
Abstract
A new wave of dual Topo I/II inhibitors was designed and synthesised via the hybridisation of spirooxindoles and pyrimidines. In situ selenium nanoparticles (SeNPs) for some derivatives were synthesised. The targets and the SeNP derivatives were examined for their cytotoxicity towards five cancer cell lines. The inhibitory potencies of the best members against Topo I and Topo II were also assayed besides their DNA intercalation abilities. Compound 7d NPs exhibited the best inhibition against Topo I and Topo II enzymes with IC50 of 0.042 and 1.172 μM, respectively. The ability of compound 7d NPs to arrest the cell cycle and induce apoptosis was investigated. It arrested the cell cycle in the A549 cell at the S phase and prompted apoptosis by 41.02% vs. 23.81% in the control. In silico studies were then performed to study the possible binding interactions between the designed members and the target proteins.
Graphical Abstract
Highlights
A new wave of dual Topo I/II inhibitors was designed and synthesised via the hybridisation of spirooxindoles and pyrimidines.
In situ selenium nanoparticles (SeNPs) for some derivatives were synthesised.
Cytotoxicity, Topo I and Topo II inhibitory assays, and DNA intercalation abilities were evaluated.
Compound 7d NPs showed the best Topo I and Topo II inhibition.
Cell cycle arrest, apoptosis induction, and molecular docking studies were performed.
Introduction
Multitarget therapies have, recently, attracted the medicinal chemists’ interest, especially in the scope of complicated diseases including inflammation, thrombotic disorders as well as cancerCitation1,Citation2. The progress of such diseases can be regulated by several factorsCitation3. The primary or acquired resistance to almost all single-target therapies is the main challenge facing the treatment of these diseases due to the activation of compensatory mechanismsCitation4–6. With respect to cancer, the currently available chemotherapeutic drugs achieved remarkable success in either curing the patients or, at least, extending their livesCitation7. However, targeting two or more of the diverse pathways that participate in cancer development gives a synergistic effect to the designed drug and weakens the resistance against itCitation4,Citation5. The construction of a multitarget drug is quite more difficult than the single-target one as it requires the overlapping of the pharmacophoric features needed by the multiple targetsCitation8,Citation9.
Apoptosis, the process of programmed cell death, has received the researchers’ attention in the last decades and became the target for almost all anticancer agents since the progress of tumours is associated with the failure of the apoptotic security pathways in deregulated cellsCitation10. Therefore, apoptosis induction in cancer cells is considered an effective pathway to eradicate uncontrolled proliferating cells. Thus, targeting DNA or, even, the enzymes involved in its metabolism is nowadays among the most promising strategies for cancer treatmentCitation11,Citation12.
Topoisomerases are a group of enzymes that resolve DNA topological problems by untangling the DNA duplexesCitation13. The later fact potentiates the topoisomerases’ role in DNA transcription and replicationCitation13. These enzymes can relax the DNA supercoiling upon the formation of transient single-stranded (topoisomerase I or Topo I) or double-stranded (topoisomerase II or Topo II) splits in DNA moleculesCitation14. Following, the DNA segment can then pass through the formed splits releasing the DNA molecule tension. However, the high levels of Topo I and Topo II enzymes in malignant cells compared to that of normal cells make the enzymes promising objectives for cancer chemotherapyCitation15. Thus, DNA Topo I and Topo II inhibitors are classified as antitumor agents that form the basis of different chemotherapy combinations and are used widely in a broad spectrum of tumoursCitation16.
However, targeting Topo II by Topo II inhibitors motivates a cell resistance mechanism via a decrease in the cellular Topo II levels. This leads, subsequently, to an elevation in the Topo I cellular levels with an increase in the cells’ sensitivity to Topo I inhibitors. The opposite phenomenon also occurs regarding resistance towards Topo I inhibitorsCitation17. Owing to this reason, targeting both Topo I and Topo II not only gives a synergistic effect but also weakens the probability of the development of resistance against such inhibitorsCitation17. Unfortunately, severe neutropenia and anaemia have been clinically reported after the co-application of Topo I and Topo II inhibitorsCitation18. An alternative approach to such a combination was the use of a dual inhibitor of both targets. Thus far, several dual Topo I/II inhibitors have been recognisedCitation19.
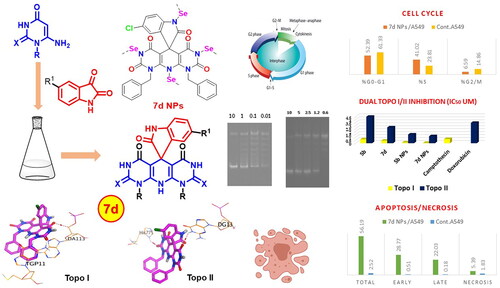
Moreover, dual Topo I/II inhibitors are classified into either DNA intercalating or DNA non-intercalating molecules according to their mechanism of action. The DNA intercalating molecules represent the majority of well-known dual Topo I/II inhibitors. Also, DNA intercalators are molecules that possess a planer (flat) aromatic system that can be sandwiched between two adjacent DNA nucleobases causing structural modifications to the DNA moleculeCitation11. Structural modification for the DNA intercalating molecules by linking their flat aromatic system to an amino group enables them to interact with Topo I/II enzymes. illustrates some reported Topo I/II inhibitors with potent in vitro anticancer effects against different types of cancersCitation17,Citation20–26.
Figure 1. Chemical structures of some reported dual Topo I/II inhibitors.
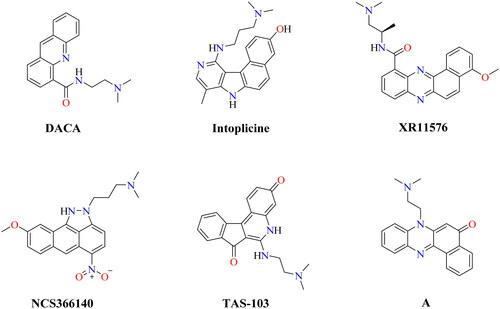
The spirooxindole-based structures are among the most vital nitrogen-containing compounds, particularly in the treatment of cancer due to the structural rigidity attributed to the spirocarbon present in these heterocyclesCitation27–29. However, spirooxindoles were reported to inhibit Topo I as well as Topo II enzymesCitation30,Citation31. In accordance with this, high interest was directed towards either discovering novel anticancer spirooxindole-based members or finding a simple and novel way to synthesise spirooxindoles. On the other hand, pyrimidine-based scaffolds also have a clear role in building several biologically active compounds, particularly in the field of cancer treatmentCitation32. The biological significance of pyrimidines may be ascribed to the ability of the nitrogen atom to bind with DNA by hydrogen bondingCitation33. However, several pyrimidine-based structures were reported to inhibit topoisomerases including, benzo[4,5]thieno[2,3-d]pyrimidinesCitation34, thiazolo[3,2-a]pyrimidinesCitation35, triazolo[4,3-a]pyrimidinesCitation36, imine-pyrazolopyrimidinonesCitation37, pyrimido[5,4-b]indolesCitation38, pyrazolo[3,4-d]pyrimidinesCitation38, thieno[2,3-d]pyrimidineCitation34, and pyrido[2,3-d:6,5-d′]dipyrimidineCitation39.
Based on the importance of spirooxindoles and pyrimidines, we decided to design a novel hybrid of DNA intercalating Topo I/II inhibitors based on the spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine] scaffold hoping to complete our efforts in searching for potent anticancer agents ().
Figure 2. Strategy for building the target hybrids.
Selenium (Se) is a crucial trace element required for different cellular functions through incorporation into selenoproteinsCitation40. The significance of selenium nanoparticles (SeNPs) stems from their interaction with proteins, bioavailability, low toxicity, and biocompatibility related to organic and inorganic selenium. At high doses, SeNPs have been shown to have potent anticancer activity. SeNPs have excellent optoelectronic and semiconducting properties and are used in various applications ranging from photovoltaic cells, semiconductors, rectifiers, and copiers to biology and medicineCitation41. SeNPs have been used in biomedical applications such as drug and targeted gene delivery, anti-inflammatory, antibacterial, and anticancerCitation42. Many reports described the synthesis of SeNPs using different methods such as microwave-assisted synthesis, laser ablation, chemosynthetic reduction, electrodeposition, solvothermal, and green synthesis, all as examples of green synthesis. However, acidic pH, harsh chemical conditions, and high temperatures limit their use in biomedical applicationsCitation43.
The molecular design rationale
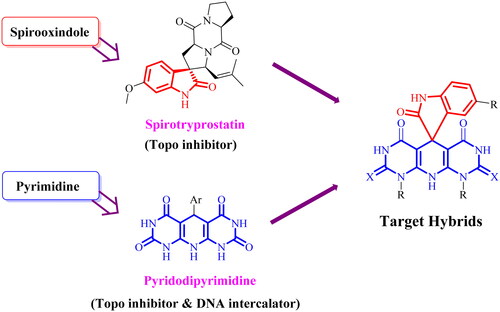
The design of a new wave of compounds that dual-target Topo I and II enzymes and intercalate onto DNA depends basically upon the overlap between the reported pharmacophoric features of both Topo inhibitors and DNA intercalators. With this respect, we designed a new series of spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine] candidates that fulfilled the requirements needed for a considerable binding with both receptors. Camptothecin, a Topo I inhibitor, and doxorubicin (Dox), a potent Topo II inhibitor and DNA intercalator, were our guides in the recent study. As so, the newly designed members achieved the following three features: (A) a planer heteroaromatic pyridodipyrimidine system to intercalate in between the DNA base pairs; (B) an NH basic centre to bind the negatively charged phosphate group of the sugar moiety; (C) a groove binder side chain to fit the DNA minor groove; and (D) a hydrogen bonding side chain to interact with both enzymes ().
Figure 3. The molecular design rationale of the new DNA intercalating and Topo I/II inhibitors.
