
Abstract
Multiple inhibitions of CA, COX-2 and 5-LOX enzymes has been recognised as a useful strategy for the development of anti-inflammatory drugs that can avoid the disadvantages of using NSAIDs alone. Here, we report new pyridazine-based sulphonamides (5a-c and 7a-f) as potential multi-target anti-inflammatory candidates. First, the furanone heterocycle in the dual CA/COX-2 inhibitor Polmacoxib was replaced with the pyridazinone one. Then, a hydrophobic tail was appended through benzylation of the 3-hydroxyl group of the pyridazinone scaffold to afford benzyloxy pyridazines 5a-c. Furthermore, the structures were adorned with the polar sulphonate functionality, in pyridazine sulphonates 7a-f, that are expected to be engaged in interactions with the hydrophilic half of the CA binding sites. All of the disclosed pyridazinones were tested for inhibitory activities against 4 hCA isoforms (I, II, IX, and XII), as well as against COX-1/2, and 5-LOX. Furthermore, in vivo anti-inflammatory and analgesic effects of pyridazinones 7a and 7b were examined.
Introduction
Carbonic anhydrases (EC 4.2.1.1) are metalloenzymes that have a metal ion in their active site.Citation1 CAs were thoroughly studied for decades and were classified as a superfamily of enzymes with eight gene families or classes to date.Citation2 There are basically several cytosolic forms (CA I-III, CA VII), four membrane-bound isozymes (CA IV, CA IX, CA XII, and CA XIV), a mitochondrial form (CA V), and a secreted CA isozyme, CA VI.Citation3 Carbonic anhydrase is involved in diverse physiological and pathological processes including lipogenesis, gluconeogenesis, and tumorigenicity.Citation4 One of the major biochemical reactions involved in proton generation in resting tissues is mediated by carbonic anhydrase (CA) enzymes.Citation5 They are known for their ability to catalyse the reversible hydration of carbon dioxide (CO2 + H2O ⟺ H+ + HCO3−) and have also been extensively demonstrated in a plethora of physiological events at the cellular and tissue levels.Citation6 Numerous CA isoforms have evolved because uncatalyzed transformation is unable to meet the physiological cells' needs.Citation7,Citation8
At the cellular level, overexpression of CA increases the concentration of ions (H+ and HCO3), and since bicarbonate ions are necessary cellular components that are immediately recovered, local extracellular acidosis is established straight away. A decrease in tissue pH is associated with inflammatory pain, and carbonic anhydrases (CAs) are primarily responsible for proton production in tissues.Citation9,Citation10 It has been well-established that the intensity of inflammation and pain-associated symptoms in diverse inflammatory disorders, such as rheumatoid arthritis, is inversely correlated with tissue pH values.Citation9,Citation10 Due to their role in the regulation of pH by reversibly catalysing the conversion of CO2 to bicarbonate and protons, CAs have been recently reported to be implicated in diverse inflammation reactions.Citation11–13 In particular, Cimaz et al.Citation14 reported that hCA IX and XII isoforms, which are widely expressed in hypoxic malignancies, are also over-expressed in the inflamed synovium of individuals with juvenile arthritis.
Lipid mediators, which include the classic eicosanoids, prostaglandins (PGs), and leukotrienes (LTs), are potent inflammatory mediators produced by local cell-type specific arachidonic acid metabolism. However, the arachidonic acid metabolism is carried out by the cyclooxygenase (COX) and lipoxygenase (LOX) families of enzymes, leading to the synthesis of prostaglandins and leukotrienes. Pain, inflammation, asthma, and allergies are all treated with drugs that target these enzymes.Citation15 Dual COX-2 and 5-LOX enzyme inhibition has recently been recognised as a useful strategy for the development of anti-inflammatory drugs that can get around the disadvantage of using NSAIDs alone.Citation16–19
Over the last few decades, sulphonamide-based small molecules were highlighted as an important class that is utilised for the management of many illnesses. Currently, there are more than 20 drugs in clinical usage, such as Acetazolamide (AAZ), Methazolamide, Diclofenamide, Furosemide, Bumetanide, Sulpiride, and Zonisamide (). It has been well-established that the sulphonamide functionality and its isosteric groups (sulfamate and sulfamide) is a crucial element for the CA and selective COX-2 inhibitory actions.Citation20
Figure 1. Examples of clinically used sulphonamide-based drugs.
Polmacoxib (Acelex, ) is a sulphonamide-tethered first-in-class dual CA/COX-2 inhibitor that was approved in 2015 by the Korean Ministry of Food and Drug Safety (MFDS) for the management of osteoarthritis.Citation21–24 Interestingly, the CA inhibitory activity has been reported for several sulphonamide-tethered COX-2 inhibitors such as Celecoxib ().Citation23,Citation25,Citation26 Besides, a proof-of-concept report has revealed that the combination of the fragments of nonsteroidal anti-inflammatory drugs (NSAIDs) and CA inhibitors could improve the effectiveness of drugs used to treat ache symptoms linked to inflammatory disorders including rheumatoid arthritis (RA).Citation27
Figure 2. Chemical structures of the dual CA/COX-2 inhibitors Polmacoxib and Celecoxib, as well as the target pyridazinones 5a-c, and 7a-f.
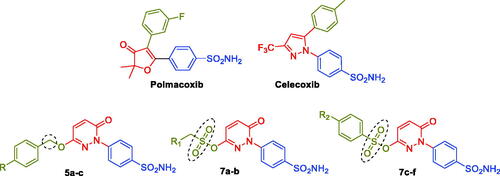
As a privileged scaffold, the pyridazinone motif has attracted a lot of interest in the realm of drug discovery. Due to its diverse range of biological actions and potential therapeutic uses, the pyridazinone moiety has been the focus of extensive investigations.Citation28 Of particular interest, diverse pyridazinone-based molecules have been reported for their anti-inflammatoryCitation29–35 and carbonic anhydrase inhibitoryCitation36,Citation37 activities.
In this report, we developed new pyridazinone-based sulphonamide derivatives (5a-c and 7a-f, ) as potential multi-target anti-inflammatory candidates, using the multi-target strategy.Citation38–40 The hCA, COX-2, and 5-LOX enzymes, which have been implicated in the inflammatory process, are anticipated to be efficiently inhibited by the designed pyridazinone-based sulphonamides (5a-c and 7a-f). Furthermore, the in vivo anti-inflammatory effects for the target pyridazinones were examined.
The design of the target pyridazinone sulphonamides is based on the bioisosteric replacement of the furanone heterocycle in the dual CA/COX-2 inhibitor Polmacoxib with the pyridazinone one, while keeping the benzenesulfonamide motif directly attached to the heterocycle. The incorporated sulfamoyl functionality is anticipated to anchor with the catalytic Zn2+ in the CA binding site, and also promotes COX-2 selectivity via hydrogen bonding within the hydrophilic sub-pocket in the COX-2 binding site.
