
Abstract
Pan-histone deacetylase (HDAC) inhibitors often have some toxic side effects. In this study, three series of novel polysubstituted N-alkyl acridone analogous were designed and synthesised as HDAC isoform-selective inhibitors. Among them, 11b and 11c exhibited selective inhibition of HDAC1, HDAC3, and HDAC10, with IC50 values ranging from 87 nM to 418 nM. However, these compounds showed no inhibitory effect against HDAC6 and HDAC8. Moreover, 11b and 11c displayed potent antiproliferative activity against leukaemia HL-60 cells and colon cancer HCT-116 cells, with IC50 values ranging from 0.56 μM to 4.21 μM. Molecular docking and energy scoring functions further analysed the differences in the binding modes of 11c with HDAC1/6. In vitro anticancer studies revealed that the hit compounds 11b and 11c effectively induced histone H3 acetylation, S-phase cell cycle arrest, and apoptosis in HL-60 cells in a concentration-dependent manner.
Introduction
Histone modifications, including acetylation and deacetylation, are considered key factors in epigenetic regulation during carcinogenesisCitation1,Citation2. Histones in cancer cells are generally in a low acetylation state, and some abnormality of histone deacetylases (HDACs) can cause the imbalance of histone acetylation, which leads to the occurrence of cancerCitation3–7. Eighteen HDAC isoforms have been found in mammals, among which, class I (HDAC1, 2, 3, 8) and class II (IIA: HDAC4, 5, 7, 9; IIB: HDAC6, 10) are Zn2+-dependent and overexpressed in a variety of cancer cellsCitation8–12. At present, there are five HDAC inhibitors (HDACIs, ) approved for marketing and clinical use in the world, namely vorinostat (SAHA), romidepsin, belinostat, panobinostat, and chidamide, which are mainly used for the treatment of peripheral T-cell lymphoma and multiple myelomaCitation13–15.
Figure 1. The chemical structures of marketed and some reported HDAC inhibitors (CAP, ZBG and Linker are marked with cyan, orange and purple, respectively).
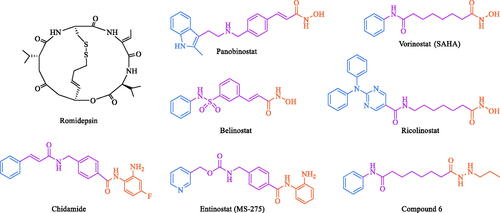
In general, the pharmacophore of HDACIs contains a zinc-binding group (ZBG), which is used to chelate zinc ions at the bottom of the HDAC pocket, and a hydrophobic surface recognition group, also known as the CAP region. Additionally, a linker is needed to connect the ZBG to the CAPCitation16–19. In the structure-activity study, hydrophobic groups such as aryl ring, quinoline, benzimidazole, purine and pyrimidine are commonly used as the CAPCitation20. When the CAP is a hydrophobic group with large steric hindrance, the molecule may exhibit isoform-selective inhibition against HDAC6Citation21,Citation22. For instance, SAHA, a pan-HDAC inhibitor, only has a structural difference from ricolinostat () in the CAP region, but ricolinostat is a HDAC6 selective inhibitorCitation23. For linker, it is divided into rigid chain and flexible chain. It was difficult to evaluate the relationship between the structure and isoform-selectivity only through the change of linkerCitation24,Citation25. For example, the marketed drugs SAHA and chidamide have a flexible aliphatic linker and a rigid linker in their structures, respectively. At present, hydroxamic acids, aminobenzamides or their derivatives, carboxylic acids and mercaptans are often used as the ZBG region of HDAC inhibitorsCitation26–28. Hydroxamic acids inhibitors such as SAHA, romidepsin, belinostat and panobinostat, generally had pan-HDAC inhibitory activityCitation7,Citation8, while those containing aminobenzamides such as chidamide and entinostat (MS-275, ), mainly targeting class I HDACsCitation4,Citation16. In addition, replacing hydroxamic acid with acylhydrazine group (compound 6, ) can also achieve significant selective inhibition against class I HDACsCitation29. Pan-HDAC inhibitors often bring some toxic side effects, which limits the clinical applicationCitation14,Citation20. Therefore, more and more attention has been paid to the development of isoform-selective HDACIs in recent yearsCitation21,Citation30.
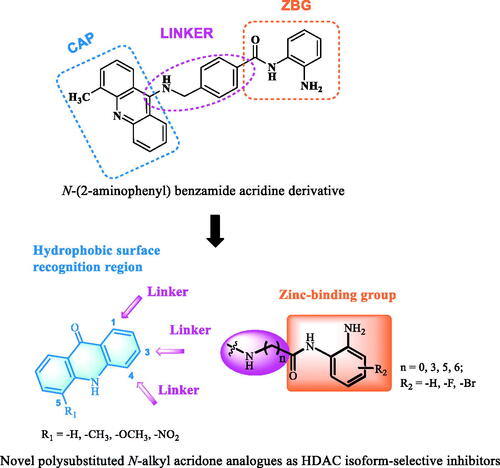
Our group previously designed and synthesised an N-(2-aminophenyl) benzamide acridine derivative (), which moderately but selectively inhibited HDAC1, with an IC50 value of 248 nM. The antiproliferative activity of this compound against leukaemia cell lines was between 120 and 350 nMCitation31,Citation32. In this study, a tricyclic conjugated acridone ring was selected as the hydrophobic region of the pharmacophore for making new HDACIs. Different ZBG structural fragments, such as benzamides with different substituents, were introduced at the C-1, C-3, and/or C-4 positions of the acridone scaffold through flexible alkyl chains to probe the resulting activities. In sum, a series of novel polysubstituted N-alkyl acridone analogues were designed and synthesised as HDAC isoform-selective inhibitors (). Reported herein are the structure-activity relationship (SAR) studies based on in vitro cancer cell antiproliferative activity and in vitro HDACs inhibition, and the selection of hit molecules after mechanistic validation.
Figure 2. The structure of an N-(2-aminophenyl) benzamide acridine derivative and our design strategy for generating novel isoform-selective HDACIs. CAP, ZBG and Linker are marked with cyan, orange and purple, respectively.
Results and discussion
Chemical synthesis
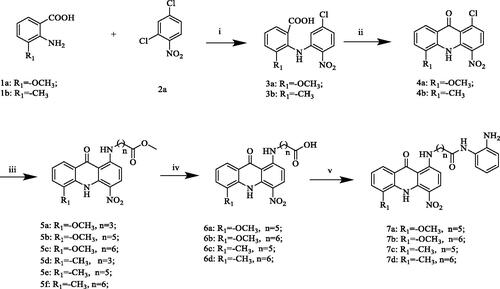
The synthetic routes for compounds 7a–d, 8a, 11a–d and 16a–b are described in Schemes 1–3. First, for the first series of compounds (Scheme 1), using different substituted anthranilic acids 1a–b and 2,4-dichloronitrobenzene 2a as starting materials, the benzoic acids 3a–b were obtained through Cu–catalyzed Ullmann reaction. Subsequently, Friedel–Crafts acylation was carried out in concentrated sulphuric acid to give acridone intermediates 4a–b, which were then converted into 5a–f by N-nucleophilic substitution reaction, and then hydrolysed under alkaline conditions to prepare 6a–d. Finally, target compounds 7a–d are obtained by amide condensation reaction between 6a–d and o-phenylenediamine derivatives at room temperature in the presence of HATU and DIPEA. Next, to explore the SAR of these compounds, we tried to introduce different flexible linkers at other positions on the acridone ring. To this end, the second and third series of compounds (Schemes 2 and Citation3), 8a, 11a–d and 16a–b were designed and synthesised respectively. The main difference in structure was that the benzamide side chain originally at the 1-position of the acridone ring was replaced at the C-3 or C-4 position of the scaffold. The synthetic methods of these two series of compounds were similar to 7a–d. Structures of all target compounds were confirmed by 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS) data.
Scheme 1. Reagents and conditions: (i) K2CO3, Cu, 130 °C, DMF, overnight; (ii) H2SO4, 80 °C, reflux, 4 h; (iii) Methyl 4-aminobutyrate hydrochloride or its derivatives, 2-ethoxyethanol, Et3N, 90–100 °C, overnight; (iv) NaOH/H2O, 80 °C, 6h; (v) HATU, DIPEA, o-phenylenediamine, DMF, DCM, room temperature, overnight.
Scheme 2. Reagents and conditions: (i) K2CO3, Cu, DMF, 130 °C, overnight; (ii) H2SO4, 80 °C, reflux, 4h; (iii) HATU, DIPEA, o-phenylenediamine derivatives, DMF, DCM, room temperature, overnight; (iv) CDI, methyl 4-aminobutyrate hydrochloride or its derivatives, DMF, DCM, room temperature, overnight; (v) NaOH/H2O, 80 °C, 6h; (vi) HATU, DIPEA, o-phenylenediamine or its derivatives, DMF, DCM, room temperature, overnight.
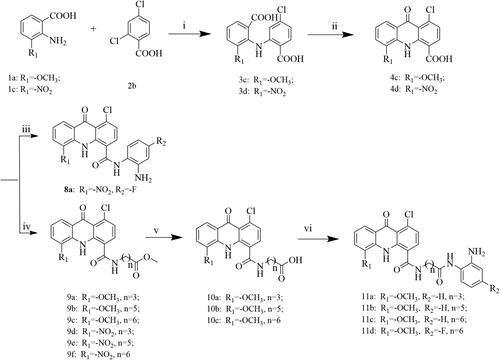
Scheme 3. Reagents and conditions: (i) DMF, K2CO3, Cu, 130 °C, overnight; (ii) H2SO4, 80 °C, reflux, 4 h; (iii) HATU, DIPEA, o-phenylenediamine or its derivatives, DMF, DCM, room temperature, overnight.