Materials and methods
Chemistry
General
SMP 30 apparatus was applied to record the melting point of the obtained congeners and the results were uncorrected. The progress of the synthetic steps was checked by the pre-coated silica gel plates (Merck 60 F254, Darmstadt, Germany) followed by visualising the applied spots with a UV lamp (254 nm). 1H NMR and 13C NMR (400 and 100 MHz, respectively) spectra were carried out by a Bruker NMR spectrometer (Billerica, MA). DMSO-d6 was the solvent used in the NMR analysis. While mass spectra were obtained using Varian MAT 311-A (70 eV).
Materials
The starting compounds, 6-aminouracil derivatives (1a–e) were synthesised following the reported protocolCitation44–46. Reagents and chemicals were supplied either from the USA or from Aldrich Chemicals Co. (St. Louis, MO). However, some reagents were obtained from commercial sources.
1′,9′-Diethyl-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′,6′,8′(3′H,7′H,9′H,10′H)-pentaone (3)
A mixture of 6-amino-1-ethyluracil (1b) (1.29 mmol) and isatin (2) (0.65 mmol) was heated under reflux for 2 h with the addition of a few traces of glacial acetic acid (6 mL). Next, the developed precipitate was filtered off. The collected product was then washed with methanol followed by a recrystallisation from DMF.
Yield: 90%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.84 (s, 1H, NH), 10.88 (s, 1H, NH), 10.72 (s, 1H, NH), 9.20 (s, 1H, NH), 7.30 (d, J = 8.1 Hz, 1H, ArH), 7.21–7.14 (m, 1H, ArH), 7.00–6.87 (m, 2H, ArH), 4.14–3.80 (m, 4H, 2CH2), 1.18 (t, J = 6.6 Hz, 6H, 2CH3); 13C NMR (100 MHz, DMSO) δ 181.76, 160.64, 157.68, 152.63, 150.38, 149.82, 146.22, 135.70, 128.11, 125.99, 123.74, 120.89, 116.80, 98.25, 83.40, 50.24, 38.27, 36.45, 13.76, 13.61 ppm. MS: m/z (%) = 422 (M+, 20), 410 (66), 359 (82), 282 (53), 186 (50), 185 (39), 121 (42), 106 (43), 105 (46), 99 (46), 91 (41), 83 (100), 77 (59); Anal. Calcd. for C20H18N6O5 (422.40): C, 56.87; H, 4.30; N, 19.90, found: C, 57.19; H, 4.41; N, 20.13.
General method for the preparation of 1′,9′-dialkyl-5-nitro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]pentaones and/or dithioxotrione (5a–e)
A solution of 6-aminouracils 1a–e (1.5 mmol) with 5-nitroisatin (4) (0.75 mmol) was refluxed for 2–2.5 h, with a few drops of glacial acetic acid (7 mL). Then, the collected precipitate was rinsed with methanol and recrystallised from DMF to yield the target members 5a–e in good to excellent yields.
1′,9′-Dimethyl-5-nitro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′,6′,8′(3′H,7′H,9′H,10′H)-pentaone (5a)
Yield: 84%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.72 (s, 1H, NH), 11.13 (s, 1H, NH), 10.82 (s, 1H, NH), 9.91 (s, 1H, NH), 8.11 (dd, J = 9.0, 2.4 Hz, 1H, ArH), 7.74 (s, 1H, ArH), 7.51 (d, J = 9.0 Hz, 1H, ArH), 3.46 (s, 3H, CH3), 3.34 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ 181.44, 160.98, 158.28, 154.36, 151.22, 150.33, 147.21, 143.42, 141.93, 124.58, 122.62, 122.04, 117.98, 98.46, 84.46, 50.45, 30.75, 29.71 ppm. MS: m/z (%) = 439 (M+, 30), 432 (58), 430 (41), 427 (54), 419 (49), 418 (55), 412 (71), 409 (86), 359 (62), 353 (48), 324 (44), 275 (61), 273 (63), 245 (74), 229 (51), 183 (61), 132 (71), 91 (65), 89 (92), 58 (81), 50 (100); Anal. Calcd. for C18H13N7O7 (439.34): C, 49.21; H, 2.98; N, 22.32, found: C, 49.40; H, 3.19; N, 22.54.
1′,9′-Diethyl-5-nitro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′,6′,8′(3′H,7′H,9′H,10′H)-pentaone (5b)
Yield: 87%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.75 (s, 1H, NH), 11.15 (s, 1H, NH), 10.84 (s, 1H, NH), 9.83 (s, 1H, NH), 8.12 (dd, J = 9.1, 2.5 Hz, 1H, ArH), 7.68 (d, J = 2.5 Hz, 1H, ArH), 7.55 (d, J = 9.1 Hz, 1H, ArH), 4.17–3.78 (m, 4H, 2CH2), 1.19 (m, 6H, 2CH3). 13C NMR (100 MHz, DMSO) δ 181.05, 160.52, 157.73, 152.96, 150.29, 149.58, 145.79, 142.92, 141.67, 124.15, 122.00, 121.68, 117.52, 98.32, 84.33, 49.93, 38.44, 36.79, 13.71 ppm. MS: m/z (%) = 467 (M+, 12), 427 (43), 388 (40), 371 (42), 249 (100), 245 (45), 186 (40), 103 (73), 97 (83); Anal. Calcd. for C20H17N7O7 (467.40): C, 51.40; H, 3.67; N, 20.98, found: C, 51.67; H, 3.84; N, 21.19.
1′,9′-Dimethyl-5-nitro-2′,8′-dithioxo-2′,3′,8′,9′-tetrahydro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,4′,6′(7′H,10′H)-trione (5c)
Yield: 85%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 12.61 (s, 1H, NH), 12.39 (s, 1H, NH), 11.96 (s, 1H, NH), 10.12 (s, 1H, NH), 8.13 (d, J = 9.0 Hz, 1H, ArH), 7.85 (d, J = 2.3 Hz, 1H, ArH), 7.59 (d, J = 9.4 Hz, 1H, ArH), 3.96 (s, 3H, CH3), 3.73 (s, 3H, CH3). 13C NMR (100 MHz, DMSO) δ 180.03, 175.83, 174.54, 161.58, 161.39, 155.22, 154.50, 146.72, 143.18, 124.52, 122.51, 120.61, 118.16, 89.91, 84.69, 50.31, 36.64, 35.92 ppm. MS: m/z (%) = 471 (M+, 34), 455 (53), 447 (40), 364 (36), 307 (40), 284 (75), 270 (100), 258 (56), 233 (50), 205 (63), 156 (67), 135 (61); Anal. Calcd. for C18H13N7O5S2 (471.47): C, 45.86; H, 2.78; N, 20.80, found: C, 46.03; H, 3.04; N, 21.07.
1′,9′-Dibenzyl-5-nitro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′,6′,8′(3′H,7′H,9′H,10′H)-pentaone (5d)
Yield: 81%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.93 (s, 1H, NH), 11.38 (s, 1H, NH), 11.03 (s, 1H, NH), 10.05 (s, 1H, NH), 8.11–8.06 (m, 1H, ArH), 7.72–7.68 (m, 1H, ArH), 7.42–7.25 (m, 11H, ArH), 5.45–4.92 (m, 4H, 2CH2). 13C NMR (100 MHz, DMSO) δ 180.98, 160.85, 157.99, 153.41, 150.48, 149.96, 146.40, 143.14, 142.76, 137.54, 136.38, 136.14, 128.81, 128.40, 127.65, 126.44, 126.33, 126.28, 124.53, 124.42, 121.99. 121.87, 121.63, 117.61, 117.32, 98.70, 84.51, 50.23, 46.12, 44.33 ppm. MS: m/z (%) = 591 (M+, 33), 583 (40), 499 (100), 498 (39), 481 (55), 428 (48), 381 (43), 342 (53), 326 (61), 310 (54), 287 (54), 263 (45), 198 (49), 166 (50), 156 (56); Anal. Calcd. for C30H21N7O7 (591.54): C, 60.91; H, 3.58; N, 16.58, found: C, 60.78; H, 3.71; N, 16.80.
5-Nitro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′,6′,8′(3′H,7′H,9′H,10′H)-pentaone (5e)Citation47
Yield: 85%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.85 (s, 1H, NH), 11.38 (s, 2H, NH), 10.84 (s, 1H, NH), 10.54 (s, 1H, NH), 10.10 (s, 1H, NH), 8.05 (dd, J = 9.0, 2.6 Hz, 1H), 7.59 (s, 1H, ArH), 7.22 (d, J = 9.0 Hz, 1H, ArH). 13C NMR (100 MHz, DMSO) δ 181.12, 162.36, 159.15, 153.24, 151.87, 150.80, 146.29, 142.88, 141.96, 124.65, 122.63, 122.46, 117.26, 97.93, 83.62, 49.42 ppm. MS: m/z (%) = 411 (M+, 45), 402 (61), 378 (43), 362 (41), 224 (29), 203 (32), 196 (33), 182 (40), 171 (46), 167 (55), 166 (62), 149 (67), 147 (100), 145 (43), 144 (75), 119 (46), 84 (61), 69 (79); Anal. Calcd. for C16H9N7O7 (411.29): C, 46.73; H, 2.21; N, 23.84, found: C, 46.97; H, 2.43; N, 23.79.
General method for the preparation of 1′,9′-dialkyl-5-chloro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]pentaones and/or dithioxotrione (7a–e)
6-Aminouracils 1a–e (1.5 mmol) and 5-chloroisatin (6) (0.75 mmol) were heated in the presence of glacial acetic acid (7 mL) for 2–2.5 h at reflux. Filtration was used to collect the precipitate, which was then washed with methanol, recrystallised from DMF, and dried in the oven to yield the desired 7a–e in excellent yields.
