
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The PD-1/PD-L1 pathway is considered as one of the most promising immune checkpoints in tumour immunotherapy. However, researchers are faced with the inherent limitations of antibodies, driving them to pursue PD-L1 small molecule inhibitors. Virtual screening followed by experimental validation is a proven approach to discover active compounds. In this study, we employed multistage virtual screening methods to screen multiple compound databases to predict new PD-1/PD-L1 ligands. 35 compounds were proposed by combined analysis of fitness scores, interaction pattern and MM-GBSA binding affinities. Enzymatic assay confirmed that 10 out of 35 ligands were potential PD-L1 inhibitors, with inhibitory rate higher than 50% at the concentration of 30 µM. Among them, ZDS20 was identified as the most effective inhibitor with low micromolar activity (IC50 = 3.27 μM). Altogether, ZDS20 carrying novel scaffold was identified and could serve as a lead for the development of new classes of PD-L1 inhibitors.
Introduction
The Immune checkpoint blockade (ICB) has revolutionised the field of cancer therapy and become one of the most valuable methods in the treatment of many late-stage cancers.Citation1 PD-L1/PD-1 blockade plays a key role in the process of immune response and has become a promising avenue in immunotherapy. Programmed cell death ligand 1 (PD-L1) is a typical immune checkpoint ligand which belongs to the B7 family of immunomodulatory ligands, highly expressed in a variety of tumour cells and immune cells, and its expression level is often directly related to the malignancy of the tumour. Programmed cell death protein 1 (PD-1), acts as immunomodulatory receptor, is expressed on the surface of immune effector cells, including T cells, B cells, natural killer T cells, monocytes, and dendritic cells. PD-L1 expressed by tumour cells plays a crucial role in the induction of inhibitory signals by interacting with programmed cell death-1 (PD-1) expressed on the T cell surface. The binding between PD-1 and PD-L1 can activate the programmed death of T cells and reduce their activity, thus leading to the downregulation of the immune system and immune evasion of tumour cells. PD-L1 immunosuppressants can prevent the binding between tumour cells and T cells, thereby enabling T cells to recognise and eliminate tumour cells.Citation2–3
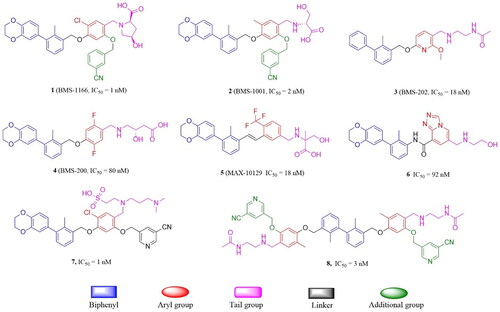
Disrupting PD-1/PD-L1 interaction has been well validated clinically by several monoclonal antibodies, these include pablizumab, navulizumab, simeplizumab, atezumab, and devaluzumab, which had gained FDA’s approval for the treatment of different types of cancer. However, antibodies have several intrinsic drawbacks, such as reduced tumour tissue permeability and severe immunogenicity issues, thus resulting in lower average response rate to the immunotherapies.Citation4–5 To address this issue, researchers are dedicated to develop small-molecule PD-1/PD-L1 inhibitors. With the aid of crystal structures of the PD-L1/inhibitor complex, researchers are actively discovering small molecule inhibitors through rational design strategies, which are then validated through a series of in vitro and in vivo experiments. Non-peptide small-molecule inhibitors targeting the PD-1/PD-L1 pathway was first reported by Bristol-Myers Squibb,Citation6 exemplified by BMS-202. However, further modifications were mainly focused on the linker or the tail group while preserving the biphenyl methanol core structure ().Citation7 Despite the great progress has been made, none of them have been approved by the FDA for clinical use. To our best knowledge, the biphenyl core is metabolic unstable, most of the inhibitors suffered from low oral availability, thus hindered their further development. Therefore, identification of unique and effective lead compounds with novel scaffolds are still in great demand.
