
Abstract
In the relentless search for new cancer treatments, organoselenium compounds, and carbonic anhydrase (CA) inhibitors have emerged as promising drug candidates. CA isoforms IX and XII are overexpressed in many types of cancer, and their inhibition is associated with potent antitumor/antimetastatic effects. Selenium-containing compounds, particularly selenols, have been shown to inhibit tumour-associated CA isoforms in the nanomolar range since the properties of the selenium atom favour binding to the active site of the enzyme. In this work, two series of selenoesters (1a–19a and 1b–19b), which gathered NSAIDs, carbo/heterocycles, and fragments from natural products, were evaluated against hCA I, II, IX, and XII. Indomethacin (17b) and flufenamic acid (19b) analogs exhibited selectivity for tumour-associated isoform IX in the low micromolar range. In summary, selenoesters that combine NSAIDs with fragments derived from natural sources have been developed as promising nonclassical inhibitors of the tumour-associated CA isoforms.
Introduction
Cancer is a major health burden that is increasing every day despite efforts in diagnosis and treatment. In 2020, there were 19.3 million new cancer cases (10.1 million males vs. 9.2 million females), plus 9.9 million cancer deaths (5.5 million males vs. 4.4 million females) worldwide.Citation1 The research of new therapeutic targets and the development of novel treatments continues unabated. The emergence of resistance to current drugs further highlights the need to bring new effective therapeutic options to the market. In this context, carbonic anhydrase (CA) inhibitors have emerged as promising anticancer drug candidates.
The CAs are a superfamily of metalloenzymes that reversibly catalyse the conversion of CO2 into carbonic acid. There are at least eight families of CA (α-ι classes) and the CA-α are present in humans. The CA enzymes found in mammals fall into four broad subgroups according to their cellular location: cytosolic isoforms (CA-I, CA-II, CA-III, CA-VII, and CA-XIII), mitochondrial isoforms (CA-VA and CA-VB), membrane-bound isoforms (CA-IV, CA-IX, CA-XII, CA-XIV, and CA-XV) and secreted isoforms (CA-VI).Citation2 CA inhibitors are clinically used mainly as diuretics and antiepileptics, but novel applications in cancer treatment have been developed. Interestingly, isoforms IX and XII are overexpressed in certain types of cancers and are associated with the growth, migration, invasion, and metastasis of tumors.Citation3–5 Therefore, they have been widely studied as new therapeutic targets for cancer.Citation6–8 CA-IX is expressed in a restricted number of normal tissues, whereas is highly expressed in many solid tumours, particularly in renal carcinoma.Citation9,Citation10 Moreover, CA-IX is a hypoxia-associated isoform that plays a crucial role in extracellular acidification of the solid tumour, contributing to tumour progression.Citation11,Citation12 On the other hand, CA-XII is overexpressed in a range of human tumours including gastric, ovarian, lung, and brain, whereas its expression in normal tissues is also restricted.Citation13–15 In addition, CA-XII expression is induced by oestrogen; hence this isoform is highly expressed in oestrogen receptor-positive breast cancer.Citation16 Therefore, it seems promising to develop inhibitors of these two CA tumour-associated isoforms for the treatment of several types of cancer, with one such compound, SLC-0111 in Phase Ib/II clinical trials as an antitumor agent.Citation17
Selenium (Se) compounds have been extensively studied as pharmaceutical drugs since they have shown antioxidant,Citation18,Citation19 antitumor,Citation18,Citation20 antiviral,Citation21,Citation22 and antimicrobialCitation23 actions. Particularly, selenols have arisen as a powerful scaffold to develop nonclassical inhibitors of the tumour-associated CA isoforms, due to the soft character of the Se atom, the polarity of the Se-H bond, and the affinity of Se-containing compounds for zinc finger proteins.Citation24 Aryl selenol derivatives have demonstrated to inhibit CA-IX at nanomolar concentrations, whereas the cytosolic CA isoforms proved to be less sensitive. Moreover, it was shown that selenol was bound directly to the Zn (II) ion from the CA active site with its usual tetrahedral geometry.Citation25 Selenol group (R-SeH) is quite nucleophilic and possess a low pKa value at physiological pH (i.e. 5.4), the main species reported is the anionic selenoate (RSe¯), which can readily react with the zinc ion of the CA active site. In addition, aliphatic selenols were synthesised and evaluated as CA inhibitors, and they demonstrated to be more potent against the cytosolic CAs rather than the membrane and tumour-associated isoforms.Citation26 However, the main limitation of selenols is that they are very unstable, showing a high tendency to be oxidised to diselenides. An approach to solve this problem is the development of selenoesters, which by the action of an esterase enzyme will release the selenol group and the corresponding carboxylic acid. The Supuran’s group has demonstrated the thioesterase,Citation27 esterase,Citation28 and selenoesteraseCitation29 activities of human CA isoforms. Therefore, it seems reasonable to design selenoesters with the aim of creating a prodrug that, after the action of CA (with selenoesterase function), releases the active selenol. This strategy also makes it possible to introduce into the molecule another active fragment, in addition to selenol, which will be released in the form of carboxylic acid.