In vitro HDAC isoforms (HDAC1/6) selectivity
To explore the potency and isoform-selectivity, we first chose class I HDAC (HDAC1) and class II HDAC (HDAC6) for in vitro screening, along with chidamide and SAHA as positive controls. As can be seen from , 7a–d and 11b–d exhibited significant selective inhibitory activity against HDAC1 with IC50 values ranging from 87 to 358 nM, but showed no inhibitory effect on HDAC6 (IC50 > 10,000 nM). This indicated that the designed compounds introducing a 2-aminobenzamide moiety (R5) as ZBG at the 1-position of the acridone scaffold via an alkyl chain (flexible linker), were potent and selective for HDAC1 inhibition. Preliminary SAR analysis suggested that the substituent group (R4) at the 5-position of the acridone scaffold had some effect on HDAC1 inhibitory activity. When the substituent R4 was a methoxy group, the activity was about two-fold higher than that of methyl-substituted compounds, suggested by the IC50 values of 7a vs. 7c, 7b vs. 7d. Secondly, the length of alkyl chains between the 4-carboxamide group and the 2-aminobenzamide moiety had a great influence on HDAC1 inhibitory activity. In general, linkers with longer lengths were found to be more suitable for maintaining potency. For instance, 7a–d and 11b–d containing five or six methylene units (n = 5 or 6) displayed higher activity than those containing three methylene units (11a, n = 3), and the IC50 values were more than 11-fold different. However, for compounds without any methylene units in the linker (n = 0), such as 8a, 16a, and 16b, the activity was completely lost. According to the results in , if the length of the linker was consistent, the introduction of the ZBG at 1- or 4-position of the acridone scaffold had little effect on the inhibitory activity, such as for 7a vs. 11b, and 7b vs. 11c. In addition, when the R5 groups were differently substituted 2-aminobenzamide moieties, the activity was also affected. For instance, when a fluorine atom was introduced into the benzamide (11d), the IC50 value was 3-fold lower than that of the unsubstituted benzamide (11c). The results indicated that 7a–d and 11b–d were novel HDAC isoform-selective inhibitors. Among them, the IC50 value of 11c against HDAC1 was 87 nM, which is lower than that observed for the positive control, chidamide (114 nM), and the selectivity index (SI) for the HDAC isoform was also superior (> 114.9 vs. > 87.8).
Table 1. In vitro HDAC isoforms (HDAC1 and HDAC6) selectivity and selectivity index of the designed compounds containing different 2-aminobenzamide groups.
Binding modes of the representative compound 11c with HDAC1/6
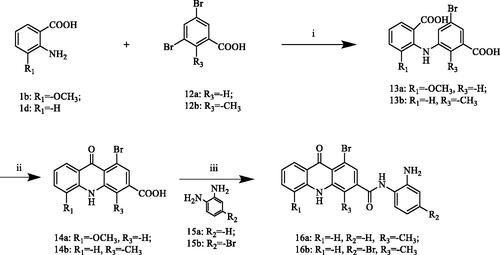
Based on the results of in vitro HDAC isoform-selectivity screening, 11c with the best inhibitory activity against HDAC1 (IC50 = 87 nM) was selected to analyse the docking modes with different isoforms of HDAC. Molecular docking studies of 11c with HDAC1 (PDB ID: 5ICN, ) showed that the oxygen atom of the amide and the Zn2+ ion formed a coordination bond and the nitrogen atom of the amide formed a hydrogen bond with Gly149. The phenylamine moiety formed hydrophobic interactions with Cys151 by Pi-sulfur interaction and its amino group was involved in hydrogen bonds with His141, Asp176 and His178. The planar conjugated aromatic acridone ring and the aliphatic flexible linker of 11c located in a hydrophobic area, which allowed it to form serval Pi-alkyl and alkyl bonds with Leu271, Phe205, Phe158 and Pro29. In addition, the oxygen atom of the carbonyl group and the acridone ring formed two Pi-anion bonds with the amino acid residue Asp99, respectively. As a pan-HDAC inhibitor, SAHA exhibited strong inhibitory activity against HDAC1/6. Therefore, we selected SAHA as the control to analyse the binding modes between the active compounds and HDAC isoforms. As can be seen from , the docking mode of SAHA and HDAC1 was similar to that of 11c. First of all, hydroxamic acid fragment formed a coordination bond with zinc ion and formed a hydrogen bond with residue TYR303. Secondly, the amino group of the of the amide formed another hydrogen bond with residue Asp99. Furthermore, the aliphatic flexible linker and the benzene ring in SAHA form multiple Pi-alkyl and alkyl bonds with residues Leu271, Phe150 and Pro29.
Figure 3. The binding mode of 11c with HDAC1 (PDB ID: 5ICN; A: 3D view and B: 2D view) and SAHA with HDAC1 (PDB ID: 5ICN; C: 3D view and D: 2D view). The carbon atoms of 11c and SAHA are represented as sticks and the carbon atoms of HDAC1 residues are shown as grey sticks, and oxygen, nitrogen and chlorine atoms are shown in red, blue and green, respectively. The active site is shown in the hydrophobicity surface. The zinc ion is represented in a gray sphere. All the non-bonded interactions are shown in dotted lines with different colours according to their different interaction properties.
The above results revealed that 11c had excellent docking modes with HDAC1. In order to better illustrate the HDAC isoform-selectivity, the docking modes of 11c with HDAC6 were further analysed in this study. As can be seen from supplemental Figure S1, compared with SAHA, the docking effect between 11c and HDAC6 was not significant, and there were only one Pi-Pi interaction and one hydrogen bond between the ligand and the receptor. Moreover, there is no interaction between ZBG and zinc ion. Therefore, these were consistent with the results of in vitro screening of target inhibition activity. In addition, Discovery Studio 2020 was used in this study to predict the binding affinity of 11c and SAHA against the target proteins HDAC1/6 through two scoring functions. It can be seen from , -CDocker Interaction Energy value of 11c for HDAC1 (59.3695 kcal/mol) was higher than that of HDAC6 (31.0782 kcal/mol), and the binding energy value of 11c for HDAC1 (−341.899 kcal/mol) was lower than that of HDAC6 (−201.810 kcal/mol). Therefore, this study reasonably elucidated the selective inhibition of 11c against HDAC isoforms by molecular docking and energy scoring functions, which were consistent with the results of in vitro HDAC1/6 selectivity screening.
Table 2. Molecular docking scores of 11c and SAHA against target proteins HDAC1/6.
In silico predictions of physicochemical properties
According to the screening results of HDAC1/6 inhibitory activity, 7a–d and 11a–d with potent HDAC1 inhibitory activity were selected to predict their physicochemical properties, to verify whether they had good drug-like properties. In this study, Molinspiration Cheminformatics 2022 was used to predict the physicochemical parameters of the selected compounds. It can be seen from that these compounds had good drug-like properties. For instance, the number of hydrogen bond acceptors (HBA) was 6–10, the number of hydrogen bond donors (HBD) was 2–5, and the octanol/water partition coefficient (clogP) was 3.19–5.04. Additionally, the molecular weight of these compounds ranged from 397.16 to 539.18. The above parameters were basically within the range specified by the Lipinski’s rule. Therefore, these predicted results suggested that 7a–d and 11a–d may have appropriate physicochemical and drugability properties, which were worthy of subsequent anti-cancer research.
Table 3. Calculated physicochemical properties of 7a–d and 11a–d.
In vitro antiproliferative activities against cancer cell lines
To evaluate whether compounds with inhibitory activity of HDAC1 have antiproliferative activity against cancer cell lines, 7a–d and 11a–d were selected for the MTT reduction assay against human promyelocytic leukaemia HL-60 cells and colon cancer HCT-116 cells, and chidamide was used as the positive control (). First of all, the first series of compounds (7a–d) showed significant anti-proliferation activity against HL-60 cells in vitro, with IC50 values ranging from 0.56 to 4.30 μM. Among them, the IC50 value of 7a was 0.62 μM, which showed a stronger anti-proliferation effect than chidamide. However, these compounds showed no inhibitory activity against solid tumour HCT-116 cells. The SAR studies have shown that compounds were more active when the R1 group was methoxy rather than methyl, such as 7a vs. 7c, 7b vs. 7d. In addition, for the HL-60 cancer cell line, when the linker contained 5 methylene units (n = 5), compounds showed the highest antiproliferative activity. However, when the linker contains six methylene units (n = 6), the activity decreases by about 2.4- to 3.9-fold, such as 7a vs. 7b, and 7c vs. 7d.
Table 4. In vitro antiproliferative activity against colon cancer HCT-116 cells and promyelocytic leukaemia HL-60 cells.
Secondly, compared with 7a–d, the second series of compounds (11b–d) exhibited significant anti-proliferation activity against both HL-60 cells and HCT-116 cells (). The SAR study indicated that keeping the R1 as a methoxy group, the chain length of the linker also had a great influence on the anticancer activity. When the linker contained 5 methylene units (n = 5), compounds had the best anticancer effect in vitro, followed by 6 methylene units (n = 6). However, when it had 3 methylene units (n = 3), the activity was significantly reduced or even lost. This was consistent with the SAR of in vitro HDACs inhibitory activity screening. For example, the IC50 value of 11a was more than 23-fold lower than that of 11b against HL-60 cells. Meanwhile, 11a had no anti-proliferation activity against HCT-116 cells. In addition, compared with 11c without any substitution on the benzene ring, the IC50 value of 11d against HCT-116 cells and HL-60 cells decreased 2-fold and 8-fold, respectively, after the introduction of the fluorine atom. We also found that the inhibitory activity of the compounds against solid tumour cells was generally weaker than that against leukaemia cells. For instance, 11b and 11c were three to five times less active against HCT-116 cells than against HL-60 cells. Among these compounds, 11b showed the highest anti-proliferation activity against HL-60 cells and HCT-116 cells, with IC50 values of 0.56 μM and 3.17 μM, respectively, which were more significant than chidamide.