5-Chloro-1′,9′-dimethyl-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′, 6′,8′(3′H,7′H,9′H,10′H)-pentaone (7a)
Yield: 91%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.52 (s, 1H, NH), 10.94 (s, 1H, NH), 10.78 (s, 1H, NH), 9.38 (s, 1H, NH), 7.29 (d, J = 8.7 Hz, 1H, ArH), 7.24 (dd, J = 8.7, 2.1 Hz, 1H, ArH), 7.04 (s, 1H, ArH), 3.38 (s, 3H, CH3), 3.32 (s, 3H, CH3). 13C NMR (100 MHz, DMSO) δ 181.62, 160.97, 158.19, 154.30, 151.33, 150.42, 147.29, 135.19, 128.71, 127.69, 126.28, 123.39, 119.08, 98.00, 83.61, 50.78, 30.73, 29.49 ppm. MS: m/z (%) = 430 (M++2, 8), 428 (M+, 29), 426 (35), 414 (43), 405 (49), 401 (83), 385 (27), 383 (76), 369 (100), 366 (94), 301 (55), 249 (54), 244 (98), 242 (62), 216 (61), 215 (54), 206 (69), 165 (82), 159 (16), 157 (58); Anal. Calcd. for C18H13ClN6O5 (428.79): C, 50.42; H, 3.06; N, 19.60, found: C, 50.71; H, 3.28; N, 19.79.
5-Chloro-1′,9′-diethyl-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′, 6′,8′(3′H,7′H,9′H,10′H)-pentaone (7b)
Yield: 92%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.53 (s, 1H, NH), 10.94 (s, 1H, NH), 10.77 (s, 1H, NH), 9.32 (s, 1H, NH), 7.34 (d, J = 8.8 Hz, 1H, ArH), 7.27 (dd, J = 8.8, 2.3 Hz, 1H, ArH), 6.93 (s, 1H, ArH), 3.95 (m, 4H, 2CH2), 1.18 (t, J = 6.7 Hz, 6H, 2CH3). 13C NMR (100 MHz, DMSO) δ 181.78, 160.97, 158.13, 153.52, 150.83, 150.14, 146.49, 135.30, 128.70, 127.60, 125.98, 123.27, 119.05, 98.16, 83.74, 50.60, 38.82, 36.99, 14.20, 14.04 ppm. MS: m/z (%) = 458 (M++2, 14), 456 (M+, 31), 368 (54), 362 (44), 359 (43), 358 (42), 353 (24), 351 (64), 342 (28), 340 (76), 339 (77), 313 (50), 233 (52), 219 (61), 218 (63), 199 (56), 197 (92), 196 (69), 188 (46), 184 (15), 183 (52), 182 (92), 163 (100), 160 (97), 128 (45), 92 (77), 72 (52); Anal. Calcd. for C20H17ClN6O5 (456.84): C, 52.58; H, 3.75; N, 18.40, found: C, 52.75; H, 3.89; N, 18.63.
5-Chloro-1′,9′-dimethyl-2′,8′-dithioxo-2′,3′,8′,9′-tetrahydro-1′H-spiro[indoline-3,5′-pyrido [2,3-d:6,5-d′]dipyrimidine]-2,4′,6′(7′H,10′H)-trione (7c)
Yield: 89%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 12.27 (s, 1H, NH), 11.95 (s, 1H, NH), 11.80 (s, 1H, NH), 9.61 (s, 1H, NH), 7.61 (s, 1H, ArH), 7.38–6.99 (m, 2H, ArH), 3.72 (s, 3H, CH3), 3.67 (s, 3H, CH3). 13C NMR (100 MHz, DMSO) δ 180.16, 174.51, 172.00, 161.55, 161.36, 158.90, 155.14, 154.47, 142.36, 134.39, 133.50, 128.64, 127.86, 89.87, 84.19, 50.49, 36.60, 35.88 ppm. MS: m/z (%) = 462 (M++2, 13), 460 (M+, 27), 405 (11), 403 (38), 354 (37), 228 (49), 221 (32), 219 (80), 193 (61), 192 (18), 190 (66), 189 (30), 187 (80), 185 (25), 183 (100), 182 (62), 178 (52), 164 (79), 159 (70), 153 (68), 146 (60), 138 (55), 123 (57), 86 (77); Anal. Calcd. for C18H13ClN6O3S2 (460.91): C, 46.91; H, 2.84; N, 18.23, found: C, 47.18; H, 3.01; N, 18.45.
1′,9′-Dibenzyl-5-chloro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′, 6′,8′(3′H,7′H,9′H,10′H)-pentaone (7d)
Yield: 88%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.71 (s, 1H, NH), 11.18 (s, 1H, NH), 10.94 (s, 1H, NH), 9.37 (s, 1H, NH), 7.39–7.19 (m, 12H, ArH), 6.89 (dd, 1H, ArH), 5.09 (s, 2H, CH2), 4.89 (s, 2H, CH2). 13C NMR (100 MHz, DMSO) δ 181.00, 160.89, 157.80, 153.68, 150.49, 149.96, 146.57, 144.78, 137.66, 136.44, 136.17, 134.65, 134.34, 128.63, 128.23, 127.62, 127.47, 127.31, 126.81, 126.38, 126.23, 122.71, 118.56, 118.32, 97.98, 81.94, 50.32, 46.05, 42.54 ppm. MS: m/z (%) = 582 (M++2, 22), 580 (M+, 36), 575 (56), 565 (62), 560 (50), 555 (54), 548 (51), 543 (100), 484 (61), 466 (78), 453 (56), 348 (54), 324 (58), 326 (45), 321 (87), 305 (85), 304 (74), 295 (54), 267 (53), 258 (48), 256 (72), 217 (63); Anal. Calcd. for C30H21ClN6O5 (580.99): C, 62.02; H, 3.64; N, 14.47, found: C, 61.97; H, 3.80; N, 14.71.
5-Chloro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′,6′,8′(3′H,7′H,-9′H,10′H)-pentaone (7e)Citation47,Citation48
Yield: 93%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.88 (s, 1H, NH), 11.06 (s, 2H, NH), 10.65 (s, 1H, NH), 10.47 (s, 1H, NH), 9.37 (s, 1H, NH), 7.22 (dd, J = 8.7, 2.4 Hz, 1H, ArH), 7.04 (d, J = 8.7 Hz, 1H, ArH), 6.80 (d, J = 2.4 Hz, 1H, ArH). 13C NMR (100 MHz, DMSO): δ 180.94, 161.98, 158.74, 152.86, 151.54, 150.17, 146.03, 134.55, 128.28, 126.64, 125.71, 123.18, 118.06, 96.84, 82.16, 49.18 ppm. MS: m/z (%) = 402 (M++2, 10), 400 (M+, 52), 399 (23), 397 (43), 396 (7), 394 (43), 382 (7), 380 (75), 367 (42), 347 (42), 341 (25), 339 (100), 323 (68), 295 (74), 263 (57); Anal. Calcd. for C16H9ClN6O5 (400.74): C, 47.96; H, 2.26; N, 20.97, found: C, 48.12; H, 2.37; N, 21.18.
General method for the preparation of 1′,9′-dialkyl-5-fluoro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]pentaones (9a,b)
6-Aminouracils 1a,e (1.5 mmol) and 5-fluoroisatin (8) (0.75 mmol) were heated in the presence of glacial acetic acid (7 mL) for 2 h under reflux. Filtration was used to collect the precipitate, which was then washed with methanol, recrystallised from DMF, and dried in the oven.
5-Fluoro-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′,6′,8′(3′H,7′H,9′H,10′H)-pentaone (9a)Citation47
Yield: 83%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.81 (s, 1H, NH), 11.01 (s, 1H, NH), 10.60 (s, 2H, NH), 10.45 (s, 1H, NH), 9.17 (s, 1H, NH), 7.04 (d, 2H, ArH), 6.65 (d, 1H, ArH). 13C NMR (100 MHz, DMSO) δ 181.32, 162.43, 159.76, 159.26, 157.45, 152.95, 150.44, 146.47, 132.43, 123.20, 118.32, 115.70, 112.99, 97.22, 81.89, 49.84 ppm. MS: m/z (%) = 384 (M+, 21), 340 (30), 337 (33), 297 (40), 294 (41), 285 (55), 275 (35), 264 (37), 247 (49), 221 (40), 169 (43), 139 (100), 138 (45); Anal. Calcd. for C16H9FN6O5 (384.28): C, 50.01; H, 2.36; N, 21.87, found: C, 50.28; H, 2.54; N, 21.98.
5-Fluoro-1′,9′-dimethyl-1′H-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-2,2′,4′,6′,8′(3′H,7′H,9′H,10′H)-pentaone (9b)
Yield: 80%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 11.50 (s, 1H, NH), 10.91 (s, 1H, NH), 10.76 (s, 1H, NH), 9.30 (s, 1H, NH), 7.33–7.25 (m, 1H, ArH), 7.08–7.03 (m, 1H, ArH), 6.90 (dd, J = 9.5, 2.7 Hz, 1H, ArH), 3.36 (s, 3H, CH3), 3.32 (s, 3H, CH3). 13C NMR (100 MHz, DMSO) δ 181.58, 161.08, 158.26, 157.78, 154.34, 151.32, 150.44, 147.42, 132.71, 123.02, 118.94, 115.76, 113.14, 97.93, 82.90, 51.07, 30.71, 29.41 ppm. MS: m/z (%) = 412 (M+, 7), 410 (29), 383 (20), 372 (45), 348 (24), 346 (45), 340 (48), 338 (45), 327 (50), 263 (34), 174 (49), 142 (100), 84 (46), 66 (44); Anal. Calcd. for C18H13FN6O5 (412.34): C, 52.43; H, 3.18; N, 20.38, found: C, 52.67; H, 3.40; N, 20.54.
In situ synthesis of SeNPs using synthetic 5b and 7d heterocyclic compounds
In a conical flask, a solution of selenious acid (H2SeO3, 0.013 g, 0.01 mmol) in deionised water (80 mL) was prepared. The later solution was then heated and added to a DMSO (10 mL) solution of members 5b or 7d (0.01 mmol). The solutions were stirred continuously for 1 h at 60 °C. Ascorbic acid (200 µL, 40 mM) was introduced to the solutions as a catalyst. The ruby red SeNPs were then suspended. Different methods were used to confirm and characterise the formation of SeNPs as described in the following subsections.