Herein, we report 38 selenoesteres (), combining active fragments from natural sources with non-steroidal anti-inflammatory drugs (NSAIDs) and carbo- and hetero-cycles derivatives as inhibitors of tumour-associated human CA. The part of the molecule corresponding to the selenol, that might be released, is an allylic (series a) or propargylic (series b) chain. This side of the molecule has been designed inspired by the active fragments present in natural sources ().
Figure 1. Chemical structures of synthetic compounds 1a–19a and 1b–19b.Citation30
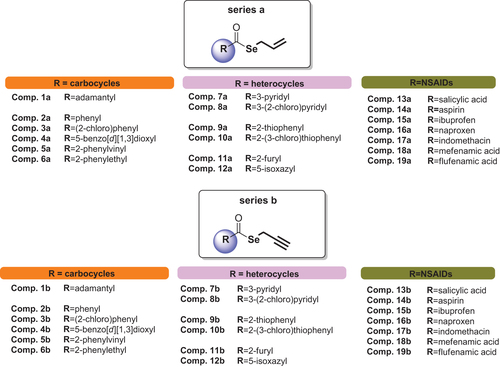
Figure 2. Allylic and propargylic molecules of natural products.Citation30
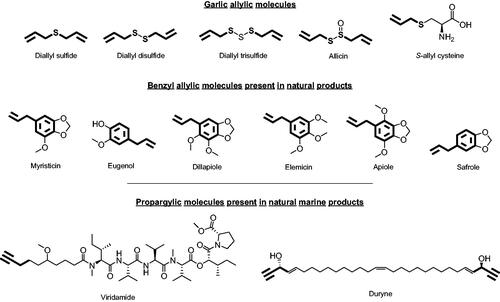
The allylic chain is present in active garlic derivatives such as diallyl selenides, allicin or S-allyl cysteine. It is also found in benzyl allylic derivatives present in several plants such as cloves, curcuma, anise, or parsley.Citation31 On the other hand, the active propargylic fragment can be found in natural marine products such as viridamide (isolated from cyanobacteria) and duryne (metabolite produced by one type of marine sponge). The other part of the molecule, which ought to be released in the form of carboxylic acid, are carbo- and hetero-cycles derivatives and NSAIDs. Inflammation is recognised as a hallmark feature of cancer development and progression,Citation32 thus the incorporation of NSAIDs into the molecule provides an extra activity that may enhance their profile as tumour-associated CA isoforms inhibitors. Moreover, NSAIDs are carboxylic acid-based compounds that have been reported as a new family of nonclassical CA inhibitors.Citation33–35 Likewise, carbo- and hetero-cycles derivatives, such as quinazoline- or benzodioxol-based compounds, have been demonstrated to inhibit some isoforms of CA, including tumour-associated IX and XII.Citation36,Citation37
Experimental
Chemistry
The studied compounds 1a–19a and 1b–19b were synthesised in our previous workCitation30 and their chemical structures are depicted in .
Carbonic anhydrase inhibition studies
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity.Citation38 Phenol red (at a concentration of 0.2 mM) has been used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as a buffer and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5 − 10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled − deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature before assay to allow for the formation of the E − I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng − Prusoff equation, as reported earlier,Citation39,Citation40 and represent the mean from at least three different determinations. The enzyme concentrations were in the range 3–11 nM. All human CA (hCA) isoforms were recombinant ones obtained in-house as reported earlier.Citation41,Citation42
Molecular modelling
The crystal structures of CA I (PDB:2NMX),Citation43 CA II (PDB:3K34),Citation44 CA IX (PDB:5FL4),Citation45 and CA XII (PDB:1JD0)Citation46 used for computational studies were downloaded by Protein Data BankCitation47 and prepared according to the Protein Preparation module in Maestro Schrödinger suite, assigning bond orders, adding hydrogens, deleting water molecules, and optimising H-bonding networks. Finally, energy minimisation with a Root Means Square Deviation (RMSD) value of 0.30 was applied using an Optimised Potential for Liquid Simulation (OPLS4) force field.Citation25,Citation48–51 Grids for docking were centred in the centroid of the complexed ligand. Docking studies were carried out with the program GlideCitation48(f) using the standard precision (SP) mode. 3D ligand structures were prepared by MaestroCitation48(a). QM geometry optimisation and atomic electrostatic charges computation were performed with JaguarCitation48(g) fitting them to an electrostatic potential calculated at the B3LYP/cc/pvtz-f level of theory. ESP atomic charges were used in docking simulations. The OPLS4 force field was modified according to Schrödinger to enable negatively charged selenate moieties for the docking procedure in Glide. Indeed, the OPLS FF does not support parameters for the Lewis structure of selenates. The best docking pose for each compound was submitted to DFT (B3LYP/LACVP*+) calculations with Jaguar within both built model systems. The QM-optimised ligands were rescored in the complete hCA macromolecular environment with GlideCitation48(f).