In addition to the bioisosteric approach followed in the design of the new ligands, a hydrophobic tail was appended to the structure in an attempt to boost the inhibitory activity and selectivity towards hCA isoforms IX and XII. This was achieved through benzylation of the 3-hydroxyl group of the pyridazinone scaffold to afford the first set of the target benzyloxy pyridazine compounds 5a-c (). In addition, the structures were adorned with the polar sulphonate functionality that is expected to be engaged in interactions within the hydrophilic half of the CAs binding site. This was achieved through sulfonation of the 3-hydroxyl group with either alkyl or aromatic sulphonyl chlorides to produce the corresponding alkyl and aromatic pyridazine sulphonates 7a-b and 7c-f, respectively.
Results and discussion
Chemistry
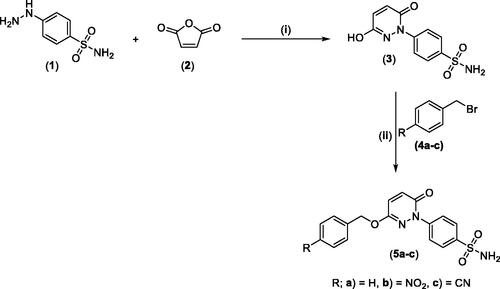
The synthesis of the target pyridazine derivatives is depicted in Schemes 1 and 2. A total of 10 compounds (3, 5a-c, and 7a-e) were synthesised, all of them novel molecules, not previously described in the literature. The syntheses followed known procedures previously described somewhere else.Citation41–43 The target compounds were obtained in an excellent isolated yield.
Reacting commercially available 4-hydrazineylbenzenesulfonamide (1) with maleic anhydride (2) under reflux conditions in water resulted in intermediate 3 in an excellent yield (92%). Next, we turned our attention to the reactivity of 4–(3-hydroxy-6-oxopyridazin-1(6H)-yl)benzenesulfonamide (3) towards an alkylation reaction, which is a very powerful tool for introducing chemical diversity. Compounds 5a-c were accessible by an optimised alkylation procedure by reacting various benzyl bromide derivatives (4a-c) with intermediate (3) to afford the corresponding pyridazine derivatives in excellent yields (Scheme 1).
Scheme 1. General Synthesis of pyridazine derivatives (2 and 5a-c); Reagents and conditions: (i) H2O, reflux, overnight; (ii) Potassium carbonate, DMF, stirring at 5 °C, 3 h.
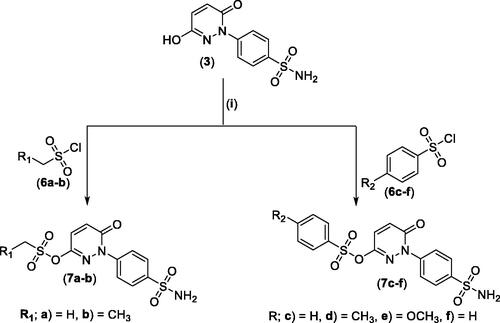
For the second set of the target pyridazine derivatives (7a-f), the synthesis was performed by reacting intermediate 3 with different aliphatic (6a-b) and aromatic (6c-f) sulphonyl chlorides in pyridine as base and solvent. The corresponding sulphonate derivatives (7a-b and 7c-f) were formed in excellent yields after stirring at 5 °C for after 1h (Scheme 2).
Scheme 2. General Synthesis of pyridazine derivatives (7a-f); Reagents and conditions: (i) Pyridine, stirring at 5 °C, 1 h.
Biological evaluation
Carbonic anhydrase inhibition
Using the stopped-flow carbon dioxide hydration assayCitation44 all the newly synthesised pyridazines (3, 5a-c and 7a-f) reported here and the standard CA inhibitor acetazolamide (AAZ) were evaluated for their inhibitory effects against hCA isoforms (I, II, IX, and XII). The tested CA isoforms were suppressed to varying degrees by the pyridazine-based benzenesulfonamides (PBS) described here, and the inhibition data are presented in . The structure-activity relationship (SAR) was obtained based on the inhibition results as KI values for the synthesised analogues.
Table 1. In vitro inhibition data of hCA I, II, IX and XII with pyridazine derivatives (3, 5a-c and 7a-f) by the stopped flow kinetic assay using AAZ as a reference drug.
The cytosolic hCA I isoform was suppressed by all pyridazine-tethered sulphonamide compounds (3, 5a-c and 7a-f) herein reported with inhibition constants (KIs) ranging from low to moderate nanomolar concentrations (i.e. between 23.5 nM and 362.8 nM). The in vitro kinetic data disclosed that the unsubstituted pyridazine derivative 3 is the most potent inhibitor within the tested compounds against the hCA I isoform (KI = 23.5 nM), followed by pyridazines 7a and 7b (KIs = 48.3 and 52.6 nM, respectively). Moderate inhibition potencies, with KI values spanning between 98.3–185.9 nM were obtained for pyridazines 5a, 5b, 7d, and 7c. Compound 7e was the least potent inhibitor (KI= 362.8 nM) compared to the standard CAI AAZ (KI= 250 nM). In general, both benzylation (series 5; KI: 98.3 − 221.5 nM) and sulfonation (series 7; KI: 48.3 − 362.8 nM) of the key intermediate 3-hydroxy-6-oxopyridazin 3 (KI = 23.5 nM) did not improve the inhibitory activities against hCA I. For the benzyloxy pyridazine series 5a-c, the substitution of the pendant phenyl motif led to a decrease in activity towards CA I (5b and 5c: KI = 116.3 and 221.5 nM vs. 5a: KI= 89.3 nM). For the sulphonate series 7, the alkyl sulphonates 7a and 7b (KI = 48.3 and 52.6 nM, respectively) possessed better inhibitory activity than the aryl sulphonates 7c-f (KI = 185.9, 123.5, 362.8 and 165.8 nM, respectively).
Regarding the inhibitory activity towards hCA II isoform, all the examined pyridazine-based sulphonamide derivatives (3, 5a-c and 7a-f) exerted good activity with inhibition constants in the range of 5.3 − 106.4 nM (). Only the benzyloxy pyridazine derivative 5a was inhibited hCA II isoform at the single-digit nanomolar range (KI = 5.3 nM). Moreover, the benzyloxy derivative 5a as well as the sulphonate derivatives 7c, 7d and 7f showed moderate activities towards hCA II isoform with inhibition constants equal 37.1, 34.2, 19.2 and 31.6 nM, respectively. Generally, benzylation (series 5) and sulfonation (series 7) of the key intermediate 3-hydroxy-6-oxopyridazin 3 (KI = 55.8 nM) have enhanced the hCA II inhibitory effect, except for pyridazines 5c, 7b and 7e (KI = 106.4, 79.1 and 88.2 nM, respectively) ().