Ultraviolet–visible (UV–vis) spectra
The 1st method to confirm the SeNPs formation is to characterise their UV–vis spectra. The Shimadzu spectrophotometer is used for the current experiment. These spectra were taken in the range of 400–700 nm.
Transmission electron microscopy (TEM)
Another confirmation method is to determine the SeNPs’ size as well as shape. High-resolution transmission electron microscopy (HRTEM), JEOL (JEM-2100 TEM; Tokyo, Japan) was applied in the current work.
Particle size distribution
A particle size analyser was used to determine the average diameter and size distribution (Nano-ZS, Malvern Instruments Ltd., Worcestershire, UK). Where the sample was sonicated before the assessment to measure size distribution for 30–60 min.
Biological testing
Cytotoxicity of different samples on five cancer cell lines
The cytotoxic activities of the constructed members were assessed via the reported MTT methodCitation49–52 (Supporting information).
The mode of cell death of the promising cytotoxic samples (5b NPs and 7d NPs)
Herein, the acridine orange/ethidium bromide (AO/EtBr) fluorescing stain and the different modes of cell death were investigated under the fluorescent microscope using the reported methodologyCitation53.
Dual topoisomerase I/II inhibitory assay
The tested members were subjected to Topo I inhibitory activity evaluation using the previously reported methodCitation54. While the Topo II inhibitory assay was done according to the method described by Patra et al.Citation55 (Supporting information).
DNA intercalation assay
The reported procedure by Atwal et al. was our guide to test the DNA intercalating ability of our candidatesCitation56 (Supporting information).
Cell cycle analysis
The propidium iodide (PI) staining strategy explained in the Supporting information was adopted in the cell cycle analysisCitation57,Citation58.
Cell apoptosis analysis
The applied procedureCitation59 used for monitoring the apoptotic effect was mentioned in detail in the Supporting information.
Molecular docking studies
Two molecular docking studies of the novel candidates (3, 5a–e, 7a–e, and 9a,b) were performed using the Schrodinger suite 2021Citation60 to investigate their potential activities against the human DNA in complex with either Topo I or Topo II targets, respectively. Collectively, each chemical structure was built in ChemDraw and transferred to the working window, and corrected. The structures were then energy minimised as discussed previouslyCitation61. Each prepared series was inserted in one database together with the native ligand in each receptor. The human DNA in complex with Topo I (1T8I) and Topo II (3QX3) target receptors were extracted. Each target protein was prepared for docking as reported previouslyCitation62. Then, two docking processes were accomplished using the corresponding database and following the default setting options and methodology as beforeCitation9. Also, PyMOL softwareCitation63 was applied for the visualisation of the selected poses. Moreover, two validation processes were performed by redocking each native ligand and the obtained RMSD values were <2 Å indicating a valid performance for the two docking processesCitation64.
Results and discussion
Chemistry
The spirooxindoles were synthesised by refluxing isatin with two equivalents of 6-aminouracil derivativesCitation44,Citation46,Citation65,Citation66 as a follow-up to our work on the uracil moiety. Bazgir and coworkersCitation67 refluxed 6-aminouracils with different isatin derivatives in ethanol and p-TSA to get spiro[pyrimido[4,5-b]quinoline-5,5′-pyrrolo[2,3-d]pyrimidine]pentaones. While, Dong et al.Citation47 synthesised spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-pentaones (5e, 7e, and 9a) via refluxing of three-component of isatins, barbituric acid, and 6-aminouracil in the presence of (CoFe2O4/BC/PDA-Ni) as a catalyst for 4 h. Furthermore, Kavyani and BaharfarCitation48 prepared 7e by the reaction of 5-chloroisatin, barbituric acid, and 6-aminouracil in the presence of Fe3O4/GO/Au-Ag as a catalyst. In our project, cyclic enaminone 1a–e underwent cyclocondensation with isatins to afford polyheterocyclic spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]-dipyrimidine]-pentaones and/or dithioxo-spiro[indoline-3,5′-pyrido[2,3-d:6,5-d′]dipyrimidine]-triones 3, 5a–e, 7a–e, and 9a,b by refluxing in acidic medium (Scheme 1). The investigation was extended to the uses of initiated isatin by electron acceptor group to yield more amounts of spiro derivatives and reduce the reaction time. All the resulting candidates were confirmed by spectral data 1H, 13C NMR, mass spectra, and elemental analysis. The 1H NMR of compounds 3, 5a, 5b, 5d, 5e, 7a, 7b, 7d, 7e, and 9a,b showed singlet signals characteristic for NH-3′, NH-7′ protons at δ 11.93–11.06 ppm, furthermore, and singlet signals at the range δ 11.38–10.47 ppm characteristic for NH-1 and δ 10.10–9.17 ppm for NH-10′. Moreover, 1H NMR of the thioxo-derivatives 5c and 7c showed characteristic singlet signals protons for NH-3′, NH-7′, NH-1, and NH-10′ at δ 12.61–12.27 ppm, δ 12.39, 11.95 ppm and at δ 10.12, 9.61 ppm, respectively. However, the disappearance of broad singlet signals of NH2-6 of uracils 1a–e around δ 6.0 ppm proved the cyclisation and the evolution of NH3. 13C NMR spectra showed C═O signals at the range δ 181.98–180.03 ppm and the appearance of signals at δ 50.94–49.42 ppm characteristic for spiro-carbons (C-5′, 3).
Scheme 1. Synthesis of spirooxindole compounds 3, 5a–e, 7a–e, and 9a,b.
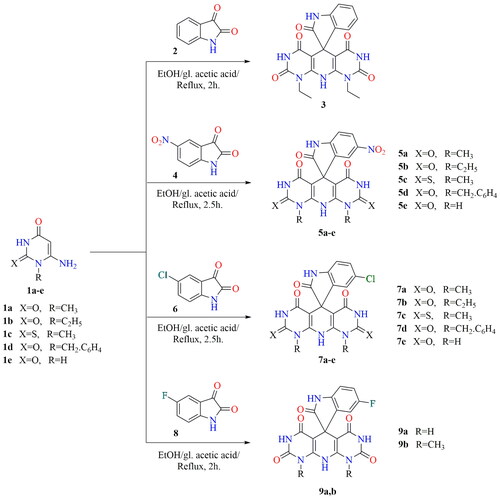
The plausible mechanistic pathway for the formation of spiroindoles may be preceded as reported that the 3-position of the isatin is more reactive towards nucleophilesCitation68–70. Based on this fact, intermediate [A] was formed via nucleophilic attack of the 5-position of uracil to the more reactive centre at the 3-position of isatin. The nonisolable acyclic intermediate [B] was formed via the nucleophilic attack of the 5-position of the second uracil molecule to the intermediate [A] with a loss of H2O. The cyclisation affording spiroindoles presumably occurred via the addition of the amino group to the C-6 of the uracil accompanied by loss of NH3 moiety affording the desired final products (Scheme 2).
Scheme 2. Mechanistic pathway for the synthesis of spirooxindole candidates.
In situ synthesis of SeNPs using synthetic heterocyclic compounds 5b and 7d
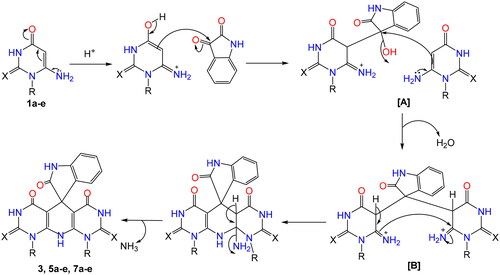
The team’s work aimed to create new SeNPs using the synthesised compounds 5b and 7d having appropriate low reducing but high stabilising properties during SeNPs preparation. Accordingly, the organic heterocycle derivatives have reduced the Se+ cation to Se0 using an ascorbic acid catalyst, which acts as an aldehyde to form SeNPs and stabilise the nano-structure of SeNPsCitation71. The organic compounds with reductive groups, such as –OH, –NH2, –NH, etc., can reduce Se+ cation. depicts the structure of the synthesised candidates.
Figure 4. Chemical structures of heterocyclic-SeNPs (5b NPs and 7d NPs).
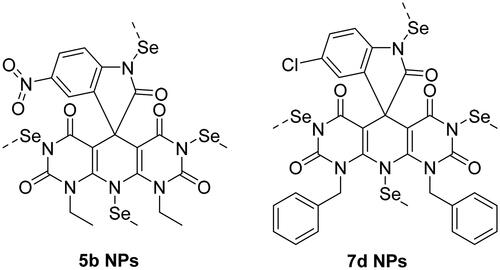
The first indication for the formation of nanoparticles is the colour of the solution that turned from pale yellow to red after adding the dissolved compounds (5b and 7d) in DMSO to the solution of H2SeO3 and stirring for one hour at 60 °C (). To easily monitor the formation of SeNPs, UV–vis spectroscopy was used (). The absorption peak of the selenium colloidal solution is at 460 nm, according to surface plasmon resonance peaks in absorption spectra.
Figure 5. (A) Formation of NPs and (B) the UV–vis absorption spectrum of SeNPs.
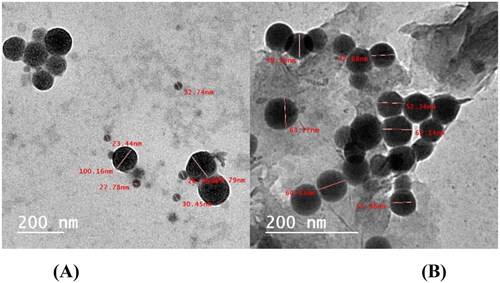
Transmission electron microscopy
It is indicating that NPs were formed and dispersed without the formation of aggregations in an aqueous solutionCitation72. shows how TEM confirmed the characterisation of selenium colloidal NPs. TEM was used to examine the prepared SeNPs, either alone or loaded onto the synthesised heterocyclic compounds. shows the presence of formed SeNPs in the tested specimens. SeNPs in spherical shapes are depicted in . Minor aggregates can be found. The size of these nanoparticles ranges from 24.98 to 60.78 nm as shown in . When SeNPs are loaded onto the investigated compounds 1, 2, 3, 4, 5, and 6, they spread uniformly through both samples (). The synthesised compounds helped to the prevention of such agglomerates in SeNPs. It appears that the nitrogen-based compounds’ stabilising effectCitation73 contributed to the existence of SeNPs separated from each other with a convenient distribution.