Based on the above discussion, we further tested the antiproliferative activity of the high active compounds 11b and 11c against human liver L-O2 cells. showed that these two compounds had low in vitro toxicity, among them, the IC50 value of 11b was about nine-fold lower than that of HL-60 cells, which was similar to that of chidamide (10.3-fold). Therefore, based on the high HDAC1 inhibitory activity and antiproliferative activity against different cancer cells as well as low toxicity in vitro, 11b and 11c were selected to evaluate the isoform-selectivity against other HDACs.
Table 5. In vitro toxicity test of compounds against normal human liver L-O2 cells.
Other HDAC isoforms (HDAC3/8/10) selectivity of 11b and 11c
To further evaluate the selective inhibitory activity against other HDAC isoforms (HDAC3, 8, 10), 11b and 11c were selected for activity screening, chidamide and SAHA were used as positive controls. As can be seen from , 11b and 11c with selective inhibitory activity of HDAC1, also exhibited significant inhibitory activity against HDAC3 (class I HDAC), with IC50 values of 418 nM and 225 nM, respectively. Moreover, similar to chidamide, these compounds showed strong inhibitory activity against HDAC10 (class II HDAC) with IC50 values of 387 nM and 177 nM, respectively. However, these two compounds showed no inhibitory activity against HDAC8 (class I HDAC). Among them, the inhibitory activity of 11c against HDAC3 and HDAC10 was more potent than that of chidamide. Therefore, 11b and 11c were selected as hit compounds in this study for subsequent anticancer mechanism studies.
Table 6. In vitro HDAC isoforms (HDAC3, 8, 10) selectivity of 11b and 11c.
Western blot analysis
Acetylation of lysine residues of histone H3 can dilate and activate chromatin, thus facilitating the binding of transcription factors to promoters of tumour suppressor genes, subsequently promoting the transcription and expression of tumour suppressor genes. In order to further investigate the effects of hit compounds 11b and 11c on the activity of HDAC enzyme, western blotting was used to verify whether the compounds affected the acetylation level of histone H3 in cancer cells. As can be seen from , 11b and 11c significantly induced histone H3 acetylation (Ac-H3) in a concentration-dependent manner after the treatment of HL-60 cells with these compounds of different concentrations for 48 h. Therefore, these results suggested that 11b and 11c effectively inhibited the intracellular HDAC1 enzyme activity and promoted the hyperacetylation of histone H3 in HL-60 cells.
Figure 4. (A) HL-60 cells were treated with different concentrations of compound 11b and 11c for 48 h, western blot analysis was used to evaluate the levels of histone H3 acetylation (Ac–H3), Rb and p-Rb. β-Actin was used as the loading control. (B) The densitometry of proteins performed on the western blotting of 11b, *p < 0.05; **p < 0.01; (C) The densitometry of proteins performed on the western blotting of 11c, *p < 0.05; **p < 0.01.
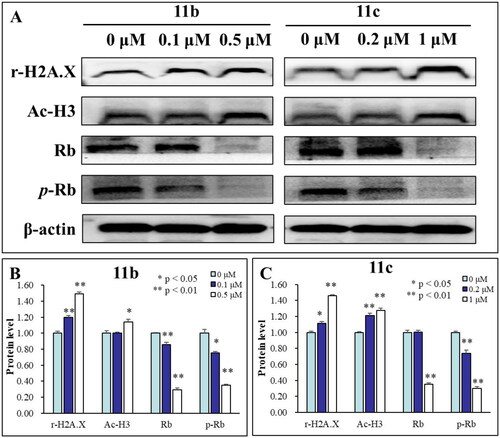
γ-H2AX is considered to be an important biomarker of DNA damageCitation33. HDAC inhibitors generally induce increases in DNA damageCitation34, therefore, it was determined whether 11b and 11c could induce the formation of γ-H2AX by immunoblot analysis. As shown in , the expression of γ-H2AX in HL-60 cells was up-regulated with the increase of compound concentration, indicating that compounds 11b and 11c induced DNA damage in HL-60 cells in a concentration-dependent manner. Phosphorylation of the retinoblastoma tumour suppressor gene (p-Rb) is an important cell cycle regulator involved in cell growth and differentiation, and the change in p-Rb expression level can be used to indicate the effect of drugs on the cell cycleCitation35,Citation36. In this study, we explored the effects of 11b and 11c on the expression levels of cell cycle-related proteins Rb and p-Rb. The results in showed that Rb and p-Rb were significantly down-regulated with the increasing concentration of 11b and 11c, which also indicated that these two compounds significantly affected the growth cycle of HL-60 cells.
S Phase arrest induced by 11b and 11c
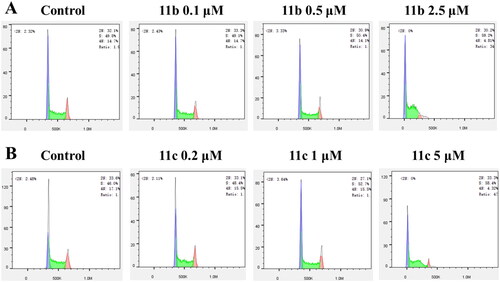
Flow cytometry was used to further analyse the effects of 11b and 11c on the cancer cell cycle. According to the IC50 value of 11b and 11c against the proliferation inhibition of HL-60 cells, different concentrations of compounds were selected to treat HL-60 cells, and the results were shown in . After 11b was incubated with cells at different concentrations of 0.1 μM, 0.5 μM and 2.5 μM for 48 h, the proportion of cells in S phase arrest increased significantly and were 49.1%, 50.4%, and 59.2%, respectively (). In addition, the proportion of cells with S phase arrest induced by 11c at different concentrations of 0.2 μM, 1 μM, and 5 μM were 48.4%, 52.7%, and 58.4%, respectively (). Therefore, the above results indicated that both 11b and 11c effectively induced S-phase arrest of HL-60 cells in a concentration-dependent manner.
Figure 5. Results of cell cycle arrest in HL-60 cells treated with compounds 11b (A) and 11c (B) for 48 h. 11b and 11c arrested the HL-60 cell cycle at the S phase.
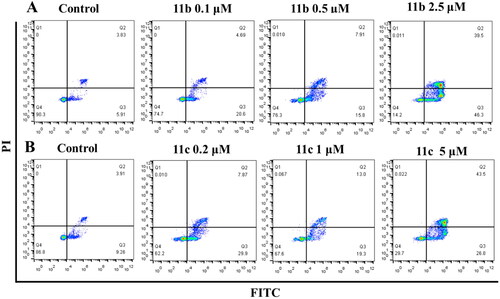
Apoptosis induced by 11b and 11c
An Annexin V-FITC/PI kit was used to detect whether 11b and 11c induced apoptosis in HL-60 cells. Annexin V-FITC can recognise early apoptotic cells and show green fluorescence. Propyridine iodide (PI) is used to identify late apoptotic and necrotic cells, showing red fluorescence. HL-60 cells were treated with different concentrations of 11b (0.1 μM, 0.5 μM, and 2.5 μM) and 11c (0.2 μM, 1 μM and 5 μM) for 48 h, respectively. As shown in , both 11b and 11c could effectively induce the apoptosis of HL-60 cells in a concentration-dependent manner. When the concentration of 11b increased from 0 μM to 2.5 μM, the proportion of apoptotic cells increased from 9.74% to 85.8% (). In addition, when the concentration of 11c increased from 0 μM to 5 μM, the proportion of apoptotic cells increased from 13.17% to 70.3% (). Therefore, combined with the activity screening results and the anticancer mechanism research, this study revealed that 11b and 11c were novel HDAC isoform-selective inhibitors, which could effectively induce apoptosis of cancer cells.
Figure 6. Flow cytometric analysis of the apoptotic rate. HL-60 cells were treated with 11b (A) and 11c (B) compounds for 48h.
Conclusions
In this study, three series of novel polysubstituted N-alkyl acridone analogues were designed and synthesised as HDAC isoform-selective inhibitors by introducing different flexible alkyl chains at the C-1, C-3, and/or C-4 positions of a tricyclic conjugated acridone scaffold, and the effects of different ZBGs on the selective inhibitory activity of HDAC isoforms were fully explored. In evaluating the preliminary SAR, we found that the compounds were most active when the linker contained 5–6 methylene units (n = 5 or 6). Additionally, compared with the relatively rigid chain structure of chidamide, compounds with more flexible linkers also exhibited potent target inhibition activity and isoform-selectivity; Furthermore, using the tricyclic conjugated acridone ring as the CAP region was shown to be feasible for drug design, according to in vitro screening results. In future follow-up studies, it will be of interest to change the conjugated tricyclic ring to a bicyclic ring (such as quinoline or isoquinoline ring) to further explore the structure and activity relationship for this class of HDAC inhibitors. It was also found that the substituent groups on the acridone ring with different electronic effects had great influence on the observed activity. For instance, the presence of electron-withdrawing groups (e.g., 5-nitro) significantly reduced the target inhibition activity. Compounds with a 2-aminobenzamide moiety (as the ZBG) generally exhibited strong HDAC isoform-selective inhibitory activity and in vitro antitumor activity. For instance, 11b and 11c selectively inhibited class I HDAC (HDAC1 and HDAC3) with IC50 values between 87 nM and 418 nM. In addition, 11b and 11c also showed strong inhibitory activity against HDAC10 (class II HDAC), with IC50 values of 387 nM and 117 nM, respectively. Among them, 11c displayed stronger inhibitory activity against HDAC isoforms than chidamide. However, these two compounds showed no inhibitory activity against HDAC6 (class II HDAC) and HDAC8 (class I HDAC). Based on these results, it is speculated that the isoform-selectivity may be caused by the introduction of 2-aminobenzamide moiety into the structure. In the MTT screening, 11b and 11c showed obvious antiproliferative activity against human promyelocytic leukaemia HL-60 cells and the colon cancer HCT-116 cells, with the IC50 values ranging from 0.56 μM to 4.21 μM. Further in vitro anticancer mechanistic studies showed that these two compounds could effectively induce histone H3 acetylation, and induce S-phase cell cycle arrest and apoptosis in HL-60 cells in a concentration-dependent manner. In conclusion, 11b and 11c can be used as novel HDAC isoform-selective inhibitors with a 2-aminobenzamide substructure for further hit-to-lead optimisation in preclinical development.