The synthesised pyridazine-based sulphonamide derivatives (3, 5a-c and 7a-f) all successfully suppressed the cancer-related hCA IX isoform (KI: 4.9 − 58.1 nM) as indicated in . The benzyloxy derivative 5c and the sulphonate derivative 7f were superior to the other derivatives and showed single-digit inhibitory activities (KI = 4.9 and 6.4 nM, respectively) against hCA IX isoform. In addition, pyridazine derivatives 5a, 5b, 7c and 7d showed potent activity with KI values equal to 14.8, 12.3, 19.4 and 22.8 nM, respectively. It is important to emphasise that the benzyloxy-tethered derivatives 5a-c (KI: 4.9 − 14.8 nM) and the aryl sulphonate derivatives 7c-f (KI: 6.4 − 58.1 nM) displayed much enhanced inhibitory activity than the intermediate 3-hydroxy-6-oxopyridazin 3 (KI = 45.1 nM) against hCA IX, whereas the alkyl sulphonate derivatives 7a-b (KI = 52.3 and 58.1 nM, respectively) showed less activity than compound 3.
In terms of the inhibitory effects against the second cancer-related hCA XII isoform tested here, all compounds (3, 5a-c and 7a-f) showed activities in the low nano-molar range (5.3 − 49.7 nM). Among the synthesised pyridazines, derivatives 3 and 7f showed superior single-digit inhibition constants equal to 5.3 and 8.7 nM, respectively. Besides, pyridazine sulphonamides 5c, 7a and 7b exerted effective hCA XII inhibitory effect (KIs = 18.4, 13.3 and 17.2 nM respectively). Notably, pyridazine 3 (KI = 5.3 nM) demonstrated greater potency than its benzyloxy (series 5; KI: 18.4 − 32.2 nM) and sulphonates (series 7; KI: 8.7 − 49.7 nM) analogs. Also, it is worth mentioning that the substitution of the terminal phenyl moiety in both series 5 and 7 resulted in an enhancement for the hCA XII inhibitory action; compounds 5b-c (KIs = 26.1 (7b) and 18.4 (7c) nM) vs. compound 5a (KI = 32.2 nM), and compounds 7d-f (KIs = 42.6 (7d), 35.9 (7e) and 8.7 (7f) nM) vs. compound 7a (KI = 49.7 nM) ().
In vitro COX-1/COX-2 and LOX inhibitory activities
In this study, further evaluation of the anti-COX-2 effect of the pyridazine-based sulphonamide derivatives (3, 5a-c and 7a-f) was carried out, in addition to evaluation of the anti-COX-1 activity in order to assess the COX-2 selectivity of the synthesised pyridazines. Celecoxib (selective COX-2 inhibitor) and Indomethacin (non-selective inhibitor) were utilised as reference COX inhibitors. IC50 values for the inhibition of COX-1 and COX-2, as well as the selectivity index (IC50 of COX-1/IC50 of COX-2) are listed in . The COX-2 enzyme was effectively inhibited by the newly reported pyridazine-based sulphonamide derivatives (3, 5a-c, and 7a-f), with IC50 values in the sub-micromolar range ranging from 0.05 to 0.14 µM, whereas COX-1 was weakly inhibited with IC50 values spanning between 5 and 12.6 µM.
Table 2. IC50 values for the in vitro COX-1/2 and LOX inhibition, as well as COX SI values of the pyridazine-based sulphonamide derivatives 3, 5a-c and 7a-f.
The methanesulfonate and ethanesulfonate pyridazine derivatives (7a and 7b) exerted the best COX-2 inhibitory effect (IC50 = 0.05 and 0.06 µM, respectively) among the tested derivatives. Both 7a and 7b were equipotent to the selective COX-2 inhibitor Celecoxib (IC50 = 0.05 µM) (). Moreover, pyridazines 3, 5c, 7c, 7d and 7e showed potent activity towards COX-2 (IC50 = 0.07, 0.07, 0.8, 0.08 and 0.09 µM for 3, 5c, 7c, 7d and 7e, respectively).
Concerning the selectivity of the target pyridazines towards COX-2 enzyme over COX-1, the findings highlighted that the pyridazine sulphonates (7a and 7b) demonstrated the highest COX-2 selectivity index of 208 and 210, respectively (). In addition, the remaining pyridazine derivatives showed considerable selectivity towards COX-2 enzyme.
Additionally, in this study, the in vitro inhibitory activity of the pyridazine-based sulphonamide derivatives 3, 5a-c, and 7a-f against the LOX enzyme was evaluated ().
Table 3. Inhibition data of LOX enzyme with pyridazine-based sulphonamide derivatives 3, 5a-c and 7a-f.
The tested pyridazines displayed potent to moderate activity against LOX enzyme with IC50 range of 2 – 7 μM. The most potent LOX inhibitors in this work were found to be 3-hydroxy-6-oxopyridazine 3 (IC50 = 2 μM) as well as methanesulfonate 7a (IC50 = 3 μM) and ethanesulfonate pyridazine derivatives 7b (IC50 = 2.5 μM) that exerted better activity than the reference LOX inhibitor Zileuton (IC50 = 3.5 µM). It is worth noting that the best COX-2 inhibitors in this study were pyridazine sulphonates (7a and 7b) (). As a result, both compounds 7a and 7b appear to be promising dual COX-2 and 5-LOX inhibitors.
In vivo biological evaluations
Analgesic activities
The acetic acid writhing test in mice has been used in this study to examine the analgesic properties of the most potent and selective COX-2 inhibitors (pyridazine sulphonates 7a and 7b).Citation45 As shown in , the frequency of writhing was counted as a pain marker.
Table 4. Analgesic impact of the tested methanesulfonate and ethanesulfonate pyridazines 7a and 7b by the use of acetic acid writhing test in mice.
A significant reduction in the total number of writhing was observed as a direct result of treatment with both pyridazine sulphonates 7a and 7b (Number of writhing = 13 and 14, respectively) in comparison to the control group (Number of writhing = 40), which highlights the analgesic effect of 7a and 7b. It’s interesting to note that pyridazines 7a and 7b had stronger analgesic effects than celecoxib ().
Anti‐inflammatory activity
Carrageen-induced rat paw edoema protocol was exploited to evaluate the anti-inflammatory effects of the pyridazine sulphonates 7a and 7b as reported by Winter et al.Citation46 using celecoxib and diclofenac as reference anti-inflammatory drugs. displays the measured percentage change of paw height.
Table 5. Results of the carrageen-induced paw edoema assay that was used to investigate the effects of the pyridazine sulphonates 7a and 7b on paw height.
The tested molecules 7a and 7b exerted significant (p < 0.05) anti-inflammatory activities via reducing the paw height and thus reduction of the paw edoema at 3 h in comparison to the control group (). The % inhibition of edoema of 7a and 7b were 56, and 38.5, respectively. Both compounds were superior to celecoxib with rapid onset of action after 1 h, and sustained duration until the third hour after the administration of the compound. Based on these findings, pyridazine 7a could be a good anti-inflammatory candidate.