Figure 6. (A) TEM of SeNPs and (B) TEM of SeNPs loaded onto (5b and 7d).
Dynamic light scattering (DLS)
The SeNPs’ particle size was estimated by DLS measurements as demonstrated in . Results revealed the compounds’ particle size fell in the range of 30.43–65.90 nm ().
Figure 7. (A) Particle size from DLS of SeNPs and (B) particle size of SeNPs loaded onto (5b and 7d).
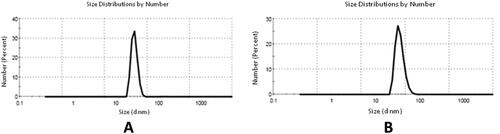
Table 1. The mean particle size of SeNPs.
Biological testing
Cytotoxicity of different samples on five cancer cell lines
The cytotoxic screening was performed using the MTT assayCitation74. The assayed samples showed diverse effects on each cell line. The most sensitive tested cell lines were HepG2 and Caco-2 cells as nine out of the tested samples exhibited considerable cytotoxicity for them. However, only five tested compounds showed cytotoxic effects on the A549 cell line. Besides, the HCT-116 one was sensitive to only two samples, while the MCF7 was sensitive to one sample only. Two standard cytotoxic drugs: Dox and 5-fluorouracil (5-FU) were also applied on the same used cancer cell lines as references. The dose–response curves for each cell line are shown in , and the respective IC50s are shown in .
Table 2. The IC50 of 3, 5a-e, 7a–e, and 9a,b compounds on five cancer cell lines and normal bone marrow cells (µg/mL).
After the cytotoxicity evaluation, some compounds were selected for the synthesis of in-situ SeNPs. The synthesised SeNPs were confirmed using the UV–vis (spectrophotometer), TEM, and particle size distribution. On the nanoscale, the toxicity was much enhanced for samples 5b NPs and 7d NPs. All the IC50 values were around 10 μg/mL and less, except for 7d NPs on the Caco-2 cell line, which reached 18.9 μg/mL.
The dose–response curves for the nanoform on each cell line are shown in the Supporting information, and the respective IC50s are shown in . The most promising cytotoxic samples were further investigated for their cytotoxic effects on normal mice bone marrow cells. The overall trending results were increasing the proliferation of the bone marrow cells at high concentrations of the samples, especially the 5b NPs nanoform sample, which reached up to 200% compared to the untreated cells. The dose–response curves on the bone marrow cells are represented in the Supporting information.
Table 3. The IC50 of samples 5b NPs and 7d NPs on five cancer cell lines (µg/mL).
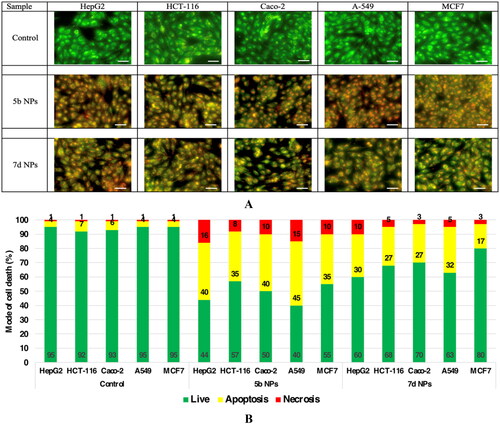
The mode of cell death of the promising cytotoxic samples (5b NPs and 7d NPs)
Using the AO/EtBr fluorescing stain, the different modes of cell death were investigated under the fluorescent microscope. The photos of samples 5b NPs and 7d NPs showed high early and late apoptotic changes with considerable necrotic changes in almost all cell lines. The mode of cell death photos and the % distribution of modes among different cell lines are illustrated in .
Figure 8. The mode of cell death of samples 5b NPs and 7d NPs on different cell lines. (A) The fluorescence stain using acridine orange/ethidium bromide stain. The photos show high early and late apoptotic changes with considerable necrotic changes. The scale bar is 50 µm and the magnification is ×20. (B) The % distribution of modes of cell death among different cell lines compared to their respective control cells.
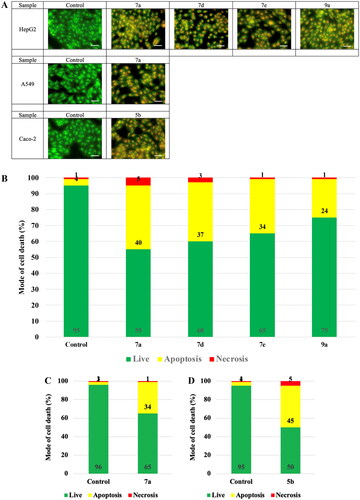
On the macro scale, the most sensitive samples on the respective cell lines are illustrated in . The photos showed the prevailed apoptotic mode of cell death with less necrotic changes.
Figure 9. The mode of cell death of the most promising samples on different cell lines. (A) The fluorescing stain is acridine orange/ethidium bromide. The photos show high early and late apoptotic changes with considerable necrotic changes. The scale bar is 50 µm and the magnification is ×20. The % distribution of modes of cell death among different samples compared to their respective control cells in (B) HepG2, (C) A549, and (D) Caco-2.
Dual topoisomerase I/II inhibitory assay
Since our main target for the recent study is to discover potent dual Topo I/II inhibitors, the highly active cytotoxic members were subjected to further assessment to measure their Topo I and Topo II inhibitory effects. Camptothecin, the potent Topo I inhibitor, and Dox, the potent Topo II inhibitor, were used as references in this study. Thus, compounds 5b and 7d in addition to their nano complexes 5b NPs and 7d NPs were evaluated for their enzyme inhibitory effects.
With regard to the Topo I enzyme, members 5b and 7d exhibited excellent inhibitory activities with similar and better IC50 values (0.651 and 0.419 µM), respectively, compared to camptothecin (0.694 µM). However, a drastic increase in the inhibitory activity was noticed regarding the nano complexes 5b NPs and 7d NPs. Compounds 5b NPs and 7d NPs were 5.8-fold and 16.5-fold more active than camptothecin with IC50 values of 0.119 and 0.042 µM, respectively ().
Figure 10. The inhibitory effects of the assessed compounds (5b, 7d, 5b NPs, and 7d NPs) against Topo I and Topo II compared to the reference drugs.
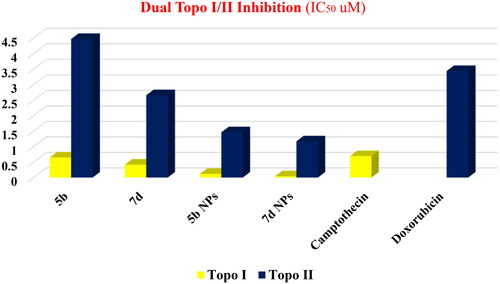
Concerning the Topo II enzyme, results also confirmed the potentiality of the tested members as congener 7d was more active than Dox with IC50 of 2.66 µM while its nano complex 7d NPs showed a potential inhibitory effect with IC50 value of 1.172 µM. In contrast, compound 5b was slightly less potent than Dox with an IC50 of 4.469 µM. Nevertheless, member 5b NPs possessed a higher inhibitory effect than that of Dox with an IC50 value of 1.464 µM ().
DNA intercalation assay
The ability of the designed members to intercalate onto the DNA molecules and increase the levels of Topo I–DNA as well as Topo II-DNA cleavage complexes by stabilising the enzymes–DNA covalent complexes were also assayed. Thus, the amount of linear DNA formed by compounds 5b and 7d, in addition to their nanoforms 5b NPs and 7d NPs, was measured. Even at low concentrations, the tested members caused considerable formation of linear DNA. As so, the designed congeners were then considered promising DNA intercalating members ().
Figure 11. DNA-fragmentation gel images of the most active compounds (5b and 7d) and their nanoforms (5b NPs and 7d NPs) at various concentrations.

Cell cycle analysis
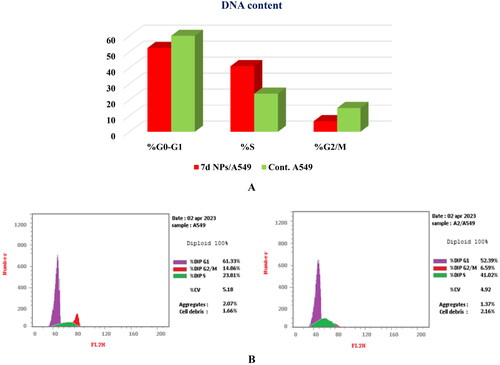
The potent cytotoxic activity of congener 7d NPs, besides its superior dual Topo I/Topo II inhibitory effects, motivated us to perform a cell cycle analysis to explore the specific phase at which 7d NPs can halt the cell cycle. With this respect, the most sensitive A549 cells were treated with compound 7d NPs at a concentration equivalent to that produced the cytotoxic effect (1.9 ± 0.1 µg/mL).
The obtained results were compatible with the cytotoxicity study. Compound 7d NPs arrested the cell cycle progression at the S phase as it caused a dramatic increase to 41.02% vs. 23.81%, compared to the untreated cells. Moreover, the percentages of cells at G0–G1 and G2/M phases were relatively reduced from 61.33% and 14.86% in the control cells to 52.39% and 6.59% after incubation with compound 7d NPs, respectively ().
Figure 12. (A) Graphical representation for cell cycle analysis of 7d NPs treated A549; (B) the histograms show the distribution of cells treated with control (left side) and 7d NPs (right side).