Experimental section
Synthesis and characterisation
See supporting information for synthetic methods and the preparation of compounds 3a–d, 10a–c, 13a–b, and 14a–b.
Materials and reagents
Anthranilic acid and its derivatives, 2,4-dichloronitrobenzene, 2,4-dichlorobenzoic acid, 2–(7-azabenzotriazole)-N,N,N',N'-tetramethylurea hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), o-phenylenediamine derivatives, CDI, N,N-dimethylethylenediamine and p-methoxybenzylamine were purchased from Energy Chemical (Shanghai, China). Methyl 4-aminobutyrate hydrochloride and its derivatives were purchased from Bide Pharmatech Ltd. (Shanghai, China). K2CO3, NaOH, Et3N, DMF and NaCl were purchased from Shanghai Macklin Biochemical Co., Ltd. (Shanghai, China). H2SO4, HCl, Cu were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). 2-ethoxyethanol and DMSO were purchased from Aladdin (Shanghai, China). Petroleum ether, ethyl acetate, methanol, ethanol and DCM were purchased from Shanghai Titan Technology Co., Ltd. (Shanghai, China). DMSO-d6 and Chloroform-d were purchased from Adamas-beta (Shanghai, China).
General procedure for compounds 5a–f
Step 1: Compound 3a–b (3.10 mmol) was stirred at 80 °C in 10 mL 98% H2SO4 for 3–5 h. After cooling to room temperature, the reaction mixture was slowly added in 100–200 mL ice water with rapid stirring for 30 min, then 20% NaOH (a.q.) was added until pH = 6–8, and the precipitate was formed. Compound 4a–b was obtained after filtration and drying, which was used without further purification. Step 2: Compound 4a–b (3.29 mmol), methyl 4-aminobutyrate or its derivatives (13.16 mmol), and 0.5 mL triethylamine were added to 20 mL 2-ethoxyethanol. The reaction was carried out at 90–110 °C overnight under nitrogen until the TLC (petroleum ether/ethyl acetate = 1/1, v/v) showed the disappearance of the starting material. The mixture was extracted with water (20 mL × 3) and dichloromethane (20 mL × 3), and the organic layer was washed with saturated sodium chloride solution (30 mL), then evaporated in vacuum to yield a crude product. Petroleum ether/ethyl acetate (3/1, v/v) were used as the eluent in column chromatography to give a pure product 5a–f.
Methyl 4-((5-methoxy-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) butanoate (5a)
Yellow solid powder, two steps yield: 42.2%, m.p.:167.2–173.9 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.75 (s, 1H), 11.93 (s, 1H), 8.43 (d, J = 9.8 Hz, 1H), 7.84 (d, J = 8.1 Hz, 1H), 7.27 (d, J = 8.0 Hz, 1H), 7.14 − 7.11 (m, 1H), 6.37 (d, J = 9.8 Hz, 1H), 4.07 (s, 3H), 3.71 (s, 3H), 3.49 (q, J = 7.0 Hz, 2H), 2.53 (t, J = 7.1 Hz, 2H), 2.14–2.08 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 173.13,158.07, 147.76, 139.51, 133.86, 129.45, 123.24, 123.06, 121.52, 116.85, 112.11,104.78, 102.06, 56.33, 51.85, 42.24, 31.10, 24.03; HR-MS(ESI): Calcd for [M + H]+ 386.1352; Found: 386.1353.
Methyl 6-((5-methoxy-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) hexanoate (5b)
Yellow solid powder, two steps yield: 37.6%, m.p.:174.4–174.5 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.76 (s, 1H), 11.87 (s, 1H), 8.41 (d, J = 9.7 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.29 − 7.23 (m, 2H), 7.12 (d, J = 7.7 Hz, 1H), 6.31 (d, J = 9.8 Hz, 1H), 4.07 (s, 3H), 3.67 (s, 3H), 3.40 (q, J = 6.6 Hz, 2H), 2.37 (t, J = 7.4 Hz, 2H), 1.82 (p, J = 7.2 Hz, 2H), 1.73 (p, J = 7.5 Hz, 2H), 1.54 (p, J = 7.8 Hz, 2H).13C NMR (151 MHz, Chloroform-d) δ 179.87, 173.92, 157.97, 147.77, 139.60, 133.81, 129.46, 123.22, 123.13, 121.34, 116.85, 112.07, 104.68,102.11, 56.33, 51.60, 42.98, 33.82, 28.53, 26.60, 24.56; HR-MS(ESI): Calcd for [M + H]+ 414.1665; Found: 414.1665.
Methyl 7-((5-methoxy-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) heptanoate (5c)
Yellow solid powder, two steps yield: 40.7%, m.p.:160.6–161.2 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.80 (s, 1H), 11.89 (s, 1H), 8.43 (d, J = 9.7 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 7.7 Hz, 1H), 6.33 (d, J = 9.8 Hz, 1H), 4.08 (s, 3H), 3.67 (s, 3H), 3.41 (q, J = 7.0 Hz, 2H), 2.34 (t, J = 7.4 Hz, 2H), 1.81 (p, J = 7.3 Hz, 2H), 1.68 (p, J = 7.5 Hz, 2H), 1.52 (p, J = 7.5 Hz, 2H), 1.42 (p, J = 7.8 Hz, 2H).13C NMR (151 MHz, Chloroform-d) δ 178.87, 173.08, 156.98, 146.76, 138.63, 132.80, 128.46, 122.20, 122.15, 120.29, 115.86, 111.06, 103.66, 101.15, 55.32, 50.52, 42.10, 32.92, 27.75, 27.61, 25.75, 23.71; HR-MS(ESI): Calcd for [M + H]+ 428.1822; Found: 428.1822.
Methyl 4-((5-methyl-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) butanoate (5d)
Yellow solid powder, two steps yield: 43.3%, m.p.: 165.1–166.9 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.72 (s, 1H), 11.97 (s, 1H), 8.44 (d, J = 9.8 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 7.1 Hz, 1H), 7.29 (d, J = 7.8 Hz, 1H), 6.41 (d, J = 9.8 Hz, 1H), 3.72(s, 3H), 3.51 (q, J = 6.9 Hz, 2H), 2.61 (s, 3H), 2.54 (t, J = 7.1 Hz, 2H), 2.13 (q, J = 7.1 Hz, 2H).13C NMR (151 MHz, Chloroform-d) δ 179.29, 172.07, 157.08, 139.14, 136.08, 133.45, 132.84, 124.23, 122.84, 122.46, 121.54, 120.38, 103.34, 101.41, 50.84, 41.24, 30.05, 23.03, 15.68; HR-MS(ESI): Calcd for [M + H]+ 370.1403; Found: 370.1404.
Methyl 6-((5-methyl-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) hexanoate (5e)
Yellow solid powder, two steps yield: 41.9%, m.p.:158.7–160.2 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.68 (s, 1H), 11.88 (s, 1H), 8.39 (d, J = 9.8 Hz, 1H), 8.16 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 7.1 Hz, 1H), 7.25 (d, J = 7.2 Hz, 1H), 6.32 (d, J = 9.8 Hz, 1H), 3.68 (s, 3H), 3.41 (q, J = 6.8 Hz, 2H), 2.58 (s, 3H), 2.37 (t, J = 7.4 Hz, 2H), 1.82 (p, J = 7.3 Hz, 2H), 1.73 (p, J = 7.5 Hz, 2H), 1.54 (p, J = 7.8 Hz, 2H).13C NMR (151 MHz, Chloroform-d) δ 180.20, 173.93, 157.94, 140.16, 137.04, 134.40, 133.73, 125.26, 123.80, 123.45, 122.57, 121.18, 104.17, 102.48, 51.62, 43.01, 33.83, 28.55, 26.60, 24.56, 16.69; HR-MS(ESI): Calcd for [M + H]+ 398.1716; Found: 398.1715.
Methyl 7-((5-methyl-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) heptanoate (5f)
Yellow solid powder, two steps yield: 39%, m.p.:150.6–153.7 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.76 (s, 1H), 11.92 (s, 1H), 8.43 (d, J = 9.8 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 7.1 Hz, 1H), 7.29 (d, J = 7.7 Hz, 1H), 6.36 (d, J = 9.8 Hz, 1H), 3.68 (s, 3H), 3.42 (q, J = 7.0 Hz, 2H), 2.61 (s, 3H), 2.34 (t, J = 7.4 Hz, 2H), 1.82 (p, J = 7.3 Hz, 2H), 1.68 (p, J = 7.5 Hz, 2H), 1.53 (p, J = 7.4 Hz, 2H), 1.43 (p, J = 7.5 Hz, 2H).13C NMR (151 MHz, Chloroform-d) δ 180.25, 174.10, 157.99, 140.24, 137.08, 134.40, 133.76, 125.25, 123.84,123.45, 122.62, 121.17, 104.20, 102.53, 51.55, 43.14, 33.93, 28.76, 28.64, 26.77, 24.73, 16.71;HR-MS(ESI): Calcd for [M + H]+ 412.1872; Found: 412.1874.