Molecular dynamics (MD) simulations were performed using Desmond Molecular Dynamics System (v.6.7)Citation48€ (Schrödinger suite) and OPLS4 force field. All systems were solvated in an orthorhombic box using simple point charge water molecules extended 15 Å away from any protein atom. The system was neutralised with 0.15 M Cl− and Na+ ions. The simulation protocol included a starting relaxation step followed by a final production phase of 100 ns. In particular, the relaxation step comprised the following: (a) a stage of 100 ps at 10 K retaining the harmonic restraints on the solute heavy atoms (force constant of 50 Kcal/mol/Å2) using the NPT ensemble with Brownian dynamics; (b) a stage of 12 ps at 10 K with harmonic restraints on the solute heavy atoms (force constant of 50 Kcal/mol/Å2), using the NVT ensemble and Berendsen thermostat; (c) a stage of 12 ps at 10 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (d) a stage of 12 ps at 300 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (e) a final 24 ps stage at 300 K and 1 atm without harmonic restraints, using the NPT Berendsen thermostat and barostat. The final production phase of MD was run using a canonical NPT Berendsen ensemble at 300 K. During the MD simulation, a time step of 2 fs was used while constraining the bond lengths of H atoms with the M-SHAKE algorithm. The atomic coordinates of the system were saved every 100 ps along the MD trajectory. Protein RMSD, ligand RMSD/RMSF (Root Mean Square Fluctuation) ligand torsions evolution, and occurrence of intermolecular H-bonds and hydrophobic contacts were provided by the Simulation Interaction Diagram (SID) implemented in Maestro along with the production phase of the MD simulation. The tool reads the MD trajectory file and identifies ligand/target interactions repeatedly occurring during the simulation time (for instance, a 60% value suggests that the interaction is maintained for 60% of the MD). The 1000 frames resulting from MDs were clustering using the Conformer Cluster tool implemented in the Schrödinger suite in 10 clusters. The representative poses of the most abundant clusters were refined with Prime MM-GBSA calculations (v.5.5)Citation48(d) with a VSGB (Variable Surface Generalised Born) solvation model considering the target flexible within 3 Å around the ligand. Figures were generated with Maestro and Chimera.Citation48,Citation52
Results and discussion
Chemistry
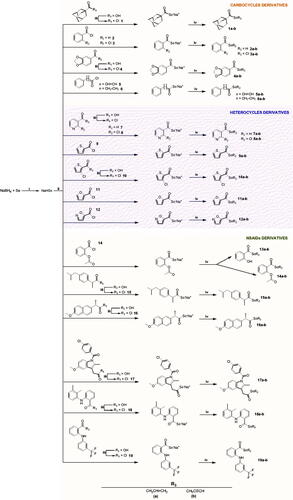
The synthesis method of the compounds is shown in Scheme 1. The synthesis procedure and the characterisation of the compounds were described in our previous study.Citation30 Briefly, hydrogen sodium selenide (NaHSe) was formed by the reduction of elemental Se with NaBH4 in water. Then, the corresponding acid chloride was added to the reaction mixture to form the corresponding sodium selenoate by a nucleophilic acyl substitution. Finally, the target compounds were obtained through a nucleophilic substitution over the allyl/propargyl bromide. All the compounds were purified by column chromatography.
Scheme 1. Synthesis procedure to yield 1a–19a and 1b–19b derivatives. Reagents and conditions: (i) H2O, 20 min, and room temperature; (ii) THF/H2O, 60 min, and room temperature; (iii) ClCOCOCl, CH2Cl2, 12 h, and room temperature; (iv) BrCH2CH = CH2 or BrCH2C≡CH, THF/H2O, 90 min, and room temperature.Citation30
Carbonic anhydrase inhibition
All the compounds 1a–19a and 1b–19b were tested in vitro for the inhibitory activity against hCA isoforms I, II, VA, IX, and XII, and their activities were compared to the standard CA inhibitor acetazolamide (AZZ) (). Among the compounds, 9 of 38 inhibited at least one CA isoforms with Ki values in the low micromolar range and the following structure-activity relationship (SAR) may be observed regarding inhibition data in :
Table 1. Inhibition of hCA I, II, VA, IX, and XII activity for compounds 1a–19a and 1b–19b.