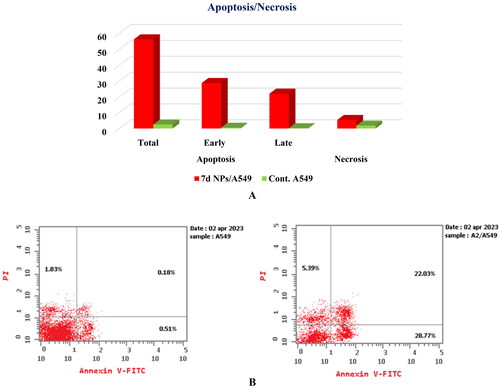
Cell apoptosis analysis
The ability of congener 7d NPs to prompt apoptosis in A549 cells was examined through the Annexin-V/PI staining assayCitation75. The present strategy depends upon the incubation of A549 cells with the tested member, 7d NPs, at the compound’s IC50 concentration (1.9 ± 0.1 µg/mL) for 24 h. Based on the given results, 7d NPs caused a marked induction of apoptosis in the treated cells by a percentage exceeding 56%. In particular, a significant increase in both early and late apoptotic populations from 0.51% and 0.18% in the control cells to 28.77% and 22.03% in the 7d NPs treated cells confirmed that its cytotoxic activity is associated with a potent apoptotic effect ().
Figure 13. (A) Graphical representation for the % of apoptotic and necrotic cells among control (A549) and 7d NPs treated cells. (B) The representative histograms show the apoptosis induction of cells treated with control (left side) and 7d NPs (right side).
Structure–activity relationship (SAR)
Observation of the obtained biological findings clarified different notes about the SAR of the designed candidates ():
Figure 14. SAR of the tested compounds (3, 5a-e, 7a–e, and 9a,b).
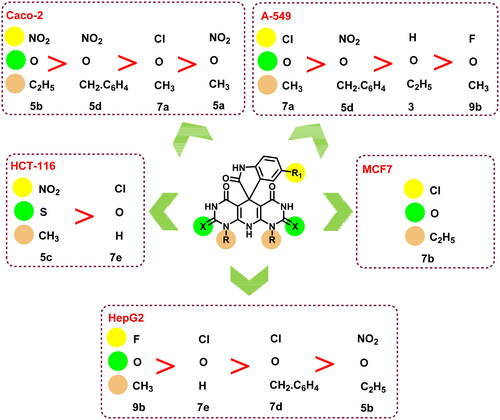
HepG2, Caco-2, and A-549 cell lines were the most sensitive cells to the examined members among the tested cell lines. As nine members out of 13 were active against HepG2 and Caco-2 cells while seven members were potent against A-549 cells.
Two compounds were active regarding HCT-116 cells and only one congener of the tested members was active against MCF7 cells.
The SeNPs complexes 5b NPs and 7d NPs exhibited a dramatic increase in the cytotoxic activity against almost all the examined cell lines.
The fluorinated member 9b possessed a superior cytotoxic activity regarding HepG2 cells followed by the chlorinated members 7d and 7e, which were more potent than the nitro compound 5b.
In contrast, the nitro derivatives 5b and 5d showed better cytotoxic activities than that of the chlorinated member 7a against Caco-2 cells.
The same observation was also reported for HCT-116 cells, as the nitro derivative 5c was more cytotoxic than the chlorinated member 7e.
Concerning A-549 cells, the descending order of activity of the tested members was 7a > 5d > 3 > 9b.
However, comparing the Topo I/Topo II inhibitory activities as well as the DNA intercalating potential of compounds 5b and 7d to the 5b NPs and 7d NPs revealed a tremendous increase in the activity of the nano complexes over the native members.
Molecular docking studies
Two molecular docking studies of the novel designed members (3, 5a–e, 7a–e, and 9a,b) were performed to investigate their potential activities against the human DNA in complex with both Topo I (https://www.rcsb.org/structure/1T8I) and Topo II (https://www.rcsb.org/structure/3QX3) targets, respectively. First, the receptor pockets of the aforementioned targets were studied to clarify the binding mode and accordingly the most important nucleotides and amino acids within each. The co-crystallised poison of the human DNA in complex with Topo I enzyme (camptothecin) was able to bind Asp533, Arg488, DT10, DA113, DC112, and TGP11. Besides, the co-crystallised poison of the human DNA in complex with Topo II enzyme (etoposide) interacted with Gln778, Met782, Asp479, DA12, and DG13. Moreover, each docked co-crystallised ligand was inserted as a reference and showed a similar binding mode as the native one indicating a valid molecular docking softwareCitation76. Herein, the superior anticancer members (5b and 7d) were selected for further investigation.
Both 5b and 7d candidates recorded very promising binding scores towards the human DNA in complex with both Topo I and Topo II targets, which were comparable to the co-crystallised ligand in each process. They showed interaction scores of (–9.08 and −7.50 kcal/mol) and (–9.73 and −8.99 kcal/mol), respectively, with respect to camptothecin (–10.24 kcal/mol) and etoposide (–10.84 kcal/mol). All root mean square deviation values were in the range of (1.09–1.56 Å) indicating valid selected posesCitation77.
Compound 5b within the Topo I binding site was able to interact with DA113 with a hydrogen bond and DC112 with a pi–pi interaction. However, compound 7d described three interactions with DA113 (hydrogen bond), DA113 (pi–pi bond), and TGP11 (pi–pi bond) as depicted in . Moreover, compound 5b within the Topo II binding site was found to bind both DA12 (hydrogen bond) and DG13 (hydrogen-pi bond). Furthermore, compound 7d bound DG13 and His775 with two hydrogen bonds as represented in .
Table 4. 2D interactions, 3D interactions, and 3D positioning of 5b and 7d within the binding pockets of the human DNA in complex with Topo I (1T8I) and Topo II (3QX3) target receptors.
According to the above results, we can observe the very promising scores and binding modes of both compounds (especially 7d) to be recommended as a potential DNA intercalator with dual Topo I/Topo II inhibitory activities.
It is worth mentioning that the C═S functional group of both 5c and 7c compounds contributed largely to the interactions with the key amino acids of Topo I and Topo II receptors indicating their great importance (Supporting information).
Conclusions
To find potent multitarget Topo I/II inhibitors, a new wave of compounds was constructed and synthesised based on the hybridisation of spirooxindoles and pyrimidines. The designed compounds were also constructed to act as DNA intercalators. Thirteen congeners were synthesised and in vitro assessed for their cytotoxic effects against five human cancer cell lines, HepG2, HCT-116, Caco-2, A-549, and MCF7. Considerable cytotoxic effects were observed regarding the tested cells. However, better cytotoxic compounds were selected for the synthesis of in situ SeNPs to enhance their activity. The nanoform compounds 5b NPs and 7d NPs exhibited excellent antitumor activities towards the tested cells (IC50 values ranging from 1.9 ± 0.1 and 18.9 ± 2.4 µg/mL). Meanwhile, the former two nanoform members were examined for their Topo I and Topo II inhibition effects. Compounds 5b NPs and 7d NPs exhibited superior activities against Topo I (IC50 = 0.119 and 0.042 µM, respectively). Also, they inhibited Topo II (IC50 values = 4.469 and 1.172 µM, respectively). Furthermore, compound 7d NPs halted the cell cycle in the S phase. Candidate 7d NPs, moreover, induced apoptosis in A549 cells. Molecular docking also showed very promising binding scores and binding modes (especially for 7d) to be recommended as a potential DNA intercalator with dual Topo I/Topo II inhibitory potentials.
Author contributions
Conceptualisation: Samar El-Kalyoubi and Ahmed A. Al‐Karmalawy; data curation: Samar El-Kalyoubi, Mahmoud T. Abo-Elfadl, Ahmed A. El-Sayed, and Ahmed A. Al-Karmalawy; visualisation: Samar El-Kalyoubi, Mohamed M. Khalifa, Mahmoud T. Abo-Elfadl, Ahmed A. El-Sayed, and Ahmed A. Al-Karmalawy; methodology: Samar El-Kalyoubi, Mahmoud T. Abo-Elfadl, Ahmed A. El-Sayed, and Ahmed A. Al-Karmalawy; validation: Mohamed M. Khalifa and Ahmed A. Al‐Karmalawy; resources and funding: Kyeong Lee and Ahmed Elkamhawy; supervision: Samar El-Kalyoubi and Ahmed A. Al‐Karmalawy; writing – review and editing: Samar El-Kalyoubi, Mohamed M. Khalifa, Mahmoud T. Abo-Elfadl, Ahmed A. El-Sayed, Ahmed Elkamhawy, Kyeong Lee, and Ahmed A. Al-Karmalawy. Finally, all authors revised and approved the final submitted manuscript.
ienz_a_2242714_sm0473.pdf
Download PDF (2.7 MB)Acknowledgements
A.E. expresses his gratitude to the Korea Institute of Science and Technology (KIST) for partially supporting this work through the "2022 KIST School Partnership Project," and he would like to thank the Technology Innovation Commercial Office (TICO) at Mansoura University for their highly effective contribution to the project’s success.
Disclosure statement
The authors declared no conflict of interest.
Additional information
Funding
References
- Kamb A, Wee S, Lengauer C. Why is cancer drug discovery so difficult? Nat Rev Drug Discov. 2007;6(2):115–120.
- Rosini M. Polypharmacology: the rise of multitarget drugs over combination therapies. Future Med Chem. 2014;6(5):485–487.
- Nussinov R, Jang H, Tsai C-J. The structural basis for cancer treatment decisions. Oncotarget. 2014;5(17):7285–7302.
- Mokhtari RB, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H. Combination therapy in combating cancer. Oncotarget. 2017;8(23):38022–38043.
- Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery: miniperspective. J Med Chem. 2014;57(19):7874–7887.
- Al-Karmalawy AA, Nafie MS, Shaldam MA, Elmaaty AA, Antar SA, El-Hamaky AA, Saleh MA, Elkamhawy A, Tawfik HO. Ligand-based design on the dog-bone-shaped BIBR1532 pharmacophoric features and synthesis of novel analogues as promising telomerase inhibitors with in vitro and in vivo evaluations. J Med Chem. 2023;66(1):777–792.