General procedure for compounds 7a–d
Step 1: Compound 5b–c or 5e–f (2.42 mmol) and NaOH (0.11 g, 2.66 mmol) were stirred in methanol (2 mL) and water (10 mL), and the mixture was stirred at 80–100 °C for 6 h. After the reaction was complete, removed the methanol, and the resulting solution was acidified with hydrochloric acid to a pH of 6–7. The precipitate was filtered and then dried to give 6a–d, which was used without further purification. Step 2: Compound 6a–d (1.25 mmol), 2–(7-azabenzotriazol-1-yl)- N,N,N',N'-tetramethyluronium hexafluorophosphate (HATU, 0.71 g, 1.88 mmol), N,N-diisopropylethylamine (DIPEA, 0.32 g, 2.50 mmol), were stirred in anhydrous N,N-dimethylformamide (DMF, 12 mL) and dichloromethane (DCM, 3 mL) for 30 min at room temperature, then added benzene-1,2-diamine (0.54 g, 5 mmol). The reaction was stirred at room temperature overnight until the TLC (ethyl acetate/ethanol = 4/1, v/v) showed the disappearance of the starting material. The mixture was extracted with DCM (4 × 20 mL) and saturated sodium chloride solution (4 × 20 mL), then dried over anhydrous MgSO4 and evaporated in vacuum to yield a crude product. Ethyl acetate/ethanol (9/1, v/v) were used as the eluent in column chromatography to give a pure product 7a–d.
N-(2-aminophenyl)-6-((5-methoxy-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) hexanamide (7a)
Yellow solid powder, two steps yield: 28.1%, m.p.: 205.5–206.8 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.58 (s, 1H), 11.78 (t, J = 5.3 Hz, 1H), 9.13 (s, 1H), 8.31 (d, J = 9.8 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.41 − 7.37 (m, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.16 (dd, J = 7.8, 1.3 Hz, 1H), 6.91 − 6.86 (m, 1H), 6.72 (dd, J = 8.0, 1.2 Hz, 1H), 6.56 (d, J = 9.9 Hz, 1H), 6.52 (td, J = 7.7, 1.3 Hz, 1H), 4.05 (s, 3H), 3.45 (q, J = 6.8 Hz, 2H), 2.37 (t, J = 7.4 Hz, 2H), 1.72 (dp, J = 29.5, 7.3 Hz, 4H), 1.49 (p, J = 7.6, 7.2 Hz, 2H).13C NMR (151 MHz, DMSO-d6) δ 179.19, 171.51, 157.90, 147.74, 142.17, 139.58, 133.76, 129.03, 126.17, 125.79, 124.07, 123.99, 122.85, 120.96, 116.73, 116.41, 113.58, 104.14, 103.73, 57.14, 42.87, 36.09, 28.59, 26.56, 25.43; HR-MS(ESI): Calcd for [M + H]+490.2090; Found: 490.2090.
N-(2-aminophenyl)-7-((5-methoxy-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) heptanamide (7b)
Yellow solid powder, two steps yield: 27.7%, m.p.: 214.8–215.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.52 (s, 1H), 11.73 (t, J = 5.2 Hz, 1H), 9.11 (s, 1H), 8.27 (d, J = 9.8 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 7.2 Hz, 1H), 7.29 (t, J = 8.0 Hz, 1H), 7.15 (dd, J = 7.8, 1.2 Hz, 1H), 6.88 (td, J = 8.1, 1.4 Hz, 1H), 6.71 (dd, J = 8.0, 1.2 Hz, 1H), 6.55 − 6.49 (m, 2H), 4.82 (s, 2H), 4.03 (s, 3H), 3.43 − 3.40 (m, 2H), 2.34 (t, J = 7.4 Hz, 2H), 1.74 − 1.68 (m, 2H), 1.64 (p, J = 7.5 Hz, 2H), 1.47 (dt, J = 14.6, 7.0 Hz, 2H), 1.41 (q, J = 7.3 Hz, 2H).13C NMR (151 MHz, DMSO-d6) δ 179.12, 171.61, 157.84, 147.70, 142.35, 139.51, 133.71, 128.96, 126.18, 125.76, 124.03, 123.94, 122.80, 120.91, 116.70, 116.65, 116.37, 113.50, 104.06, 103.64, 57.10, 42.91, 36.17, 28.79, 28.65, 26.69, 25.67; HR-MS(ESI): Calcd for [M + H]+ 504.2247; Found: 504.2245.
N-(2-aminophenyl)-6-((5-methyl-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) hexanamide (7c)
Yellow solid powder, two steps yield: 25.4%, m.p.: 148.9–150.1 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.34 (s, 1H), 11.68 (t, J = 5.2 Hz, 1H), 9.11 (s, 1H), 8.23 (d, J = 9.8 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 7.1 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 7.16 (dd, J = 7.8, 1.3 Hz, 1H), 6.88 (td, J = 8.0, 1.4 Hz, 1H), 6.70 (dd, J = 8.0, 1.2 Hz, 1H), 6.53 − 6.47 (m, 2H), 4.83 (s, 2H), 3.40 (q, J = 6.7 Hz, 2H), 2.44 (s, 3H), 2.37 (t, J = 7.4 Hz, 2H), 1.71 (dp, J = 15.3, 7.4 Hz, 4H), 1.49 (p, J = 7.7, 7.3 Hz, 2H).13C NMR (151 MHz, DMSO-d6) δ 178.23, 170.40, 156.61, 156.59, 141.27, 138.79, 135.73, 133.66, 132.46, 125.08, 124.70, 124.62, 122.89, 122.70, 122.42, 121.08, 119.68, 115.49, 115.22, 102.76, 102.42, 41.80, 35.01, 27.49, 25.49, 24.36, 15.27; HR-MS(ESI): Calcd for [M + H]+ 474.2141; Found: 474.2141.
N-(2-aminophenyl)-7-((5-methyl-4-nitro-9-oxo-9,10-dihydroacridin-1-yl) amino) heptanamide (7d)
Yellow solid powder, two steps yield: 22.9%, m.p.: 169.4–173.5 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.35 (s, 1H), 11.68 (t, J = 5.1 Hz, 1H), 9.11 (s, 1H), 8.23 (d, J = 9.8 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 7.1 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 7.16 (d, J = 7.8 Hz, 1H), 6.89 (t, J = 8.2 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 6.55 − 6.51 (m, 1H), 6.48 (d, J = 9.9 Hz, 1H), 4.82 (s, 2H), 3.39 (q, J = 6.7 Hz, 2H), 2.44 (s, 3H), 2.35 (t, J = 7.4 Hz, 2H), 1.68 (dp, J = 29.8, 7.2 Hz, 4H), 1.45 (ddt, J = 33.8, 13.9, 8.0 Hz, 4H).13C NMR (151 MHz, DMSO-d6) δ 178.25, 170.49, 156.60, 141.27, 138.79, 135.73, 133.66, 132.47, 125.07, 124.66, 124.62, 122.94, 122.69, 122.43, 121.08, 119.69, 115.54, 115.26, 102.73, 102.43, 41.84, 35.09, 27.72, 27.56, 25.63, 24.59, 15.27; HR-MS(ESI): Calcd for [M + H]+ 488.2298; Found: 488.2289.
N-(2-amino-4-fluorophenyl)-1-chloro-5-nitro-9-oxo-9,10-dihydroacridine-4-carboxamide (8a)
Step 1: Compound 3d (3.10 mmol) was stirred at 80 °C in 10 mL 98% H2SO4 for 3–5 h. After cooling to room temperature, the reaction mixture was slowly added in 100–200 mL ice water with rapid stirring for 30 min, then 20% NaOH (a.q.) was added until pH = 6–8, and the precipitate was formed. Compound 4d was obtained after filtration and drying, which was used without further purification. Step 2: Compound 4d (0.5 g, 1.57 mmol), HATU (0.9 g, 2.36 mmol), DIPEA (0.40 g, 3.14 mmol), were stirred in DMF (12 mL) and DCM (3 mL) for 30 min at room temperature, then added 4-fluorobenzene-1,2-diamine (0.79 g, 0.63 mmol). The reaction was stirred at room temperature overnight until the TLC (ethyl acetate/ethanol = 3/1, v/v) showed the disappearance of the starting material. The precipitate was formed after adding ethyl acetate to the reaction mixture, then the solid was washed with hot methanol solution three times to give a pure compound 8a. Gray solid powder, two steps yield: 53.2%, m.p.: 216.2–217.4 °C. 1H NMR (600 MHz, DMSO-d6) δ 14.23 (s, 1H), 10.08 (s, 1H), 8.75 − 8.70 (m, 1H), 8.65 − 8.61 (m, 1H), 8.46 (d, J = 8.3 Hz, 1H), 7.53 (d, J = 8.3 Hz, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.18 − 7.12 (m, 1H), 6.57 (dd, J = 11.3, 2.8 Hz, 1H), 6.41 (td, J = 8.5, 2.7 Hz, 1H), 5.44 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 174.20, 165.70, 161.78, 160.19, 145.57 (d, J = 12.0 Hz), 141.75, 136.80, 134.28 (d, J = 7.6 Hz), 133.88, 133.47, 131.22, 128.68 (d, J = 10.7 Hz), 123.93, 123.53, 120.41, 117.70, 117.23, 117.18, 101.34 (d, J = 22.8 Hz), 100.63 (d, J = 25.3 Hz). HR-MS(ESI): Calcd for [M + H]+ 427.0609; Found: 427.0692.