Inflammatory mediator’s measurements
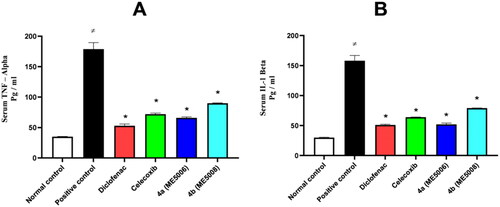
Based on the encouraging anti-inflammatory and analgesic properties of pyridazine sulphonates 7a and 7b stated above, an ELISA assay was used to further assess the serum level of two inflammatory mediators namely, tumour necrosis factor-alpha (TNF-α) and interleukin one beta (IL-1β).
As can be seen in , the levels of the inflammatory mediators in the positive control group were significantly higher at the end of the paw edoema test as compared to the levels in the normal control group. On the other hand, the studied sulphonates 7a and 7b significantly lowered TNF-α and IL-1β levels when compared to the positive control group.
Figure 3. Serum level of the inflammatory mediators (TNF-α and IL-1β) after paw edoema test. The value is expressed as mean ± SD (n = 5). *Significantly different from the positive control group. A) Tumour necrosis factor – alpha (TNF-α). B) Interleukin one beta (IL-1β).
Ulcerogenic effects
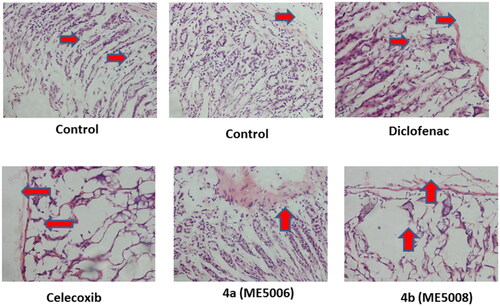
The degree of inflammation or ulceration of the examined pyridazine sulphonates (7a and 7b) in the gastric tissues was evaluated and confirmed by histopathological examinations, following the previously described protocol.Citation47
As shown in , the examination of the fasted rat stomach disclosed normal texture for pyridazine 7a as well as the reference drug celecoxib. On the other hand, pyridazine 7b displayed a mild inflammation as presented in (cf. red arrows). Diclofenac, also, showed inflammation and damage to the layers of the stomach (red arrows), which indicated some ulcerogenic side effects. These data suggested the safety profile of pyridazine 7a on the stomach tissues.
Figure 4. Ulcerogenic effects of the tested pyridazine sulphonates (7a and 7b), diclofenac and celecoxib on the gastric tissue of rats. Histopathological examination was performed using H & E stain and the magnification power of the images was 40X. The red arrows indicated the normal or damaged parts.
Molecular modelling
The most selective pyridazines against the tumour-associated isoforms CA IX and XII versus the ubiquitous cytosolic enzymes CA I and II, 5c and 7f, were investigated in silico to predict their binding mode within the active site of these four isozymes.
As expected, all docking poses showed that the benzenesulfonamide moiety bonded to the zinc ion with the deprotonated nitrogen atom of the sulphonamide group (SO2NH−) in a tetracoordinated geometry. Moreover, the sulphonamide-metal coordination is stabilised by two H-bonds with the side chain OH and the backbone NH of T199, and by hydrophobic interactions that occur between the benzene ring and hydrophobic residues such as V143, L198 and W205 (). In all four enzyme isoforms (CA I, II, IX, XII), the 4-nitrophenyl moiety was oriented towards a lipophilic cleft where the mutation of some amino acid residues occurs among the different CA isoforms, is responsible for the different sizes of the pocket. Consequently, the positioning and stabilisation of the ligands are different. In the CA I, the lipophilic area is lined by Y20, L131, A132, A135, P201, P202 and Y204. Here the 4-nitrophenyl moiety of 5c engages in vdW contacts with A132, P202 and Y204, and a π-π stacking interaction with the Y204 side chain (). In CA II the Y204L, L131F and S136Q mutations allow the NO2 group to orient towards the Q136 and to establish an H-bond with the side chain NH2 (). The ligand is further stabilised by the hydrophobic contacts with F131, G132, L135 and L204 and by π-π stacking contact involving the aromatic pyridazin-3(2H)-one linker and the F131 side chain.
Figure 5. Predicted binding mode of 5c (purple) and 7f (cyan) within A) CA I, B) CA II, C) CA IX and D) CA XII active site.
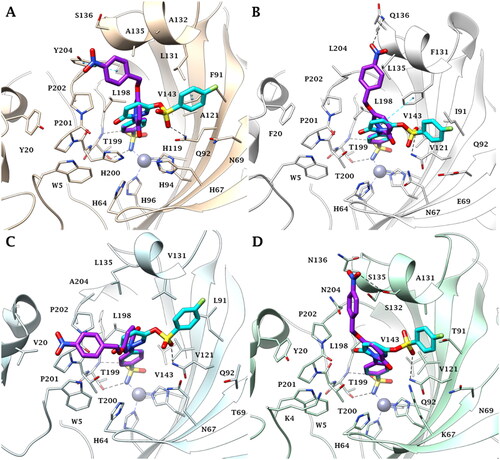
As a result of the CA I (or II)/IX mutations at Y20V (CA I/IX), Y204A (CA II/IX) and F131V in CA IX, the aryl pendant extends towards V20 making vdW interactions with this residue and W5, P201 and P202 (). In CA XII, the nitro group faces the same area as in CA II. However, due to the mutation Q136N (CA II/XII) and the shorter length of the amino acidic side chain, the group forms an H-bond with the side chain OH of S132 (). The larger lipophilic pocket of the tumour-associated isoforms (CA IX and XII) compared to the off-target (CA I and II) ones, allows 5c a good fit and complementarity to the binding site, which is probably the reason for its better inhibitory profile observed experimentally.
In contrast to 5c, the tail of 7f is oriented towards the hydrophilic half of all investigated active sites. In particular, the sulphonic linker engages in an H-bond with the side chain NH2 of the conserved residues Q92 in CA I, II, IX and XII (). The steric hindrance of F91 in CA I and, to less extent, F131 in CA II, led the 4-fluorophenyl group to move much closer to the hydrophilic residues N69 and E69 of the enzyme isoforms (). Moreover, the aromatic ring form π-π stacking interactions with F91 (CA I), while the pyridazin-3(2H)-one linker of 7f forms π-π stacking interaction with the F131 side chain in CA II. Similarly, the larger lipophilic area of the tumour-associated isoforms allows the aryl moiety to engage many contact points with the targets CA IX/CA XII (L91/T91, V121, V131/A131 and Q92), thus binding a larger surface as compared to the other enzyme isoforms that might account for the activity profile (i.e. CA IX = CA XII > CA II ≫ CA I).