- Djulbegovic B, Kumar A, Soares HP, Hozo I, Bepler G, Clarke M, Bennett CL. Treatment success in cancer: new cancer treatment successes identified in phase 3 randomized controlled trials conducted by the National Cancer Institute – Sponsored Cooperative Oncology Groups, 1955 to 2006. Arch Intern Med. 2008;168(6):632–642.
- Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48(21):6523–6543.
- Ragab MA, Eldehna WM, Nocentini A, Bonardi A, Okda HE, Elgendy B, Ibrahim TS, Abd-Alhaseeb MM, Gratteri P, Supuran CT, et al. 4-(5-Amino-pyrazol-1-yl)benzenesulfonamide derivatives as novel multi-target anti-inflammatory agents endowed with inhibitory activity against COX-2, 5-LOX and carbonic anhydrase: design, synthesis, and biological assessments. Eur J Med Chem. 2023;250:115180.
- Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014;2014:150845.
- El-Helby A-GA, Sakr H, Ayyad RR, Mahdy HA, Khalifa MM, Belal A, Rashed M, El-Sharkawy A, Metwaly AM, Elhendawy MA, et al. Design, synthesis, molecular modeling, in vivo studies and anticancer activity evaluation of new phthalazine derivatives as potential DNA intercalators and topoisomerase II inhibitors. Bioorg Chem. 2020;103:104233.
- Farouk F, Elmaaty AA, Elkamhawy A, Tawfik HO, Alnajjar R, Abourehab MAS, Saleh MA, Eldehna WM, Al‐Karmalawy AA. Investigating the potential anticancer activities of antibiotics as topoisomerase II inhibitors and DNA intercalators: in vitro, molecular docking, molecular dynamics, and SAR studies. J Enzyme Inhib Med Chem. 2023;38(1):2171029.
- Forterre P, Gribaldo S, Gadelle D, Serre M-C. Origin and evolution of DNA topoisomerases. Biochimie. 2007;89(4):427–446.
- Khalifa MM, Al-Karmalawy AA, Elkaeed EB, Nafie MS, Tantawy MA, Eissa IH, Mahdy HA. Topo II inhibition and DNA intercalation by new phthalazine-based derivatives as potent anticancer agents: design, synthesis, anti-proliferative, docking, and in vivo studies. J Enzyme Inhib Med Chem. 2022;37(1):299–314.
- Vann KR, Sedgeman CA, Gopas J, Golan-Goldhirsh A, Osheroff N. Effects of olive metabolites on DNA cleavage mediated by human type II topoisomerases. Biochemistry. 2015;54(29):4531–4541.
- Xu X, Wu Y, Liu W, Sheng C, Yao J, Dong G, Fang K, Li J, Yu Z, Min X, et al. Discovery of 7‐methyl‐10‐hydroxyhomocamptothecins with 1, 2, 3‐triazole moiety as potent topoisomerase I inhibitors. Chem Biol Drug Des. 2016;88(3):398–403.
- Salerno S, Da Settimo F, Taliani S, Simorini F, La Motta C, Fornaciari G, Marini AM. Recent advances in the development of dual topoisomerase I and II inhibitors as anticancer drugs. Curr Med Chem. 2010;17(35):4270–4290.
- Van Gijn R, Lendfers R, Schellens J, Bult A, Beijnen J. Dual topoisomerase I/II inhibitors. J Oncol Pharm Pract. 2000;6(3):92–108.
- Denny WA, Baguley BC. Dual topoisomerase I/II inhibitors in cancer therapy. Curr Top Med Chem. 2003;3(3):339–353.
- Mišković K, Bujak M, Baus Lončar M, Glavaš-Obrovac L. Antineoplastic DNA-binding compounds: intercalating and minor groove binding drugs. Arh Hig Rada Toksikol. 2013;64(4):593–602.
- Riou J-F, Fossé P, Nguyen CH, Larsen AK, Bissery M-C, Grondard L, Saucier J-M, Bisagni E, Lavelle F. Intoplicine (RP 60475) and its derivatives, a new class of antitumor agents inhibiting both topoisomerase I and II activities. Cancer Res. 1993;53(24):5987–5993.
- Finlay G, Riou J-F, Baguley B. From amsacrine to DACA (N-[2-(dimethylamino) ethyl] acridine-4-carboxamide): selectivity for topoisomerases I and II among acridine derivatives. Eur J Cancer. 1996;32A(4):708–714.
- Poddevin B, Riou J-F, Lavelle F, Pommier Y. Dual topoisomerase I and II inhibition by intoplicine (RP-60475), a new antitumor agent in early clinical trials. Mol Pharmacol . 1993;44(4):767–774.
- Adjei AA, Charron M, Rowinsky EK, Svingen PA, Miller J, Reid JM, Sebolt-Leopold J, Ames MM, Kaufmann SH. Effect of pyrazoloacridine (NSC 366140) on DNA topoisomerases I and II. Clin Cancer Res. 1998;4(3):683–691.
- Utsugi T, Aoyagi K, Asao T, Okazaki S, Aoyagi Y, Sano M, Wierzba K, Yamada Y. Antitumor activity of a novel quinoline derivative, TAS‐103, with inhibitory effects on topoisomerases I and II. Jpn J Cancer Res. 1997;88(10):992–1002.
- Yao B-L, Mai Y-W, Chen S-B, Xie H-T, Yao P-F, Ou T-M, Tan J-H, Wang H-G, Li D, Huang S-L, et al. Design, synthesis and biological evaluation of novel 7-alkylamino substituted benzo [a] phenazin derivatives as dual topoisomerase I/II inhibitors. Eur J Med Chem. 2015;92:540–553.
- Gjorgjieva M, Tomašič T, Barančokova M, Katsamakas S, Ilaš J, Tammela P, Peterlin Mašič L, Kikelj D. Discovery of benzothiazole scaffold-based DNA gyrase B inhibitors. J Med Chem. 2016;59(19):8941–8954.
- Patravale AA, Gore AH, Kolekar GB, Deshmukh MB, Choudhari PB, Bhatia MS, Prabhu S, Jamdhade MD, Patole MS, Anbhule PV. Synthesis, biological evaluation and molecular docking studies of some novel indenospiro derivatives as anticancer agents. J Taiwan Inst Chem Eng. 2016;68:105–118.
- Haider K, Shafeeque M, Yahya S, Yar MS. A comprehensive review on pyrazoline based heterocyclic hybrids as potent anticancer agents. Eur J Med Chem Rep. 2022;5:100042.
- Korrapati SB, Yedla P, Pillai GG, Mohammad F, Ch VRR, Bhamidipati P, Amanchy R, Syed R, Kamal A. In-silico driven design and development of spirobenzimidazo-quinazolines as potential DNA gyrase inhibitors. Biomed Pharmacother. 2021;134:111132.
- Wu G, Ouyang L, Liu J, Zeng S, Huang W, Han B, Wu F, He G, Xiang M. Synthesis of novel spirooxindolo-pyrrolidines, pyrrolizidines, and pyrrolothiazoles via a regioselective three-component [3 + 2] cycloaddition and their preliminary antimicrobial evaluation. Mol Divers. 2013;17(2):271–283.
- Chiacchio MA, Iannazzo D, Romeo R, Giofrè SV, Legnani L. Pyridine and pyrimidine derivatives as privileged scaffolds in biologically active agents. Curr Med Chem. 2019;26(40):7166–7195.
- Özkay Y, Işıkdağ İ, İncesu Z, Akalın G. Synthesis of 2-substituted-N-[4-(1-methyl-4, 5-diphenyl-1H-imidazole-2-yl) phenyl] acetamide derivatives and evaluation of their anticancer activity. Eur J Med Chem. 2010;45(8):3320–3328.
- El-Metwally SA, Khalil AK, El-Sayed WM. Design, molecular modeling and anticancer evaluation of thieno [2, 3-d] pyrimidine derivatives as inhibitors of topoisomerase II. Bioorg Chem. 2020;94:103492.
- Sekhar T, Thriveni P, Venkateswarlu A, Daveedu T, Peddanna K, Sainath SB. One-pot synthesis of thiazolo [3, 2-a] pyrimidine derivatives, their cytotoxic evaluation and molecular docking studies. Spectrochim Acta A Mol Biomol Spectrosc. 2020;231:118056.
- Nemr MT, AboulMagd AM. New fused pyrimidine derivatives with anticancer activity: synthesis, topoisomerase II inhibition, apoptotic inducing activity and molecular modeling study. Bioorg Chem. 2020;103:104134.
- Baviskar AT, Banerjee UC, Gupta M, Singh R, Kumar S, Gupta MK, Kumar S, Raut SK, Khullar M, Singh S, et al. Synthesis of imine-pyrazolopyrimidinones and their mechanistic interventions on anticancer activity. Bioorg Med Chem. 2013;21(18):5782–5793.
- Zhao M, Yang K, Zhu X, Gao T, Yu W, Liu H, You Z, Liu Z, Qiao X, Song Y. Design, synthesis and biological evaluation of dual Topo II/HDAC inhibitors bearing pyrimido [5, 4-b] indole and pyrazolo [3, 4-d] pyrimidine motifs. Eur J Med Chem. 2023;252:115303.
- Farag B, Agili F, El‐Kalyoubi S, Said SA, Youssif S, Shehta W. Synthesis, molecular docking and anticancer activity of some 5‐aryl‐5, 10‐dihydropyrido [2, 3‐d: 6, 5‐d′] dipyrimidine‐2, 4, 6, 8‐tetraone derivatives and pyrido [2, 3‐d] pyrimidines. ChemistrySelect. 2022;7(7):e202103834.
- Skalickova S, Milosavljevic V, Cihalova K, Horky P, Richtera L, Adam V. Selenium nanoparticles as a nutritional supplement. Nutrition. 2017;33:83–90.