General procedure for compounds 9a–f
Step 1: Compound 3c–d (3.10 mmol) was stirred at 80 °C in 10 mL 98% H2SO4 for 3–5 h. After cooling to room temperature, the reaction mixture was slowly added in 100–200 mL ice water with rapid stirring for 30 min, then 20% NaOH (a.q.) was added until pH = 6–8, and the precipitate was formed. Compound 4c–d was obtained after filtration and drying, which was used without further purification. Step 2: Compound 4c–d (1.65 mmol) and 1,1′-Carbonyldiimidazole (CDI, 0.48 g, 2.97 mmol) were stirred in DMF (12 mL) and DCM (3 ml) for 30 min at room temperature, then added methyl 4-aminobutanoate or its derivative (6.6 mmol). The reaction was stirred at room temperature overnight until the TLC (petroleum ether/ethyl acetate = 1/1, v/v) showed the disappearance of the starting material. The mixture was extracted with DCM (4 × 20 mL) and saturated sodium chloride solution (4 × 20 mL), then dried over anhydrous MgSO4 and evaporated in vacuum to yield a crude product. Petroleum ether/ethyl acetate (3/1, v/v) were used as the eluent in column chromatography to give a pure product 9a–f.
Methyl 4–(1-chloro-5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxamido) butanoate (9a)
Yellow solid powder, two steps yield: 46.2%, m.p.: 213.4–214.3 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.83 (s, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.33 (s, 1H), 7.19 (t, J = 8.0 Hz, 1H), 7.10 (d, J = 7.4 Hz, 2H), 4.08 (s, 3H), 3.70 (s, 3H), 3.60 (q, J = 6.3 Hz, 2H), 2.55 (t, J = 6.7 Hz, 2H), 2.05 (p, J = 6.6 Hz, 2H).13C NMR (151 MHz, Chloroform-d) δ 177.24, 174.76, 168.10, 147.79, 142.65, 139.80, 130.66, 122.97, 122.77, 121.75, 118.59, 117.95,116.57, 111.94, 56.32, 52.05, 40.20, 32.10, 23.79; HR-MS(ESI): Calcd for [M + H]+ 403.1061; Found: 403.1057.
Methyl 6–(1-chloro-5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxamido) hexanoate (9b)
Yellow solid powder, two steps yield: 43.5%, m.p.: 178.7–179.3 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.68 (s, 1H), 7.94 (d, J = 8.1 Hz, 1H), 7.70 (d, J = 8.1 Hz, 1H), 7.19 (t, J = 8.0 Hz, 1H), 7.10 (d, J = 7.6 Hz, 1H), 7.04 (d, J = 8.0 Hz,1H), 6.82 (s, 1H), 4.07 (s, 3H), 3.68 (s, 3H), 3.55 (q, J = 6.8 Hz, 2H), 2.36 (t, J = 7.2 Hz, 2H), 1.71 (h, J = 7.6 Hz, 4H), 1.48 (p, J = 7.7 Hz, 2H).13C NMR (151 MHz, Chloroform-d) δ 177.27, 174.22, 168.08, 147.80, 142.46, 139.56, 130.69, 130.66, 122.93, 122.64, 121.78, 118.49, 117.89, 117.09, 111.95, 56.33, 51.63, 39.78, 33.75, 28.87, 26.34, 24.18; HR-MS(ESI): Calcd for [M + H]+ 431.1374; Found: 431.1369.
Methyl 7–(1-chloro-5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxamido) heptanoate (9c)
Yellow solid powder, two steps yield: 24.4%, m.p.: 165.5–165.8 °C. 1H NMR (600 MHz, Chloroform-d) δ 12.64 (s, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.71 − 7.62 (m, 1H), 7.18 (t, J = 7.8 Hz, 1H), 7.10 (d, J = 7.7 Hz, 1H), 7.03 (d, J = 4.9 Hz, 1H), 6.73 (s, 1H), 4.07 (s, 3H), 3.66 (s, 3H), 3.53 (s, 2H), 2.33 (t, J = 7.4 Hz, 2H), 1.75 − 1.65 (m, 4H), 1.48 − 1.37 (m, 4H).13C NMR (151 MHz, Chloroform-d) δ 177.26, 174.23, 168.06, 147.80, 142.45, 139.61, 130.66, 130.61, 122.95,122.67, 121.80, 118.52, 117.92, 117.12, 111.96, 56.34, 51.57, 40.12, 33.92, 29.22, 28.71, 26.63, 24.74; HR-MS(ESI): Calcd for [M + H]+ 445.1530; Found: 445.1530.
Methyl 4–(1-chloro-5-nitro-9-oxo-9,10-dihydroacridine-4-carboxamido) butanoate (9d)
Orange solid powder, two steps yield: 54.1%, m.p.: 224.3–224.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 14.39 (s, 1H), 9.04 (t, J = 5.4 Hz, 1H), 8.71 (dd, J = 8.1, 1.6 Hz, 1H), 8.59 (dd, J = 7.8, 1.3 Hz, 1H), 8.19 (d, J = 8.3 Hz, 1H), 7.48 − 7.43 (m, 2H), 3.60 (s, 3H), 3.38 (q, J = 6.8 Hz, 2H), 2.45 (t, J = 7.4 Hz, 2H), 1.86 (p, J = 7.2 Hz, 2H).13C NMR (151 MHz, DMSO-d6) δ 175.23, 173.61, 167.22,142.70, 137.66, 135.40, 135.25, 134.57, 133.90, 132.22, 125.01, 124.60, 121.36,118.44, 118.32, 51.78, 39.18, 31.22, 24.56; HR-MS(ESI): Calcd for [M + H]+ 418.0806; Found: 418.0807.
Methyl 6–(1-chloro-5-nitro-9-oxo-9,10-dihydroacridine-4-carboxamido) hexanoate (9e)
Orange solid powder, two steps yield: 50.7%, m.p.: 188.6–188.7 °C. 1H NMR (600 MHz, Chloroform-d) δ 14.29 (s, 1H), 8.76 (dd, J = 7.8, 1.3 Hz, 1H), 8.70 (dd, J = 8.1, 1.5 Hz, 1H), 7.83 (d, J = 8.2 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.29 (d, J = 8.2 Hz, 1H), 6.65 (s, 1H), 3.67 (s, 3H), 3.60 (q, J = 6.9 Hz, 2H), 2.36 (t, J = 7.1 Hz, 2H), 1.71 (h, J = 7.4 Hz, 4H), 1.47 (p, J = 7.7 Hz, 2H).13C NMR (151 MHz, Chloroform-d) δ 175.81, 174.28, 167.03, 142.53, 139.51, 135.68, 135.02, 134.63, 131.72, 131.52, 124.85, 124.79, 120.67, 118.94, 118.46, 51.63, 39.80, 33.66, 28.78, 26.20, 24.01; HR-MS(ESI): Calcd for [M + H]+ 446.1119; Found: 446.1111.
Methyl 7–(1-chloro-5-nitro-9-oxo-9,10-dihydroacridine-4-carboxamido) heptanoate (9f)
Orange solid powder, two steps yield: 29.8%, m.p.: 193.0–193.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 14.40 (s, 1H), 8.99 (t, J = 5.5 Hz, 1H), 8.71 (dd, J = 8.1, 1.6 Hz, 1H), 8.58 (dd, J = 7.8, 1.3 Hz, 1H), 8.17 (d, J = 8.3 Hz, 1H), 7.47 − 7.42 (m,2H), 3.57 (s, 3H), 3.35 (d, J = 7.0 Hz, 2H), 2.31 (t, J = 7.4 Hz, 2H), 1.57 (dp, J = 22.1, 7.3 Hz, 4H), 1.35 (td, J = 17.8, 8.8, 5.2 Hz, 4H).13C NMR (151 MHz, DMSO-d6) δ 175.23, 173.83, 167.03, 142.69, 137.56, 135.39, 135.25, 134.57, 133.81, 132.20, 125.01, 124.60, 121.33, 118.61, 118.31, 51.64, 40.52, 33.68, 29.03, 28.67, 26.62, 24.86; HR-MS(ESI): Calcd for [M + H]+ 460.1275; Found: 460.1269.
General procedure for compounds 11a–d
Compound 10a–c (1.29 mmol), HATU (0.73 g, 1.93 mmol), and DIPEA (0.33 g, 2.58 mmol) were stirred in DMF (12 ml) and DCM (3 ml) for 30 min at room temperature, then added benzene-1,2-diamine or its derivative (5.16 mmol). The reaction was stirred at room temperature overnight until the TLC (ethyl acetate/ethanol = 3/1, v/v) showed the disappearance of the starting material. The precipitate was formed after adding ethyl acetate to the reaction mixture, then the solid was washed with hot methanol solution 3 times to give a pure compound 11a–d.