Surprisingly, the allylic chain (series a) presence in all the compounds leads to a loss of activity in the tested CA isoforms. On the contrary, the introduction of the propargylic chain (series b) seems important for the compounds’ inhibitory capacity since Ki values lower than 10 µM have been observed in some cases.
Among the compounds of series b, the following SAR may be noted:
The cytosolic CA I was inhibited by the aromatic derivative 6b which has a two-carbon atom spacer between the benzene ring and the carbonyl group (Ki = 3.1 μM). This compound was 7.7 and 26 times more potent than its analog with the doble-bond spacer (compound 5b: Ki = 24.1 μM) and with the analog without the spacer (compound 2b: Ki = 80.5 μM), respectively. In addition, the presence of a chlorine atom in the ortho position (3b) or the presence of an oxygen atom in both 3 and 4 positions (4b) improved the inhibition capacity of the benzene-derived compounds (compound 3b and 4b: Ki = 42.1 and 31.3 μM, respectively). However, the substitution of the benzene ring by a carbocycle, such as an adamantane, led to the loss of activity (compound 1b: Ki >100 μM). On the other hand, the inhibition potency increased with the presence of heterocycles such as pyridine (7b), thiophene (9b), and furan (11b) (Ki = 8.5–30.2 μM). It is curious how the pyridine derivative with one chlorine atom in the ortho position increased the inhibitory activity 13.7 times (compound 8b: Ki = 2.2 μM) compared to unsubstituted pyridine, while the ortho-substituted thiophene derivative with one chlorine atom lost the activity (compound 10b: Ki >100 μM). The substitution of the furan ring (11b) by the isoxazole one (12b) decreased the inhibitory capacity nearly 7-fold (Ki = 8.5 and 58.4 μM, respectively). Finally, among the NSAID derivatives, the aspirin analog (14b) was the most active, with a Ki value of 5.0 μM, followed by the salicylic derivative (13b), Ki = 23.3 μM. The remaining analogs showed no inhibition capacity (Ki > 77 μM).
The dominant cytosolic human isoform, CA II, was less sensitive to the selenoester derivatives. As in CA isoform I, the aromatic derivative 6b, which has a two-carbon atom spacer between the benzene ring and the carbonyl group, and the compound 8b, with a chlorine atom substituted pyridine derivative, inhibited CA II in the low micromolar range (Ki = 9.5 and 8.6 μM, respectively). The adamantane derivative (1b), as well as the benzene analogue (2b), did not inhibit this isoform. The benzene derivatives substituted with the chlorine atom (3b) and with the oxygen atoms (4b) were less active in this isoform, as was the derivative with the double bond spacer (5b). Derivatives containing unsubstituted heterocycles inhibited CA II in the medium micromolar range (compounds 7b, 9b, and 11b: Ki = 66.3, 46.6, and 49.9 μM, respectively). The presence of the isoxazole ring (12b) led to the loss of inhibition activity (Ki > 100 μM). Likewise, the aspirin derivative (14b) inhibited CA II 11 times less than against CA I and the salicylic analog (13b) was not able to inhibit this CA isoform. The rest of the NSAID derivatives did not inhibit CA II, except for the mefenamic acid analog (18b), which exhibited 22-fold greater inhibition potency than for CA I isoforms (Ki = 4.5 μM).
The tumour-associated transmembrane CA IX was inhibited by compounds 1b in the low micromolar range (Ki = 9.1 μM). This adamantane derivative was not able to inhibit cytosolic isoforms I and II. Similarly, the benzene analog (2b), which had not inhibited isoforms I and II, exhibited a potent inhibition action with a Ki value of 7.8 μM (nearly 13 and 10 times higher than for isoforms I and II, respectively). The remaining benzene derivatives (3b and 4b–6b) inhibited CA IX in the medium-high micromolar range (Ki = 25.0–79.3 μM). Furthermore, the unsubstituted thiophene derivative (9b), which inhibited CA I in the low micromolar range, did not inhibit CA IX. In addition, the presence of the thiophene ring (9b) did not provide inhibition activity, in contrast to what happened in isoform I (Ki = 9.2 μM), while the presence of the furan ring (11b) maintained the inhibition potency in the low micromolar range (Ki = 7.1 μM). On the other hand, the NSAID derivatives showed greater activity against this isoform (Ki = 9.5–58.5 μM), except for the aspirin analog (14b) which exhibited no inhibition capacity. The phenamate derivatives (18b and 19b) stood out with Ki values of 9.6 and 9.5 μM, respectively.
