-trimethoxyphenyl) acetamide. J Mol Struct 2016;1113:133–45.
- Alaa A-M, Abou-Zeid LA, ElTahir KEH, et al. Design, synthesis of 2,3-disubstitued 4(3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg Med Chem 2016;24:3818–28.
- Abdel-Sayed MA, Bayomi SM, El-Sherbeny MA, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibition activities and molecular docking study of pyrazoline derivatives. Bioorg Med Chem 2016;24:2032–42.
- El-Gamal MI, Bayomi SM, El-Ashry SM, et al. Synthesis and anti-inflammatory activity of novel (substituted)benzylidene acetone oxime ether derivatives: molecular modeling study. Euro J Med Chem 2010;45:1403–14.
- Goda FE, Alaa A-M, Ghoneim HA. Synthesis and biological evaluation of novel 6-nitro-5-substituted aminoquinolines as local anesthetic and anti-arrhythmic agents: molecular modeling study. Bioorg Med Chem 2005;13:3175–83.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 2002;277:46265–72.
- MOE. 2008.10 of Chemical Computing Group. Inc. Available from: http://www.chemcomp.com/press_releases/2008-11-04.htm.