N-(4-((2-aminophenyl) amino)-4-oxobutyl)-1-chloro-5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxamide (11a)
Yellow solid powder, yield: 44.2%, m.p.: 146.1–147.7 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.29 (s, 1H), 9.14 (s, 1H), 9.07 (t, J = 5.4 Hz, 1H), 8.20 (d, J = 8.3 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.35 (dd, J = 15.6, 8.0 Hz, 2H), 7.24 (t, J = 8.0 Hz, 1H), 7.17 − 7.14 (m, 1H), 6.91 − 6.87 (m, 1H), 6.71 (dd, J = 8.0, 1.2 Hz, 1H), 6.53 (td, J = 7.8, 1.2 Hz, 1H), 4.85 (s, 2H), 4.04 (s, 3H), 3.43 (q, J = 6.8 Hz, 2H), 2.45 (t, J = 7.4 Hz, 2H), 1.93 (p, J = 7.2 Hz, 2H).13C NMR (151 MHz, DMSO-d6) δ 175.00, 170.18, 166.83, 146.91, 141.84, 141.43, 136.91, 131.97, 129.38, 125.18, 124.84, 122.78, 122.09, 121.55, 121.30, 116.88, 116.40, 115.99, 115.47, 115.18, 112.24, 55.88, 38.45, 32.60, 24.31; HR-MS(ESI): Calcd for [M + H]+ 479.1486; Found: 479.1484.
N-(6-((2-aminophenyl) amino)-6-oxohexyl)-1-chloro-5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxamide (11b)
Yellow solid powder, yield: 36.8%, m.p.: 176.7–181.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.28 (s, 1H), 9.17 (s, 1H), 9.03 (t, J = 5.5 Hz, 1H), 8.18 (d, J = 8.3 Hz, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.37 (dd, J = 7.9, 0.9 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.15 (dd, J = 7.8, 1.2 Hz, 1H), 6.93 − 6.87 (m, 1H), 6.75 (d, J = 7.7 Hz, 1H), 6.56 (t, J = 7.4 Hz, 1H), 5.11 (s, 2H), 4.04 (s, 3H), 3.38 (d, J = 6.8 Hz, 2H), 2.35 (t, J = 7.4 Hz, 2H), 1.65 (dp, J = 15.3, 7.4 Hz, 4H), 1.42 (p, J = 7.8 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 176.09, 171.59, 167.78, 147.99, 142.90, 142.13, 137.93, 132.98, 130.47, 126.16, 125.75, 124.12, 123.20, 122.63, 122.38, 117.96, 117.48, 117.13, 116.76, 116.45, 113.33, 56.97, 40.52, 36.16, 29.06, 26.65, 25.52; HR-MS(ESI): Calcd for [M + H]+ 507.1799; Found: 507.1800.
N-(7-((2-aminophenyl) amino)-7-oxoheptyl)-1-chloro-5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxamide (11c)
Yellow solid powder, yield: 31.3%, m.p.: 180.9–182.1 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.27 (s, 1H), 9.09 (s, 1H), 9.01 (t, J = 5.4 Hz, 1H), 8.18 (d, J = 8.3 Hz, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.37 (dd, J = 7.9, 1.0 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.14 (dd, J = 7.8, 1.3 Hz, 1H), 6.88 (td, J = 7.9, 1.4 Hz, 1H), 6.71 (dd, J = 8.0, 1.3 Hz, 1H), 6.53 (td, J = 7.7, 1.3 Hz, 1H), 4.81 (s, 2H), 4.04 (s, 3H), 3.38 − 3.34 (m, 2H), 2.33 (t, J = 7.4 Hz, 2H), 1.62 (dp, J = 14.8, 7.4 Hz, 4H), 1.43 − 1.37 (m, 4H).13C NMR (151 MHz, DMSO-d6) δ 176.09, 171.61, 167.78, 147.98, 142.88, 142.35, 137.91, 132.98, 130.47, 126.16, 125.74, 124.04, 123.20, 122.63, 122.38, 117.95, 117.49, 117.17, 116.64, 116.36, 113.33, 56.97, 40.52, 36.21, 29.16, 28.93, 26.80, 25.75; HR-MS(ESI): Calcd for [M + H]+ 521.1956; Found: 521.1953.
N-(7-((2-amino-4-fluorophenyl) amino)-7-oxoheptyl)-1-chloro-5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxamide (11d)
Yellow solid powder, yield: 44.8%, m.p.: 218.7–218.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.26 (s, 1H), 9.01 (d, J = 8.4 Hz, 2H), 8.17 (d, J = 8.3 Hz, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.34 (dd, J = 16.4, 8.0 Hz, 2H), 7.23 (t, J = 8.0 Hz, 1H), 7.08 (dd, J = 8.6, 6.4 Hz, 1H), 6.48 (dd, J = 11.2, 2.9 Hz, 1H), 6.29 (td, J = 8.5, 2.8 Hz, 1H), 5.13 (s, 2H), 4.03 (s, 3H), 3.38 − 3.34 (m, 2H), 2.31 (t, J = 7.4 Hz, 2H), 1.61 (h, J = 7.0 Hz, 4H), 1.42 − 1.35 (m, 4H).13C NMR (151 MHz, DMSO-d6) δ 176.07, 171.85, 167.76, 160.92 (d, J = 238.4 Hz), 147.97, 144.70 (d, J = 11.7 Hz), 142.88, 137.91, 132.96, 130.45, 127.52 (d, J = 10.2 Hz), 123.18, 122.62, 122.35, 120.04, 117.94, 117.47, 117.13, 113.29, 102.35 (d, J = 22.3 Hz), 101.92 − 101.71 (m), 56.95, 40.51, 36.10, 29.15, 28.94, 26.81, 25.67 .HR-MS(ESI): Calcd for [M + H]+ 539.1861; Found: 539.1858.
General procedure for compounds 16a–b
Compound 14a–b (1.44 mmol), HATU (0.82 g, 2.16 mmol), DIPEA (0.37 g, 2.88 mmol), were stirred in DMF (12 mL) and DCM (3 mL) for 30 min at room temperature, then added benzene-1,2-diamine or its derivative 15a–b (5.16 mmol). The reaction was stirred at room temperature overnight until the TLC (ethyl acetate/ethanol = 1/1, v/v) showed the disappearance of the starting material. The precipitate was formed after adding DCM to the reaction mixture, then the solid was washed with hot methanol solution three times to give a pure compound 16a–b.
N-(2-aminophenyl)-1-bromo-4-methyl-9-oxo-9,10-dihydroacridine-3-carboxamide (16a)
Light gray solid powder, yield: 62.7%, m.p.: 215.3–217.1 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.56 (s, 1H), 9.83 (s, 1H), 8.19 (d, J = 7.3 Hz, 1H), 7.96 (d, J = 7.7 Hz, 1H), 7.75 (s, 1H), 7.60 (s, 1H), 7.32 (d, J = 6.9 Hz, 2H), 7.01 − 6.96 (m, 1H), 6.80 (d, J = 7.2 Hz, 1H), 6.65 − 6.60 (m, 1H), 4.98 (s, 2H), 2.59 (s, 3H).13C NMR (151 MHz, DMSO-d6) δ 176.38, 166.60, 143.05, 142.49, 141.91, 140.74, 134.00, 127.10, 126.52, 126.42, 126.11, 123.03, 122.99, 122.54, 121.77, 118.67, 118.31, 117.54, 116.69, 116.58, 15.44; HR-MS(ESI): Calcd for [M + H]+ 422.0504; Found: 422.0578.
N-(2-amino-4-bromophenyl)-1-bromo-4-methyl-9-oxo-9,10-dihydroacridine-3- carboxamide (16b)
Light gray solid powder, yield 66.4%, m.p.: 223.9–225.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.54 (s, 1H), 9.81 (s, 1H), 8.19 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.76 (t, J = 7.5 Hz, 1H), 7.64 (s, 1H), 7.34 − 7.27 (m, 2H), 6.99 − 6.95 (m, 1H), 6.74 (s, 1H), 5.31 (s, 2H), 2.57 (s, 3H).13C NMR (151 MHz, DMSO-d6) δ 176.40, 166.72, 144.93, 142.38, 141.67, 140.64, 134.06, 128.31, 126.42, 126.16, 122.99, 122.60, 122.15, 121.75, 119.35, 118.77, 118.58, 118.33, 118.19, 117.59, 15.41.HR-MS(ESI): Calcd for [M + H]+ 499.9609; Found: 499.9553.
Molecular docking
The HDAC1 crystal structure (PDB ID: 5ICN) and HDAC6 crystal structure (PDB ID: 8D9B) were downloaded from the RCSB protein data bank. The CDOCKER procedure of Discovery Studio 2020 software has been used to explore the binding mode of the 11c and SAHA with HDAC1/6. These two targets were prepared for docking by the automatic “prepare protein” procedure, including removing water molecules, adding hydrogen atoms, and protonating amino acid resides at the specified pH of 7.4. The 3D structures of 11c and SAHA were built and prepared by the “prepare ligands” procedure at the specified pH of 7.4. The pose with the lowest CDOCKER Interaction Energy was selected as the most probable binding conformation.
In silico physicochemical properties analysis
Physicochemical parameters for selected compounds were predicted in silico using Molinspiration Cheminformatics 2022 (https://www.molinspiration.com/).
Biological evaluation
Cell culture
HCT-116 cells (adherent cell line, purchased from the Chinese Academy of Sciences Cell Bank) was cultured in DMEM, with 10% foetal bovine serum (FBS) in humidified air at 37 °C with 5% CO2. HL-60 cells (suspension cells line, purchased from the Chinese Academy of Sciences Cell Bank) and L-O2 cells (adherent cell line, purchased from the Chinese Academy of Sciences Cell Bank) were cultured in RPIM-1640, with 10% foetal bovine serum (FBS) in humidified air at 37 °C with 5% CO2.