As in CA isoforms I and II, both aromatic derivative 6b, which has a two-carbon atom spacer between the benzene ring and the carbonyl group, and compound 8b, with a chlorine atom substituted pyridine derivative, inhibited tumour-associated CA XII in the low micromolar range (Ki = 7.0 and 9.6 μM respectively). It is very remarkable the fact that the introduction of a double bond in the two-carbon spacer (5b) led to the loss of CA XII inhibition activity. The introduction of the adamantane carbocycle (1b) or the unsubstituted or chloro-substituted benzene ring (2b and 3b respectively) maintained the inhibition potency in the medium micromolar range (Ki = 28.4–55.7 μM). However, the replacement of the benzene ring by pyridine (7b) or benzodioxol (4b) resulted in the loss of inhibition activity. In addition, thiophene derivatives (9b and 10b) did not inhibit CA IX and furan and isoxazole derivatives (11b and 12b, respectively) did so in the medium-high micromolar range (Ki = 59.6 and 66.6 μM respectively). No NSAID derivative was able to inhibit CA IX, except for the 18b compound, the mefenamic acid analog, which did so in the medium micromolar range (Ki = 46.3 μM).
An interesting inhibition profile was observed for indomethacin and flufenamic acid analogs (17b) and (19b), respectively, which exhibited selectivity for tumour-associated isoform IX. Likewise, adamantane analog (1b) showed selectivity for tumour-associated isoforms IX and XII. On the other hand, salicylic (13b), naproxen (16b), and ibuprofen (15b) analogs exhibited selectivity for cytosolic CA I isoform and tumour-associated CA IX. Another interesting point was that the unsubstituted (7b) and chlorine substituted (8b) pyridine and furane (11b) heterocyclic derivatives, as well as the benzene derivative with the two-carbon spacer (6b), showed inhibition activity in all CA isoforms. Compounds 8b and 6b inhibited all CA isoforms in the low-medium micromolar range (Ki = 2.2–48.5 μM).
Molecular modelling
In silico studies were applied to get insight into the different inhibitory activities of derivatives in towards the CA I and II (off-target) and the tumour-associated isoforms CA IX and CA XII. As already reported, the investigated compounds act as prodrugs that, thanks to the esterase activity of CAs, release selenol and carboxylic fragments. Thus, interaction studies were carried out on the active allyl (a) and propargyl (b) selenol moieties of compounds 1–19 using the available 3D coordinates of the targets.
According to the literature,Citation25,Citation53 all docking solutions have shown the deprotonated Se atom bound to the zinc atom in tetragonal coordination in the active site of the investigated CA isoforms ().
Figure 3. Predicted binding mode of propargyl selenol (violet) and allyl selenol (cyan) within the (A) CA I (yellow), (B) CA II (white), (C) CA IX (blue) and (D) CA XII (green) active site.
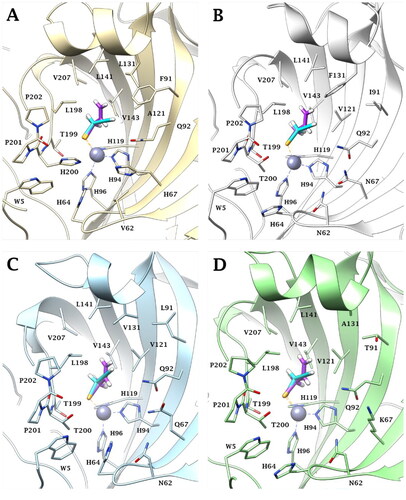
Both a and b selenol actives are positioned in an area featured by conserved amino acid residues in the different CA isoforms, positioning in the same manner in all the studied isoenzymes. In detail, the allyl selenol obtained from the hydrolysis of selenoesters of series a, orients in a cleft, lined by hydrophobic residues (A121 (CA I)/V121 (CA II, IX, XII); L131 (CA I)/F131 (CA II)/V131 (CA IX)/A131 (CA XII); L141, V143, L198 and V207), while the propargyl end points towards the hydrophilic half of the CAs active sites, defined by residues Q92, H67 (CA I)/N67 (CA II)/Q67 (CA IX)/K67 (CA XII), V62 (CA I)/N62 (CA II, IX, XII), H64 and W5.
As is evident by , the geometric parameters (bond length and angles) of the experimental (X-ray data) and the in silico (docking poses) solutions of the a (allyl) and b (propargyl) actives, are in very good agreement (). 100 ns long molecular dynamics simulations confirm the stability over the time of the docking solutions.