- Chhabria S, Desai K. Selenium nanoparticles and their applications. In: Encyclopedia of nanoscience and nanotechnology. Vol. 20; 2016. p. 1–32. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Selenium+nanoparticles+and+their+applications.+In%3A+Encyclopedia+of+nanoscience+and+nanotechnology.&btnG=.
- Chaudhary S, Umar A, Mehta S. Surface functionalized selenium nanoparticles for biomedical applications. J Biomed Nanotechnol. 2014;10(10):3004–3042.
- Ferro C, Florindo HF, Santos HA. Selenium nanoparticles for biomedical applications: from development and characterization to therapeutics. Adv Healthc Mater. 2021;10(16):e2100598.
- El-Kalyoubi S, Agili F, Adel I, Tantawy MA. Novel uracil derivatives depicted potential anticancer agents: in vitro, molecular docking, and ADME study. Arab J Chem. 2022;15(4):103669.
- El-Kalyoubi S, Agili F. Synthesis, in silico prediction and in vitro evaluation of antitumor activities of novel pyrido [2, 3-d] pyrimidine, xanthine and lumazine derivatives. Molecules. 2020;25(21):5205.
- El-Kalyoubi SA, Taher ES, Ibrahim TS, El-Behairy MF, Al-Mahmoudy AM. Uracil as a Zn-binding bioisostere of the allergic benzenesulfonamide in the design of quinoline–uracil hybrids as anticancer carbonic anhydrase inhibitors. Pharmaceuticals. 2022;15(5):494.
- Dong LN, Wang YM, Zhang WL, Mo LP, Zhang ZH. Nickel supported on magnetic biochar as a highly efficient and recyclable heterogeneous catalyst for the one‐pot synthesis of spirooxindole‐dihydropyridines. Appl Organomet Chem. 2022;36(5):e6667.
- Kavyani S, Baharfar R. Design and characterization of Fe3O4/GO/Au–Ag nanocomposite as an efficient catalyst for the green synthesis of spirooxindole‐dihydropyridines. Appl Organomet Chem. 2020;34(4):e5560.
- El-Adl K, El-Helby A-GA, Ayyad RR, Mahdy HA, Khalifa MM, Elnagar HA, Mehany ABM, Metwaly AM, Elhendawy MA, Radwan MM, et al. Design, synthesis, and anti-proliferative evaluation of new quinazolin-4(3H)-ones as potential VEGFR-2 inhibitors. Bioorg Med Chem. 2021;29:115872.
- Eissa IH, El-Helby A-GA, Mahdy HA, Khalifa MM, Elnagar HA, Mehany ABM, Metwaly AM, Elhendawy MA, Radwan MM, ElSohly MA, et al. Discovery of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors: design, synthesis, and anti-proliferative evaluation. Bioorg Chem. 2020;105:104380.
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89(2):271–277.
- Thabrew MI, Hughes RD, McFarlane IG. Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J Pharm Pharmacol. 1997;49(11):1132–1135.
- El-Garawani IM, Hassab El-Nabi S. Increased sensitivity of apoptosis detection using direct DNA staining method and integration of acridine orange as an alternative safer fluorescent dye in agarose gel electrophoresis and micronucleus test. Can J Pure Appl Sci. 2016;102(2):3865–3871.
- Kardile RA, Sarkate AP, Borude AS, Mane RS, Lokwani DK, Tiwari SV, Azad R, Burra PV, Thopate SR. Design and synthesis of novel conformationally constrained 7, 12-dihydrodibenzo [b, h][1, 6] naphthyridine and 7H-chromeno [3, 2-c] quinoline derivatives as topoisomerase I inhibitors: in vitro screening, molecular docking and ADME predictions. Bioorg Chem. 2021;115:105174.
- Patra N, De U, Kang J-A, Kim JM, Ahn MY, Lee J, Jung JH, Chung HY, Moon HR, Kim HS. A novel epoxypropoxy flavonoid derivative and topoisomerase II inhibitor, MHY336, induces apoptosis in prostate cancer cells. Eur J Pharmacol. 2011;658(2–3):98–107.
- Atwal M, Swan RL, Rowe C, Lee KC, Lee DC, Armstrong L, Cowell IG, Austin CA. Intercalating TOP2 poisons attenuate topoisomerase action at higher concentrations. Mol Pharmacol. 2019;96(4):475–484.
- Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci. 2000;113(Pt 5):753–757.
- Belal A, Elanany MA, Santali EY, Al-Karmalawy AA, Aboelez MO, Amin AH, Abdellattif MH, Mehany ABM, Elkady H. Screening a panel of topical ophthalmic medications against MMP-2 and MMP-9 to investigate their potential in keratoconus management. Molecules. 2022;27(11):3584.
- Sabt A, Abdelhafez OM, El-Haggar RS, Madkour HM, Eldehna WM, El-Khrisy EE-DA, Abdel-Rahman MA, Rashed LA. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: synthesis, in vitro biological evaluation, and QSAR studies. J Enzyme Inhib Med Chem. 2018;33(1):1095–1107.
- Maestro-Desmond interoperability tools. New York (NY): Schrödinger; 2017.
- Ma C, Taghour MS, Belal A, Mehany AB, Mostafa N, Nabeeh A, Eissa IH, Al-Karmalawy AA. Design and synthesis of new quinoxaline derivatives as potential histone deacetylase inhibitors targeting hepatocellular carcinoma: in silico, in vitro, and SAR studies. Front Chem. 2021;9:725135.
- Khayat MT, Ahmed HEA, Omar AM, Muhammad YA, Mohammad KA, Malebari AM, Khayyat AN, Halawa AH, Abulkhair HS, Al-Karmalawy AA, et al. A novel class of phenylpyrazolone-sulphonamides rigid synthetic anticancer molecules selectively inhibit the isoform IX of carbonic anhydrases guided by molecular docking and orbital analyses. J Biomol Struct Dyn. 2023;1–19.
- Yuan S, Chan HS, Hu Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip Rev Comput Mol Sci. 2017;7(2):e1298.
- Raslan MA, Taher RF, Al-Karmalawy AA, El-Ebeedy D, Metwaly AG, Elkateeb NM, Ghanem A, Elghaish RA, Abd El Maksoud AI. Cordyline fruticosa (L.) A. Chev. leaves: isolation, HPLC/MS profiling and evaluation of nephroprotective and hepatoprotective activities supported by molecular docking. New J Chem. 2021;45(47):22216–22233.
- El-Kalyoubi S, El-Sebaey SA, Rashad A, Al-Ghulikah HA, Ghorab MM, Elfeky SM. Synthesis, DFT calculations, and anti-proliferative evaluation of pyrimidine and selenadiazolopyrimidine derivatives as dual topoisomerase II and HSP90 inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):2198163.
- El-Kalyoubi S, Agili F, Zordok WA, El-Sayed AS. Synthesis, in silico prediction and in vitro evaluation of antimicrobial activity, DFT calculation and theoretical investigation of novel xanthines and uracil containing imidazolone derivatives. Int J Mol Sci. 2021;22(20):10979.
- Dabiri M, Azimi SC, Khavasi HR, Bazgir A. A novel reaction of 6-amino-uracils and isatins. Tetrahedron. 2008;64(30–31):7307–7311.
- Emami S, Valipour M, Kazemi Komishani F, Sadati-Ashrafi F, Rasoulian M, Ghasemian M, Tajbakhsh M, Honarchian Masihi P, Shakiba A, Irannejad H, et al. Synthesis, in silico, in vitro and in vivo evaluations of isatin aroylhydrazones as highly potent anticonvulsant agents. Bioorg Chem. 2021;112:104943.
- Varun, Sonam, Kakkar R. Isatin and its derivatives: a survey of recent syntheses, reactions, and applications. MedChemComm. 2019;10(3):351–368.
- Kumar S, Nair AS, Abdelgawad MA, Mathew B. Exploration of the detailed structure–activity relationships of isatin and their isomers as monoamine oxidase inhibitors. ACS Omega. 2022;7(19):16244–16259.
- Kapur M, Soni K, Kohli K. Green synthesis of selenium nanoparticles from broccoli, characterization, application and toxicity. Adv Tech Biol Med. 2017;5(1):2379.
- Ranjitha V, Ravishankar V. Extracellular synthesis of selenium nanoparticles from an actinomycetes Streptomyces griseoruber and evaluation of its cytotoxicity on HT-29 cell line. Pharm Nanotechnol. 2018;6(1):61–68.
- Khalil AM, Hassan ML, Ward AA. Novel nanofibrillated cellulose/polyvinylpyrrolidone/silver nanoparticles films with electrical conductivity properties. Carbohydr Polym. 2017;157:503–511.
- Mourad MAE, Abo Elmaaty A, Zaki I, Mourad AAE, Hofni A, Khodir AE, Aboubakr EM, Elkamhawy A, Roh EJ, Al-Karmalawy AA. Novel topoisomerase II/EGFR dual inhibitors: design, synthesis and docking studies of naphtho[2′,3′:4,5]thiazolo[3,2-a]pyrimidine hybrids as potential anticancer agents with apoptosis inducing activity. J Enzyme Inhib Med Chem. 2023;38(1):2205043.
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995;184(1):39–51.
- Al-Muntaser SM, Al-Karmalawy AA, El-Naggar AM, Ali AK, Abd El-Sattar NEA, Abbass EM. Novel 4-thiophenyl-pyrazole, pyridine, and pyrimidine derivatives as potential antitumor candidates targeting both EGFR and VEGFR-2; design, synthesis, biological evaluations, and in silico studies. RSC Adv. 2023;13(18):12184–12203.
- Hamed AA, El-Shiekh RA, Mohamed OG, Aboutabl EA, Fathy FI, Fawzy GA, Al-Taweel AM, Elsayed TR, Tripathi A, Al-Karmalawy AA. Cholinesterase inhibitors from an endophytic fungus Aspergillus niveus Fv-er401: metabolomics, isolation and molecular docking. Molecules. 2023;28(6):2559.