Cell growth inhibition assay
HCT-116, HL-60, and L-O2 cells were seeded into 96-well plates at 0.8–1.2*104 cells/well, treated with the synthesised compounds and positive controls. After 72 h treatment, the cells were incubated with 15 μL MTT (3–(4, 5-dimethyl-thiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide from Sigma) solution (5 mg/mL) for 4 h at 37 °C with 5% CO2. The formazan precipitates were dissolved in 100 mL DMSO. At 490 nm, the absorbance was measured by SpectraMax® Paradigm® Multi-Mode Microplate Reader (Molecure Devices, USA).
HDACs inhibition assays
This experiment was conducted by Shanghai ChemPartner Co., Ltd. In vitro HDACs assays (HDAC1, 3, 6, 8 and 10, BPS) based on the HDAC kit instruction, chidamide and SAHA were used as the positive controls. The fluorescence generated was monitored at 355 nm (excitation) and 460 nm (emission) using a Synergy MX plate reader (PerkinElmer Life Sciences, Boston, MA). IC50 values were calculated from the inhibitory curves.
Western blot analysis
HL-60 cells were cultured in 6 cm dishes, followed by treatment with 11b and 11c for different concentration periods for 48 h. Protein concentrations in the supernatant were determined using bicinchonininc acid (BCA). Lysate proteins were subjected to 12% sodium dodecylsulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), and electrophoretically transferred to PVDF membrane (amcBiobind NT-200). After blotting, the membrane was blocked in 5% milk for 1 h, and incubated with the specific primary antibody overnight at 4 °C. Protein bands were detected using the BIO-RAD GelDoc XR after hybridisation with the antibody. γ-H2AX, Rb, p-Rb antibodies were purchased from ProteintechGroup, Inc. (Wuhan, China).
Cell cycle analysis
HL-60 cells were seeded in a six-well plate and incubated for 12 h following treated with graded concentrations of 11b and 11c for 48 h. Cells were collected and washed twice by PBS, then fixed in ice-cold 70% ethanol. Cells were stained with 4 mg/mL PI and 0.1 mg/mL RNaseA in PBS. After being incubated in the dark at room temperature for 30 min, samples were subjected to flow cytometric analysis. Cell cycle and apoptosis analysis kit was purchased from Beyotime Biotechnology (Shanghai, China).
Cell apoptosis detection
HL-60 cells were seeded in a six-well plate and incubated for 12 h following treated with graded concentrations of 11b and 11c for 48 h. Phosphatidylserine externalisation was determined with the scheme which is designed as the instructions of the manufacturer by AnnexinV-FITC/PI apoptosis detection kit (Beyotime Company).
Supplemental Material
Download PDF (2.6 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, Han J, Wei X. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62–100.
- Bennett RL, Licht JD. Targeting epigenetics in cancer. Annu Rev Pharmacol Toxicol. 2018;58:187–207.
- Zagni C, Floresta G, Monciino G, Rescifina A. The search for potent, small-molecule HDACIs in cancer treatment: a decade after vorinostat. Med Res Rev. 2017; 37(6):1373–1428.
- Ramaiah MJ, Tangutur AD, Manyam RR. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021;277:119504.
- Botchkarev VA, Sharov AA. Histone deacetylases in the control of epidermal homeostasis: from chromatin biology toward therapy. J Invest Dermatol. 2022;142(1):12–14.
- Qiu X, Zhu L, Wang H, Tan Y, Yang Z, Yang L, Wan L. From natural products to HDAC inhibitors: an overview of drug discovery and design strategy. Bioorg Med Chem. 2021;52:116510.
- Roche J, Bertrand P. Inside HDACs with more selective HDAC inhibitors. Eur J Med Chem. 2016;121:451–483.
- Milazzo G, Mercatelli D, Di Muzio G, Triboli L, De Rosa P, Perini G, Giorgi FM. Histone deacetylases (HDACs): Histone Deacetylases (HDACs): evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes. 2020;11(5):556–604.
- Yoon S, Eom GH. HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J. 2016;52(1):1–11.
- Goutas D, Theocharis S, Tsourouflis G. Unraveling the epigenetic role and clinical impact of histone deacetylases in neoplasia. Diagnostics. 2021;11(8):1346–1362.
- Zhang N, Sun P, Jin HZ, Yang YQ, Zhao QY, Zhou L, Guo LL, Yang XH, Lu LM. Chidamide combined with paclitaxel effectively reverses the expression of histone deacetylase in lung cancer. Anticancer Drugs. 2020;31(7):702–708.
- Kollar J, Frecer V. Selective inhibitors of zinc-dependent histone deacetylases. Therapeutic targets relevant to cancer. Curr Pharm Des. 2015; 21(11):1472–1502.
- Gong P, Wang Y, Jing Y. Apoptosis induction by histone deacetylase inhibitors in cancer cells: role of Ku70. Int J Mol Sci. 2019;20(7):1601–1615.
- Schobert R, Biersack B. Multimodal HDAC inhibitors with improved anticancer activity. Curr Cancer Drug Targets. 2018;18(1):39–56.
- Yang F, Zhao N, Hu Y, Jiang C-S, Zhang H. The development process: from SAHA to hydroxamate HDAC inhibitors with branched CAP region and linear linker. Chem Biodivers. 2020;17(1):e1900427.
- Qin HT, Li HQ, Liu F. Selective histone deacetylase small molecule inhibitors: recent progress and perspectives. Expert Opin Ther Pat. 2017;27(5):621–636.
- Peng X, Sun Z, Kuang P, Chen J. Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatment. Eur J Med Chem. 2020;208:112831.
- Schaker HL, Haschemi R, Buch T, Kraft FB, Brumme B, Scholer A, Jenke R, Meiler J, Aigner A, Bendas G, et al. Balancing histone deacetylase (HDAC) inhibition and drug-likeness: biological and physicochemical evaluation of class I selective HDAC inhibitors. ChemMedChem. 2022;17(9):e202100755.
- Ibrahim HS, Abdelsalam M, Zeyn Y, Zessin M, Mustafa AM, Fischer MA, Zeyen P, Sun P, Bulbul EF, Vecchio A, et al. Synthesis, molecular docking and biological characterization of pyrazine linked 2-aminobenzamides as new class I selective histone deacetylase (HDAC) inhibitors with anti-leukemic activity. Int J Mol Sci. 2021;23(1):369–402.
- Melesina J, Simoben CV, Praetorius L, Bulbul EF, Robaa D, Sippl W. Strategies to design selective histone deacetylase inhibitors. ChemMedChem. 2021;16(9):1336–1359.
- Yang F, Zhao N, Ge D, Chen Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019;9(34):19571–19583.
- Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem. 2012; 4(11):1439–1460.
- Wang XX, Wan RZ, Liu ZP. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur J Med Chem. 2018;143:1406–1418.
- Linciano P, Benedetti R, Pinzi L, Russo F, Chianese U, Sorbi C, Altucci L, Rastelli G, Brasili L, Franchini S. Investigation of the effect of different linker chemotypes on the inhibition of histone deacetylases (HDACS). Bioorg Chem. 2021;106:104462.
- Sangwan R, Rajan R, Mandal PK. HDAC as onco target: reviewing the synthetic approaches with SAR study of their inhibitors. Eur J Med Chem. 2018;158:620–706.
- Zhang L, Zhang J, Jiang Q, Zhang L, Song W. Zinc binding groups for histone deacetylase inhibitors. J Enzyme Inhib Med Chem. 2018;33(1):714–721.
- Thaler F, Mercurio C. Towards selective inhibition of histone deacetylase isoforms: what has been achieved, where we are and what will be next. ChemMedChem. 2014;9(3):523–526.
- Wagner FF, Weїwer M, Lewis MC, Holson EB. Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics. 2013;10(4):589–604.
- Yue K, Qin M, Huang C, James CC, Jiang Y, Li X. Comparison of three zinc binding groups for HDAC inhibitors - a potency, selectivity and enzymatic kinetics study. Bioorg Med Chem Lett. 2022;70:128797.
- Vaidya GN, Rana P, Venkatesh A, Chatterjee DR, Contractor D, Satpute DP, Nagpure M, Jain A, Kumar D. Paradigm shift of “classical” HDAC inhibitors to “hybrid” HDAC inhibitors in therapeutic interventions. Eur J Med Chem. 2021;209:112844.
- Zhang Q, Wang Z, Chen X, Qiu H, Gu Y, Wang N, Wang T, Wang Z, Ma H, Zhao Y, et al. 8a, a new acridine antiproliferative and pro-apoptotic agent targeting HDAC1/DNMT1. Int J Mol Sci. 2021;22(11):5516–5532.
- Zhang B, Zhang QT, Liu ZD, Wang N, Jin HX, Liu F, Zhang C, He S. Synthesis and Anticancer Research of N-(2-aminophenyl) benzamide acridine derivatives as dual topoisomerase i and isoform-selective HDAC inhibitors. Chemistryselect. 2020; 5(27):8311–8318.
- Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol. 2012;920:613–626.
- Carine R, Feyruz VR. Chapter three - HDAC inhibitors: roles of DNA damage and repair. Adv Cancer Res. 2012;116:87–129.
- Eto S, Saeki K, Yoshitake R, Yoshimoto S, Shinada M, Ikeda N, Kamoto S, Tanaka Y, Kato D, Maeda S, et al. Anti-tumor effects of the histone deacetylase inhibitor vorinostat on canine urothelial carcinoma cells. Plos One. 2019;14(6):e0218382.
- Hye YP, Young JL, Taehyung K, Jaewon L, Sungpil Y, Wahnsoo C, Chang-Seon M, Hyungsik K. Effects of trichostatin A, a histone deacetylase inhibitor, on the regulation of apoptosis in H-ras-transformed breast epithelial cells. Int J Mol Med. 2008;22:605–611.