Conclusion
We present the design and synthesis of a novel set of pyridazinone-based sulphonamide derivatives (5a-c and 7a-f) as multi-carbonic anhydrase, COX-2, and 5-LOX inhibitors with the primary objective of developing new effective multitarget anti-inflammatory candidates. The furanone heterocycle in the dual CA/COX-2 inhibitor Polmacoxib was replaced with the pyridazinone one while keeping the benzenesulfonamide motif directly attached to the heterocycle. Then, a hydrophobic tail was appended through benzylation of the 3-hydroxyl group of the pyridazinone scaffold to afford benzyloxy pyridazine compounds 5a-c. Also, the structures were adorned with the polar sulphonate functionality, in pyridazine sulphonates 7a-f, that expected to be engaged in interactions with the hydrophilic half of all the CA active site. Most the synthesised pyridazine-based sulphonamides successfully suppressed the examined carbonic anhydrase isoforms in a variable degree; KI ranges: 23.5 − 362.8 nM, 5.3 − 106.4 nM, 4.9 − 58.1 nM and 5.3 − 49.7 nM for CA I, II, IX and XII respectively, also, they effectively inhibited the COX-2 enzyme with IC50 values in the sub-micromolar range ranging from 0.05 to 0.14 µM, whereas COX-1 was weakly inhibited with IC50 values spanning between 5 and 12.6 µM. In particular, the methanesulfonate and ethanesulfonate pyridazine derivatives (7a and 7b) exerted the most potent (IC50 = 0.05 and 0.06 µM) and selective (SI = 208 and 210) COX-2 inhibitory activity. Moreover, the tested pyridazines displayed potent to moderate activity against LOX enzyme with IC50 range of 2–7 μM. Further in vivo investigations for pyridazine sulphonates 7a and 7b revealed their ability to reduce the total number of writhing in mice, the rat paw edoema, and the serum levels of the inflammatory mediators (TNF-α and IL-1β), which highlights their analgesic and anti-inflammatory activities.
Experimental
Chemistry
General remarks
1H and 13C NMR spectra were collected by a Bruker Avance 400 MHz NMR spectrometer at 400 MHz (1H), or 101 MHz (13C), respectively. Chemical shifts are reported in parts per million (ppm) relative to the deuterated solvent, i.e. DMSO, δ 1H: 2.49 ppm; 13C: 39.7 ppm. PLC-HRMS analyses were performed on reverse phase gradient using Agilent (Santa Clara, CA) analyses series binary pump (G1312B), waters XTerra MS C18 (3.5 um; 2.1 × 150 mm) + Phenomenex C18 security guard column (2 × 4 mm) using 0.2% acetic acid in H2O/methanol as mobile phases; wavelength = 254 nm; and mass spectrometry was done with 6220 Agilent (Santa Clara, CA) TOF in electrospray ionisation (ESI) mode with the positive and negative method in both Profile and Centroid mode.
4–(3-Hydroxy-6-oxopyridazin-1(6H)-yl)benzenesulfonamide (3)
In 50 ml screw cap vial, maleic anhydride 2 (2 g, 20.4 mmol) was added to a solution of 4-hydrazineylbenzenesulfonamide 1 (4.56 g, 20.4 mmol) in boiling water (20 ml). The reaction mixture was refluxed overnight, and then cooled to room temperature. The produced solid was collected by filtration, washed with hot water and dried to yield the desired compound 3 as a pale-yellow powder (92%); 1H NMR (400 MHz, DMSO-d6) δ 11.47 (s, 1H, OH), 7.96 − 7.86 (m, 2H), 7.86 − 7.77 (m, 2H), 7.44 (s, 2H, SO2NH2), 7.20 (d, J = 9.8 Hz, 1H), 7.05 (d, J = 9.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 157.86 (C = O), 153.16, 143.83, 142.49, 134.13, 128.19, 126.05, 125.54. HRMS m/z for C10H9N3O4S [M + Na]+. Calcd 290.020598, found 290.020620.
General procedure for the synthesis of benzyloxy pyridazine compounds (5a-c)
The appropriate benzyl bromide derivative 4a-c (0.432 mmol) was added drop-wise to a solution of 4–(3-hydroxy-6-oxopyridazin-1(6H)-yl)benzenesulfonamide 3 (0.393 mmol) and potassium carbonate (0.432 mmol) in DMF (1 ml) at 5 °C, the reaction mixture was allowed to stir for 3 h and monitored with TLC. The reaction mixture was poured into cold water and extracted with ethyl acetate (3 × 15 ml). The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure to yield pyridazines 3a-c.
4–(3-(Benzyloxy)-6-oxopyridazin-1(6H)-yl)benzenesulfonamide (5a)
White powder (89%); 1H NMR (400 MHz, DMSO-d6) δ 7.89 (q, J = 8.8 Hz, 4H), 7.51 − 7.33 (m, 8H), 7.13 (d, J = 9.8 Hz, 1H), 5.24 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 157.97 (C=O), 152.40, 143.77, 142.52, 136.02, 134.18, 128.45, 128.24, 127.87, 126.09, 125.15, 68.59. HRMS m/z for C17H15N3O4S [M + Na]+ calcd 380.057548, found 380.067428.
4–(3-((4-Nitrobenzyl)oxy)-6-oxopyridazin-1(6H)-yl)benzenesulfonamide (5b)
White powder (91%); 1H NMR (400 MHz, DMSO-d6) δ 8.34 − 8.21 (m, 2H), 7.95 − 7.68 (m, 6H), 7.50 − 7.38 (m, 3H), 7.16 (d, J = 9.8 Hz, 1H), 5.41 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 157.97, 152.07, 147.15, 143.94, 143.65, 142.57, 134.33, 128.63, 127.73, 126.10, 125.13, 123.59, 67.28. HRMS m/z for C17H14N4O6S [M + Na]+ calcd 425.052626, found 425.052865.
4–(3-((4-Cyanobenzyl)oxy)-6-oxopyridazin-1(6H)-yl)benzenesulfonamide (5c)
White powder (90%);1H NMR (400 MHz, DMSO-d6) δ 7.90 (dd, J = 8.5, 6.9 Hz, 4H), 7.86 − 7.81 (m, 2H), 7.70 − 7.64 (m, 2H), 7.43 (d, J = 10.1 Hz, 3H), 7.15 (d, J = 9.8 Hz, 1H), 5.35 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 157.98, 152.13, 143.67, 142.57, 141.85, 134.29, 132.42, 128.47, 127.79, 126.11, 125.14, 118.73, 110.74, 67.56. HRMS m/z for C18H14N4O4S [M + Na]+ calcd 405.062797, found 405.062819.
General procedure for the synthesis of sulphonate pyridazine compounds (7a-f)
The appropriate sulphonyl chloride derivative 6a-f (0.374 mmol) was added to a solution of 4–(3-hydroxy-6-oxopyridazin-1(6H)-yl)benzenesulfonamide 3 (0.374 mmol) in pyridine (1 ml) at 5 °C and stirred for 1 h. The mixture was poured into 2 N HCl solution and then extracted with ethyl acetate (3 × 15 ml). The combined organic layers were washed with water and brine. The organic layer was dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure to yield compounds 7a-f.