Figure 4. (A) Superposition of (A) X-ray solved structures of phenyl selenol-CA II (red, PDB:6HX5)Citation25 and benzyl selenol-CA II (green, PDB:7QBH)Citation53 complex. (B) Superposition of ligands benzyl selenol (green), propargyl selenol (cyan), and allyl selenol (violet). (C) Superposition of X-ray solved structures of benzyl selenol-CA II (green)Citation53 and predicted binding mode of allyl (violet)/propargyl (cyan) selenol within CA II active site.
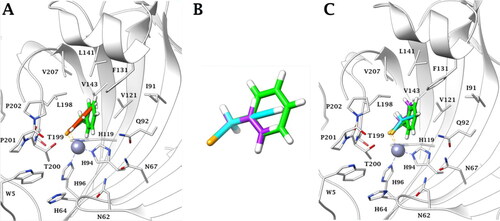
Table 2. Geometric parameters of docking and crystallography data.
The wider network of interactions of the b compared to a active does not, as such, explain the large different activity pointed out by inhibition studies, thereby supporting the previous hypothesis that the difference in the activity between derivatives of the two studied series is due to a different hydrolysis rate of the compounds in the CA isozymes. The more compact conformation assumed by derivatives 1–19b could allow an easier entry into the active sites, promoting faster hydrolysis and leading to a higher inhibitory activity compared to analogs of series a.
Conclusions
Novel 38 Se-analogs which present an innovative structural design have been tested as CA inhibitors. Their design takes advantage of the selenoesterase activity that CA has demonstrated,Citation29 as they combine two active fragments via a selenoester functional group which will be release after the action of CA. In this work, propargyl derivatives have exhibited greater in vitro inhibitory activities against hCA I, hCA II, hCA XI, and hCA XII, rather than the allyl derivatives. Indomethacin (17b) and flufenamic acid (19b) analogs exhibited selectivity for tumour-associated isoform IX in the low micromolar range. Likewise, adamantane analog (1b) showed selectivity for tumour-associated isoforms IX and XII. These results seem to suggest that molecules containing carbocycles are more selective than the ones which present heterocycles, providing a new approach for the investigation of future CA inhibitors.
Molecular modelling was also performed to get insight into the different inhibitory activities of derivatives towards the CA I and II (off-target) and the tumour-associated isoforms CA IX and CA XII. Selenols moieties of allyl and propargyl were evaluated in silico as they were cleaved upon CA action. The compounds of series a and b showed a very good agreement between the geometrical parameters (bond length and bond angles) of the experimental solutions (X-ray data) and the in silico data (docking positions) of active a (allyl) and b (propargyl). However, these results do not explain the large different activity exhibited in vitro. Thus, it seems that the different hydrolysis rate of the compounds in the CA isozymes would be related to the difference in the activity between derivatives of the two studied series. The more compact conformation assumed by derivatives 1–19b could allow an easier entry into the active sites, promoting faster hydrolysis and leading to a higher inhibitory activity compared to analogs of series a.
Acknowledgements
This research was financed by the Italian Ministry for Education and Science (MIUR), grant PRIN: rot. 2017XYBP2R and by Ente Cassa di Risparmio di Firenze (ECRF), grant CRF2020.1395 (to CTS).
Disclosure statement
All authors except CTS declare no conflict of interest. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J. 2016;473(14):2023–2032.
- Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs. 2018;27(12):963–970.
- Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review. Expert Opin Ther Pat. 2018;28(10):729–740.
- Supuran CT, Alterio V, Di Fiore A, D’ Ambrosio K, Carta F, Monti SM, De Simone G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev. 2018;38(6):1799–1836.
- Chiche J, Ilc K, Laferrière J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouysségur J, Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009;69(1):358–368.
- Kciuk M, Gielecińska A, Mujwar S, Mojzych M, Marciniak B, Drozda R, Kontek R. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J Enzyme Inhib Med Chem. 2022;37(1):1278–1298.
- Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol. 2020;12:603–617.
- Courcier J, de la Taille A, Nourieh M, et al. Carbonic anhydrase IX in renal cell carcinoma, implications for disease management. Int J Mol Sci. 2020;21(19):7146.
- Daunys S, Petrikaitė V. The roles of carbonic anhydrases IX and XII in cancer cell adhesion, migration, invasion and metastasis. Biol Cell. 2020;112(12):383–397.
- Queen A, Bhutto HN, Yousuf M, et al. Carbonic anhydrase IX: a tumor acidification switch in heterogeneity and chemokine regulation. Semin Cancer Biol. 2022;86:899–913.
- Lee SH, Griffiths JR. How and why are cancers acidic? carbonic anhydrase IX and the homeostatic control of tumour extracellular pH. Cancers. 2020;12(6):1616.