Figure 1. Structures of representative small-molecule PD-1/PD-L1modulators.
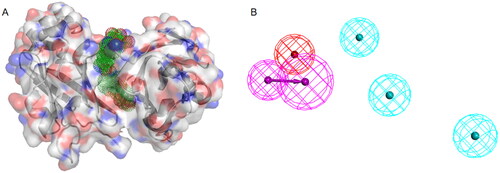
In recent years, computer-aided drug design (CADD) technology has been instrumental in the development of drugs. Virtual screening particularly has become a powerful tool for identifying hit compounds due to its faster computational speed and lower cost compared to high throughput screening. While it is challenging to design small-molecule drugs to competitively bind to the large, flat surfaces of PD-1/PD-L1,Citation8 small molecule inhibitors that form stable PD-L1 protein dimers by occupying the deep hydrophobic channels of PD-L1 protein were able to impede its recognition with PD-1. Recently, Lin’s group constructed a well-defined pharmacophore model based on a variety of small molecules inhibitors that promote PD-L1 protein dimerisation ().Citation9 Leveraging this hydrophobic channel as an entry point, we focused on reversing the immunosuppression by promoting the formation of PD-L1 dimer proteins. With the aim of identifying new inhibitors of PD-1/PD-L1, we applied a multi-stage virtual screening methods based on the shape of ligand, pharmacophore, molecular docking, molecular dynamics, and MM-GBSA caculations to screen the ZINC3D (http://zinc.docking.org), Topscience (http://www.tsbiochem.com) and ChemDiv (http://chemdiv.com) database in this study. Upon chemical synthesis and bioactivity evaluation, ten novel PD-L1 inhibitors were discovered and ZDS20 was identified as the most potential hits with tetrahydroisoquinoline and thiazolidinedione-quinazoline-5-ketone frameworks. These compounds demonstrate the reliability of the virtual screening workflow for drug discovery. Collectively, a novel PD-L1 small molecular inhibitor was identified which can provide a valuable guide for the further development of PD-L1 inhibitors.
Figure 2. (A) PD-L1 dimeric protein, green dots indicate hydrophobic channels. (B) Pharmacophore model. As the figure shows, red indicate positive ionisable point, purple indicate hydrogen bond donor, blue indicate hydrophobic point.
Materials and methods
Virtual screening
Preparation of receptor and ligands
The crystallographic structure of PD-L1 (PDB ID: 6R3K) was retrieved from the Protein Data Bank database. The protein preparation task was performed by modelling missing loop regions, repairing the missing side chain residues, deleting the redundant C and D chain, and removing water and ligand with the help of SYBYL-X-2.0.Citation10 The structure was further optimised for 10,000 steps with an energy iteration of 0.005 kcal/(mol/Å). The force field parameter of the protein was generated by using AMBER ff99 force field,Citation11 and partial atomic charges were calculated using the Gasteiger-Hückel charge method. Polar hydrogens were added to the protein by AutoDock Tools,Citation12 the protein file was prepared and saved in pdbqt format for further analysis.
The compounds in Chemdiv database (approximately 1.6 million molecules) ZINC 3D library (21.54 million molecules) and Topscience database (approximately 47,102 molecules) were converted to SDF format for further analysis. The co-crystallised ligand BMS1166 was extracted from the 6R3K protein and used as a template molecule for shape screening. Energy minimizations and conformational analysis of the ligands were performed with Tripos force field and Gasteiger-Hückel charge by means of SYBYL-X-2.0 package.
Shape and pharmacophore screening
Shape and pharmacophore screening was performed by an online server Pharmit (http://pharmit.csb.pitt.edu). A predefined pharmacophore model provided by Lin’s group was inputted, as well as BMS1166 was defined as a template molecule for shape query. Only compounds exhibiting favourable fit values in the pharmacophore and shape models were subjected to further molecular docking analysis.