6-Oxo-1–(4-sulfamoylphenyl)-1,6-dihydropyridazin-3-yl methanesulfonate (7a)
White powder (94%); 1H NMR (400 MHz, DMSO-d6) δ 7.95 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 9.9 Hz, 1H), 7.49 (s, 2H), 7.32 (d, J = 9.9 Hz, 1H), 3.61 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 158.38, 146.14, 143.53, 142.88, 134.91, 129.62, 126.32, 125.88, 39.21. HRMS m/z for C11H11N3O6S2 [M + Na]+ calcd. 367.998148, found 367.998341.
6-Oxo-1–(4-sulfamoylphenyl)-1,6-dihydropyridazin-3-yl ethanesulfonate (7b)
White powder (95%); 1H NMR (400 MHz, DMSO-d6) δ 7.95 (d, J = 8.5 Hz, 2H), 7.85 − 7.78 (m, 2H), 7.69 − 7.60 (m, 1H), 7.49 (s, 2H), 7.31 (dd, J = 9.9, 0.8 Hz, 1H), 3.75 (q, J = 7.3 Hz, 2H), 1.39 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 158.35, 146.10, 143.51, 142.88, 134.87, 129.71, 126.34, 125.82, 46.30, 7.97. HRMS m/z for C12H13N3O6S2 [M + Na]+ calcd. 382.013798, found 382.013848.
6-Oxo-1–(4-sulfamoylphenyl)-1,6-dihydropyridazin-3-yl benzenesulfonate (7c)
White powder (92%); 1H NMR (400 MHz, DMSO-d6) δ 8.08 − 7.99 (m, 2H), 7.88 (dd, J = 8.6, 2.2 Hz, 3H), 7.84 − 7.79 (m, 1H), 7.73 (t, J = 7.7 Hz, 2H), 7.58 (d, J = 9.9 Hz, 1H), 7.48 (t, J = 4.3 Hz, 4H), 7.28 (d, J = 9.9 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 158.22, 153.15, 145.78, 143.46, 142.56, 135.54, 135.00, 134.51, 130.00, 128.63, 126.17, 125.42. HRMS (EI) m/z for C16H13N3O6S2 [M + Na]+ calcd. 430.013798, found 430.014189.
6-Oxo-1–(4-sulfamoylphenyl)-1,6-dihydropyridazin-3-yl 4-methylbenzenesulfonate (7d)
White powder (96%); 1H NMR (400 MHz, DMSO-d6) δ 7.91–7.87 (m, 4H), 7.59 − 7.42 (m, 7H), 7.27 (d, J = 9.9 Hz, 1H), 2.45 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 158.21, 146.59, 145.76, 143.61, 142.52, 134.92, 131.46, 130.40, 130.35, 129.51, 128.72, 128.70, 126.13, 125.43, 124.64, 21.20. HRMS m/z for C17H15N3O6S2 [M + Na]+ calcd. 444.029448, found 444.029379.
6-Oxo-1–(4-sulfamoylphenyl)-1,6-dihydropyridazin-3-yl 4-methoxybenzenesulfonate (7e)
White powder (91%); 1H NMR (400 MHz, DMSO-d6) δ 7.92 (d, J = 9.1 Hz, 2H), 7.89 − 7.81 (m, 4H), 7.60 − 7.43 (m, 5H), 7.19 (d, J = 9.1 Hz, 1H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.22, 164.57, 158.24, 145.84, 143.51, 142.62, 132.21, 132.10, 131.23, 126.17, 125.52, 115.12, 56.09. HRMS m/z for C17H15N3O7S2 [M + Na]+ calcd 460.024362; found 460.024876.
6-Oxo-1–(4-sulfamoylphenyl)-1,6-dihydropyridazin-3-yl 4-fluorobenzenesulfonate (7f)
White powder (89%); 1H NMR (400 MHz, DMSO-d6) δ 8.13 (dd, J = 8.9, 5.0 Hz, 1H), 7.94 (d, J = 8.9 Hz, 1H), 7.89 (dd, J = 8.6, 3.8 Hz, 2H), 7.62 − 7.47 (m, 6H), 7.30 − 7.20 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 164.57, 158.24, 145.84, 145.73, 143.51, 142.62, 132.21, 132.10, 131.23, 126.17, 125.52, 115.12. HRMS (EI) m/z for C16H12FN3O6S2 [M + Na]+ calcd 448.004376, found 448.004591.
Biological evaluations
COX-1, COX-2 and LOX inhibition assay
The ability of the examined pyridazine-based sulphonamide derivatives 3, 5a-c and 7a-f to inhibit both COX‐1/COX‐2 has been in vitro investigated by the use of COX Inhibitor screening ELISA assay kit (cat. no. 560131, Cayman, USA) according to the supplier’s recommendations and as described previously.Citation35 Besides, LOX inhibitory screening assay kit (Cat. No. 760700, Cayman, USA) was exploited to evaluate the 5-LOX inhibitory activity of the examined pyridazine-based sulphonamide derivatives 3, 5a-c and 7a-f according to the manufacturer’s instructions and as previously reported.Citation48
Determination of the CA inhibitory activities
The experimental methodology utilised to evaluate the CA inhibitory action of the pyridazine-based sulphonamide derivatives 3, 5a-c and 7a-f disclosed here was previously described.Citation49–53
In vivo assays
The experimental procedures adopted to perform the acetic acid-induced writhing assay for the analgesic activities,Citation45 Paw edoema assay for the anti-inflammatory activities,Citation46 as well as the acute ulcerogenic testCitation47 have been carried out as described previously.
Biochemical determination for certain inflammatory mediators
ELISA assays were used to assess the serum level for the inflammatory mediators TNF-α (Kit Cat. No MBS355371, MyBioSource, USA), and IL-1β (Kit Cat. No MBS8825017, MyBioSource, USA) based on the manufacture instructions.Citation54
Molecular Modelling
For the computational docking study, crystal structures of CA I, II, IX and XII (PDBs: 6Y00, 3K34, 5FL4 and 5LL5, respectivelyCitation55–58) have been downloaded from Protein Data BankCitation59 and prepared using the Protein Preparation Wizard tool implemented in the Schrödinger suite. The detailed procedures exploited in this analysis are previously reported.Citation54
Supplemental Material
Download PDF (2.3 MB)Disclosure statement
CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J. 2016;473(14):2023–2032.
- Nocentini A, Supuran CT, Capasso C. An overview on the recently discovered iota-carbonic anhydrases. J Enzyme Inhib Med Chem. 2021;36(1):1988–1995.
- Supuran CT. Carbonic anhydrases-an overview. Curr Pharm Des. 2008; 14(7):603–614.
- Eldehna WM, Nocentini A, Elsayed ZM, Al-Warhi T, Aljaeed N, Alotaibi OJ, Al-Sanea MM, Abdel-Aziz HA, Supuran CT. Benzofuran-based carboxylic acids as carbonic anhydrase inhibitors and antiproliferative agents against breast cancer. ACS Med Chem Lett. 2020;11(5):1022–1027.
- Angeli A, Carta F, Nocentini A, Winum J-Y, Zalubovskis R, Akdemir A, Onnis V, Eldehna WM, Capasso C, Simone GD, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites. 2020;10(10):412.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem. 2016;31(3):345–360.