- Tonissen KF, Poulsen SA. Carbonic anhydrase XII inhibition overcomes P-glycoprotein-mediated drug resistance: a potential new combination therapy in cancer. Cancer Drug Resist. 2021;4(2):343–355.
- Waheed A, Sly WS. Carbonic anhydrase XII functions in health and disease. Gene. 2017;623:33–40.
- Haapasalo J, Nordfors K, Haapasalo H, Parkkila S. The expression of carbonic anhydrases II, IX and XII in brain tumors. Cancers. 2020;12(7):1723.
- Barnett DH, Sheng S, Charn TH, Waheed A, Sly WS, Lin C-Y, Liu ET, Katzenellenbogen BS. Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Res. 2008;68(9):3505–3515.
- McDonald PC, Chafe SC, Supuran CT, Dedhar S. Cancer therapeutic targeting of hypoxia induced carbonic anhydrase IX: from bench to bedside. Cancers. 2022;14(14):3297.
- Calvo-Martín G, Plano D, Encío I, et al. Novel N,N'-disubstituted selenoureas as potential antioxidant and cytotoxic agents. Antioxidants. 2021;10(5):777.
- Ruberte AC, Ramos-Inza S, Aydillo C, et al. Novel N,N'-disubstituted acylselenoureas as potential antioxidant and cytotoxic agents. Antioxidants. 2020;9(1):55.
- Bartolini D, Sancineto L, Fabro de Bem A, Tew KD, Santi C, Radi R, Toquato P, Galli F. Selenocompounds in cancer therapy: an overview. Adv Cancer Res. 2017;136:259–302.
- Chu CK, Ma L, Olgen S, Pierra C, Du J, Gumina G, Gullen E, Cheng YC, Schinazi RF. Synthesis and antiviral activity of oxaselenolane nucleosides. J Med Chem. 2000;43(21):3906–3912.
- Mangiavacchi F, Botwina P, Menichetti E, et al. Seleno-functionalization of quercetin improves the non-covalent inhibition of M(pro) and its antiviral activity in cells against SARS-CoV-2. Int J Mol Sci. 2021;22(13):7048.
- Witek K, Nasim M, Bischoff M, Gaupp R, Arsenyan P, Vasiljeva J, Marć M, Olejarz A, Latacz G, Kieć-Kononowicz K, et al. Selenazolinium salts as “small molecule catalysts” with high potency against ESKAPE bacterial pathogens. Molecules. 2017;22(12):2174.
- Blessing H, Kraus S, Heindl P, Bal W, Hartwig A. Interaction of selenium compounds with zinc finger proteins involved in DNA repair. Eur J Biochem. 2004;271(15):3190–3199.
- Angeli A, Tanini D, Nocentini A, Capperucci A, Ferraroni M, Gratteri P, Supuran CT. Selenols: a new class of carbonic anhydrase inhibitors. Chem Commun. 2019;55(5):648–651.
- Tanini D, Capperucci A, Ferraroni M, Carta F, Angeli A, Supuran CT. Direct and straightforward access to substituted alkyl selenols as novel carbonic anhydrase inhibitors. Eur J Med Chem. 2020;185:111811.
- Tanc M, Carta F, Scozzafava A, Supuran CT. α-Carbonic anhydrases possess thioesterase activity. ACS Med Chem Lett. 2015;6(3):292–295.
- Lopez M, Vu H, Wang CK, Wolf MG, Groenhof G, Innocenti A, Supuran CT, Poulsen S-A. Promiscuity of carbonic anhydrase II. Unexpected ester hydrolysis of carbohydrate-based sulfamate inhibitors. J Am Chem Soc. 2011;133(45):18452–18462.
- Angeli A, Carta F, Donnini S, Capperucci A, Ferraroni M, Tanini D, Supuran CT. Selenolesterase enzyme activity of carbonic anhydrases. Chem Commun. 2020;56(32):4444–4447.
- Astrain-Redin N, Talavera I, Moreno E, Ramírez MJ, Martínez-Sáez N, Encío I, Sharma AK, Sanmartín C, Plano D. Seleno-analogs of scaffolds resembling natural products a novel warhead toward dual compounds. Antioxidants. 2023;12(1):139.
- Astrain-Redin N, Sanmartin C, Sharma A, et al. From natural sources to synthetic derivatives: allyl motif as a powerful tool for fragment-based design in cancer. J Med Chem. 2023;Submitted for publication.
- Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014; Oct15(11):e493-503–e503.
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of mammalian isoforms I-XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg Med Chem. 2008;16(15):7424–7428.
- Cau Y, Vullo D, Mori M, Dreassi E, Supuran C, Botta M. Potent and selective carboxylic acid inhibitors of tumor-associated carbonic anhydrases IX and XII. Molecules. 2017;23(1):17.