- Tanc M, Carta F, Scozzafava A, Supuran CT. α-Carbonic anhydrases possess thioesterase activity. ACS Med Chem Lett. 2015;6(3):292–295.
- Supuran CT. Carbonic anhydrase inhibitors: An update on experimental agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig. 2021;1–12.
- Farr M, Garvey K, Bold AM, Kendall MJ, Bacon PA. Significance of the hydrogen ion concentration in synovial fluid in rheumatoid arthritis. Clin. Exp. Rheumatol. 1985;3:99–104.
- Steen KH, Steen AE, Reeh PW. A dominant role of acid pH in inflammatory excitation and sensitization of nociceptors in rat skin, in vitro. J Neurosci. 1995;15(5 Pt 2):3982–3989.
- Chang X, Han J, Zhao Y, Yan X, Sun S, Cui Y. Increased expression of carbonic anhydrase I in the synovium of patients with ankylosing spondylitis. BMC Musculoskelet Disord. 2010;11(1):1–11.
- Zheng Y, Wang L, Zhang W, Xu H, Chang X. Transgenic mice over-expressing carbonic anhydrase I showed aggravated joint inflammation and tissue destruction. BMC Musculoskelet Disord. 2012;13(1):19.
- Liu C, Wei Y, Wang J, Pi L, Huang J, Wang P. Carbonic anhydrases III and IV autoantibodies in rheumatoid arthritis, systemic lupus erythematosus, diabetes, hypertensive renal disease, and heart failure. Clin Dev Immunol. 2012;2012:354594.
- Margheri F, Ceruso M, Carta F, Laurenzana A, Maggi L, Lazzeri S, Simonini G, Annunziato F, Del Rosso M, Supuran CT, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem. 2016;31(sup4):60–63.
- Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011;96(1-4):27–36.
- Gökşen US, Kelekçi NG. A new avenue in anti-inflammatory therapy: Dual inhibitors of cyclooxygenase and 5-lipoxygenase. Hacettepe Univ Eczacilik Fak Derg. 2010;1:81–118.
- P JJ, Manju SL, Ethiraj KR, Elias G. Safer anti-inflammatory therapy through dual COX-2/5-LOX inhibitors: A structure-based approach. Eur J Pharm Sci. 2018;121:356–381.
- Koeberle A, Werz O. Multi-target approach for natural products in inflammation. Drug Discov Today. 2014;19(12):1871–1882.
- Meshram MA, Bhise UO, Makhal PN, Kaki VR. Synthetically-tailored and nature-derived dual COX-2/5-LOX inhibitors: Structural aspects and SAR. Eur J Med Chem. 2021;225:113804.
- Peerzada MN, Hamel E, Bai R, Supuran CT, Azam A. Deciphering the key heterocyclic scaffolds in targeting microtubules, kinases and carbonic anhydrases for cancer drug development. Pharmacol Ther. 2021;225:107860.
- Lee C, Yang J, Kang S, Seo H, Kim C, Kim S, Park M, Chang H, Schmidt W, Cho J, et al. CG100649, a novel dual-acting COX-2 and carbonic anhydrase inhibitor: Preclinical pharmacology. J. Pain. 2008;9(4):5.
- Singh O, Kakularam KR, Reddanna P, Aparoy P. Understanding the dual inhibition of COX-2 and carbonic anhydrase-II by Celecoxib and CG100649 using density functional theory calculations and other molecular modelling approaches. Protein Pept Lett. 2015;22(10):903–912.
- Kim HT, Cha H, Hwang KY. Structural insight into the inhibition of carbonic anhydrase by the COX-2-selective inhibitor polmacoxib (CG100649). Biochem Biophys Res Commun. 2016; 478(1):1–6.
- Hirankarn S, Barrett JS, Alamuddin N, FitzGerald GA, Skarke C. GCG100649, A novel cyclooxygenase-2 Inhibitor, exhibits a drug disposition profile in healthy volunteers compatible with high affinity to carbonic anhydrase-I/II: Preliminary dose-exposure relationships to define clinical development strategies. Clin Pharmacol Drug Dev. 2013; 2(4):379–386.
- Weber A, Casini A, Heine A, Kuhn D, Supuran CT, Scozzafava A, Klebe G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. J Med Chem. 2004;47(3):550–557.
- Fiore AD, Pedone C, D'Ambrosio K, Scozzafava A, De Simone G, Claudiu TS. Carbonic anhydrase inhibitors: valdecoxib binds to a different active site region of the human isoform II as compared to the structurally related cyclooxygenase II ‘selective’ inhibitor celecoxib. Bioorg Med Chem Lett. 2006;16(2):437–442.
- Bua S, Di Cesare Mannelli L, Vullo D, Ghelardini C, Bartolucci G, Scozzafava A, Supuran CT, Carta F. Design and synthesis of novel nonsteroidal anti-Inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem. 2017;60(3):1159–1170.
- Akhtar W, Shaquiquzzaman M, Akhter M, Verma G, Faraz Khan M, Alam MM. The therapeutic journey of pyridazinone. Eur J Med Chem. 2016;123:256–281.
- Hassan MS, Ahmed EM, El‐Malah AA, Kassab AE. Anti‐inflammatory activity of pyridazinones: A review. Arch Pharm (Weinheim) . 2022;355(8):e2200067.
- Singh J, Sharma D, Bansal R. Pyridazinone: an attractive lead for anti-inflammatory and analgesic drug discovery. Future Med Chem. 2017;9(1):95–127.
- Saini M, Kumar Mehta D, Das R, Saini G. Recent advances in anti-inflammatory potential of pyridazinone derivatives. Mini Rev Med Chem. 2016;16(12):996–1012.
- Cantini N, Schepetkin IA, Danilenko NV, Khlebnikov AI, Crocetti L, Giovannoni MP, Kirpotina LN, Quinn MT. Pyridazinones and structurally related derivatives with anti-inflammatory activity. Molecules. 2022;27(12):3749.
- Ahmed EM, Kassab AE, El-Malah AA, Hassan MS. Synthesis and biological evaluation of pyridazinone derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents. Eur J Med Chem. 2019;171:25–37.
- Barberot C, Moniot A, Allart-Simon I, Malleret L, Yegorova T, Laronze-Cochard M, Bentaher A, Médebielle M, Bouillon J-P, Hénon E, et al. Synthesis and biological evaluation of pyridazinone derivatives as potential anti-inflammatory agents. Eur J Med Chem. 2018;146:139–146.
- Loksha YM, Abd‐Alhaseeb MM. Synthesis and biological screening of some novel 6‐substituted 2‐alkylpyridazin‐3 (2H)‐ones as anti‐inflammatory and analgesic agents. Arch Pharm Chem Life Sci. 2020;353(3):1900295.
- Krasavin M, Shetnev A, Baykov S, Kalinin S, Nocentini A, Sharoyko V, Poli G, Tuccinardi T, Korsakov M, Tennikova TB, et al. Pyridazinone-substituted benzenesulfonamides display potent inhibition of membrane-bound human carbonic anhydrase IX and promising antiproliferative activity against cancer cell lines. Eur J Med Chem. 2019;168:301–314.
- Yaseen R, Ekinci D, Senturk M, Hameed AD, Ovais S, Rathore P, Samim M, Javed K, Supuran CT. Pyridazinone substituted benzenesulfonamides as potent carbonic anhydrase inhibitors. Bioorg Med Chem Lett. 2016;26(4):1337–1341.
- Alcaro S, Bolognesi ML, García-Sosa AT, Rapposelli S. Multi-target-directed ligands (MTDL) as challenging research tools in drug discovery: From design to pharmacological evaluation. Front Chem. 2019;7:71.
- Supuran CT. Multitargeting approaches involving carbonic anhydrase inhibitors: hybrid drugs against a variety of disorders. J Enzyme Inhib Med Chem. 2021;36(1):1702–1714.
- Medina-Franco JL, Giulianotti MA, Welmaker GS, Houghten RA. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today. 2013;18(9-10):495–501.
- Elagawany M, Ibrahim MA, Ali Ahmed HE, El-Etrawy ASh, Ghiaty A, Abdel-Samii ZK, El-Feky SA, Bajorath J. Design, synthesis, and molecular modelling of pyridazinone and phthalazinone derivatives as protein kinases inhibitors. Bioorg Med Chem Lett. 2013; 23(7):2007–2013.
- Elagawany M, Schmitt M, Ghiaty A, El-Etrawy ASh, Ibrahim MA, Bihel F, Sbardelotto AB, Pessoa C, Nguyen TL, Hamel E, et al. Synthesis and antiproliferative effects of 5,6-disubstituted Pyridazin-3(2H)-ones designed as conformationally constrained combretastatin A-4 Analogues. Anticancer Agents Med Chem. 2013; 13(7):1133–1140.
- Elagawany M, Ibrahim M, Ibrahim T, Al-Mahmoudy A, Moawad A, Ghoneim M, Radwan M, Panda S. Synthesis and anticancer studies of novel N-benzyl pyridazinone derivatives. LDDD. 2017;14(9):1008–1013.
- Khalifah RG. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase: I. stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- Gawade S. Acetic acid induced painful endogenous infliction in writhing test on mice. J Pharmacol Pharmacother. 2012;3(4):348.
- Winter CA, Risley EA, Nuss GW. Carrageenin-induced edema in hind paw of the rat as an assay for antiinflammatory drugs. Proc Soc Exp Biol Med. 1962;111(3):544–547.
- Ibrahim TH, Loksha YM, Elshihawy HA, Khodeer DM, Said MM. Synthesis of Some Novel 2,6-Disubstituted Pyridazin-3(2H)-one Derivatives as Analgesic, Anti-Inflammatory, and Non-Ulcerogenic Agents. Arch Pharm Chem Life Sci. 2017;350(9):e1700093.
- Ahmed AHH, Mohamed MF, Allam RM, Nafady A, Mohamed SK, Gouda AE, Beshr EA. Design, synthesis, and molecular docking of novel pyrazole-chalcone analogs of lonazolac as 5-LOX, iNOS and tubulin polymerization inhibitors with potential anticancer and anti-inflammatory activities. Bioorg Chem. 2022;129:106171.
- Alkhaldi AAM, Al-Sanea MM, Nocentini A, Eldehna WM, Elsayed ZM, Bonardi A, Abo-Ashour MF, El-Damasy AK, Abdel-Maksoud MS, Al-Warhi T, et al. 3-Methylthiazolo [3,2-a] benzimidazole-benzenesulfonamide conjugates as novel carbonic anhydrase inhibitors endowed with anticancer activity: Design, synthesis, biological and molecular modeling studies. Eur J Med Chem. 2020;207:112745.
- Shaldam M, Eldehna WM, Nocentini A, Elsayed ZM, Ibrahim TM, Salem R, El-Domany RA, Capasso C, Abdel-Aziz HA, Supuran CT. Development of novel benzofuran-based SLC-0111 analogs as selective cancer-associated carbonic anhydrase isoform IX inhibitors. Eur J Med Chem. 2021;216:113283.
- Petreni A, Bonardi A, Lomelino C, Osman SM, ALOthman ZA, Eldehna WM, El-Haggar R, McKenna R, Nocentini A, Supuran CT. Inclusion of a 5-fluorouracil moiety in nitrogenous bases derivatives as human carbonic anhydrase IX and XII inhibitors produced a targeted action against MDA-MB-231 and T47D breast cancer cells. Eur J Med Chem. 2020;190:112112.
- Fares M, Eladwy RA, Nocentini A, Abd El Hadi SR, Ghabbour HA, Abdel-Megeed A, Eldehna WM, Abdel-Aziz HA, Supuran CT. Synthesis of bulky-tailed sulfonamides incorporating pyrido [2, 3-d][1, 2, 4] triazolo [4, 3-a] pyrimidin-1 (5H)-yl) moieties and evaluation of their carbonic anhydrases I, II, IV and IX inhibitory effects. Bioorg Med Chem. 2017;25(7):2210–2217.
- Al-Sanea MM, Elkamhawy A, Paik S, Bua S, Ha Lee S, Abdelgawad MA, Roh EJ, Eldehna WM, Supuran CT. Synthesis and biological evaluation of novel 3-(quinolin-4-ylamino) benzenesulfonamides as carbonic anhydrase isoforms I and II inhibitors. J Enzyme Inhib Med Chem. 2019;34(1):1457–1464.
- Ragab MA, Eldehna WM, Nocentini A, Bonardi A, Okda HE, Elgendy B, Ibrahim TS, Abd-Alhaseeb MM, Gratteri P, Supuran CT, et al. 4-(5-Amino-pyrazol-1-yl) benzenesulfonamide derivatives as novel multi-target anti-inflammatory agents endowed with inhibitory activity against COX-2, 5-LOX and carbonic anhydrase: design, synthesis, and biological assessments. Eur J Med Chem. 2023;250:115180.
- Ali M, Bozdag M, Farooq U, Angeli A, Carta F, Berto P, Zanotti G, Supuran CT. Benzylaminoethyureido-tailed benzenesulfonamides: design, synthesis, kinetic and X-ray investigations on human carbonic anhydrases. IJMS. 2020;21(7):2560.
- Behnke CA, Le Trong I, Godden JW, Merritt EA, Teller DC, Bajorath J, Stenkamp RE. Atomic resolution studies of carbonic anhydrase II. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 5):616–627.
- Leitans J, Kazaks A, Balode A, Ivanova J, Zalubovskis R, Supuran CT, Tars K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem. 2015;58(22):9004–9009.
- Whittington DA, Waheed A, Ulmasov B, Shah GN, Grubb JH, Sly WS, Christianson DW. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA. 2001;98(17):9545–9550.
- RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021;49:D437–D451.