- Lomelino CL, McKenna R. Carbonic anhydrase II in complex with carboxylic acid-based inhibitors. Acta Crystallogr F Struct Biol Commun. 2019;75(Pt 3):166–170.
- Falsini M, Squarcialupi L, Catarzi D, Varano F, Betti M, Di Cesare Mannelli L, Tenci B, Ghelardini C, Tanc M, Angeli A, et al. 3-Hydroxy-1H-quinazoline-2,4-dione as a new scaffold to develop potent and selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. J Med Chem. 2017;60(14):6428–6439.
- Altıntop MD, Sever B, Akalın Çiftçi G, Kucukoglu K, Özdemir A, Soleimani SS, Nadaroglu H, Kaplancıklı ZA. Synthesis and evaluation of new benzodioxole-based dithiocarbamate derivatives as potential anticancer agents and hCA-I and hCA-II inhibitors. Eur J Med Chem. 2017;125:190–196.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- Del Prete S, Angeli A, Ghobril C, Hitce J, Clavaud C, Marat X, Supuran CT, Capasso C. Sulfonamide inhibition profile of the β-carbonic anhydrase from Malassezia restricta, an opportunistic pathogen triggering scalp conditions. Metabolites. 2020;10(1):39.
- Vullo D, Del Prete S, Nocentini A, Osman SM, AlOthman Z, Capasso C, Bozdag M, Carta F, Gratteri P, Supuran CT, et al. Dithiocarbamates effectively inhibit the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa. Bioorg Med Chem. 2017;25(3):1260–1265.
- Akgul O, Di Cesare Mannelli L, Vullo D, Angeli A, Ghelardini C, Bartolucci G, Alfawaz Altamimi AS, Scozzafava A, Supuran CT, Carta F, et al. Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the management of rheumatoid arthritis. J Med Chem. 2018;61(11):4961–4977.
- Bua S, Di Cesare Mannelli L, Vullo D, Ghelardini C, Bartolucci G, Scozzafava A, Supuran CT, Carta F. Design and synthesis of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem. 2017;60(3):1159–1170.
- Vomasta D, Innocenti A, König B, Supuran CT. Carbonic anhydrase inhibitors: two-prong versus mono-prong inhibitors of isoforms I, II, IX, and XII exemplified by photochromic cis-1,2-alpha-dithienylethene derivatives. Bioorg Med Chem Lett. 2009;19(5):1283–1286.
- Behnke CA, Le Trong I, Godden JW, Merritt EA, Teller DC, Bajorath J, Stenkamp RE. Atomic resolution studies of carbonic anhydrase II. Acta Crystallogr D Biol Crystallogr. 2010 May;66(Pt 5):616–627.
- Leitans J, Kazaks A, Balode A, Ivanova J, Zalubovskis R, Supuran CT, Tars K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem. 2015;58(22):9004–9009.
- Whittington DA, Waheed A, Ulmasov B, Shah GN, Grubb JH, Sly WS, Christianson DW. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci U S A. 2001;98(17):9545–9550.
- Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, Crichlow GV, Christie CH, Dalenberg K, Di Costanzo L, Duarte JM, et al. RCSB protein data bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021;49(D1):D437–D451.
- Schrödinger Suite Release 2022–3. New York (NY): Schrödinger, LLC; 2022:(a) Maestro v.12.9. (b) Epik, v.5.7. (c) Impact, v.9.2. (d) Prime, v.5.5. (e) Macromodel v.13.3. (f) Glide, v.9.2. (g) Jaguar, v.11.3. (h) Desmond, v.6.7.
- Ibrahim HS, Allam HA, Mahmoud WR, Bonardi A, Nocentini A, Gratteri P, Ibrahim ES, Abdel-Aziz HA, Supuran CT. Dual-tail arylsulfone-based benzenesulfonamides differently match the hydrophobic and hydrophilic halves of human carbonic anhydrases active sites: selective inhibitors for the tumor-associated hCA IX isoform. Eur J Med Chem. 2018;152:1–9.
- Lu C, Wu C, Ghoreishi D, Chen W, Wang L, Damm W, Ross GA, Dahlgren MK, Russell E, Von Bargen CD, et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. 2021;17(7):4291–4300.
- Nocentini A, Bonardi A, Gratteri P, Cerra B, Gioiello A, Supuran CT. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem. 2018;33(1):1453–1459.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612.
- Angeli A, Ferraroni M, Capperucci A, Tanini D, Costantino G, Supuran CT. Selenocarbamates as a prodrug-based approach to carbonic anhydrase inhibition. ChemMedChem. 2022;17(11):e202200085.