Structure-Based virtual screening
Molecules obtained from shape and pharmacophore screening were converted to pdbqt format by OpenBabel.Citation13 Co-crystallised ligand BMS-1166 was utilised to identify the binding site of the receptor, the grid box was generated by Autodock tools and defined as 24 Å × 24 Å × 24 Å. Screening was performed using semi-flexible docking through AutoDock Vina. The num_modes were set at nine, and the exhaustiveness was set at eight. PyMOL (PyMOL | pymol.org) was used to visualise the results. The top 2000 compounds based on scoring rank were retained and clustered based on structural similarities.
Molecular dynamics simulation
To get a deeper understanding of the PD-L1 dynamic behaviour in the presence of ligands, molecular dynamics simulations were performed for all candidate molecules including BMS1166. The initial structures of PD-L1-ligands complex for the MD simulations were retrieved from AutoDock Vina docking. The Antechamber program of AMBER16Citation14 was utilised to apply BCC charge and GAFF2 force fieldCitation15 to the candidate molecules, supplementary position parameters were verified using ParmCHK. A 15 Å × 15Å × 15Å solvent cubic box was constructed by the LEAP program, and the TIP3P water modelCitation16 was employed to fill the box. To maintain the system’s electroneutrality, four Na+ ions were added to the ligand-receptor-solvent system. Periodic boundary conditions were implemented to eliminate any boundary effects. At the beginning, the limiting potentials of the protein, ligand, and counterbalancing ions were all constrained by force constants of 200 kcal/(mol·Å2) to achieve a low energy state. Then, the protein skeleton was limited by a force constant of 20 kcal/(mol·Å2), permitting only side chains to relax during energy minimisation. The system was then minimised without any restriction. To ensure that the system was in equilibrium, the NPT ensemble (isothermal and isobaric) was employed to gradually heat the system from 10 k to 300 k at a constant volume over a period of 100 ps. Finally, a 100-ns NPT simulation was implemented to generate the production trajectory.
MM-GBSA binding free energy Calculation
The molecular mechanics generalised born surface area (MM-GBSA) method was used to determine the binding free energy (ΔGbind) of the receptor-ligand complexes and the energy contribution by each residue based on the trajectory generated by the MD simulation. The energy of each small molecule forming a complex with the protein was expressed using the following formula.Citation17
ΔEMM refers to the MM energy of gas phase during the binding process, whereas ΔGsol and -TΔS represent the change in the solvation free energy and the conformational entropy of the ligand binding, respectively. T represents the absolute temperature, and ΔS represents the molecular entropy value. To circumvent the computational complexity involved in the analysis of TΔS via simple positive wavelength methods, only amino acids that are positioned within the vicinity of the ligand were considered.
ΔEinternal pertains specifically to the energies attributed to the bonds, angles, and dihedra, which are comprised of both the electrostatic energy and the van der Waals force ΔEvdw, forming ΔEM. The precise calculation formula for this energy contribution is as follows:
The energy contribution of ΔGsol entails the combined effect of electrostatic solvation energy (polar contribution) ΔGGB and the non-electrostatic solvation component ΔGSA (non-polar contribution). The nonpolar contributions were acquired via the utilisation of the solvent accessible surface area (SASA) and the linear combination pairwise overlap (LCPO) models. The precise calculation formula for this energy contribution is as follow:
Chemistry
All reagents and solvents were purchased from Bide Pharmatech Co., Ltd (Shanghai, China) and used without further purification. Reactions were monitored by TLC on GF254 silica gel coated plates (Qingdao Ocean Chemical) and UV light (254 and 365 nm). NMR spectra of 1H NMR (600 MHz) and 13C NMR (100 or 150 MHz) were calibrated with residual untriturated solvent (chloroform or DMSO) or with tetramethylsilane (TMS) as internal standard. The chemical shifts are measured in parts per million, and the coupling constants are all measured in Hertz. High-resolution mass spectrograms (HRMS) were obtained from Agilent Technologies 6224 TOF LC/MS.
General procedures for preparation of ZDS1
Tert-butyl ((1H-indol-5-yl)methyl)carbamate (9)
5-(aminomethyl) indole (100 mg, 0.69 mmol) and two drops of triethylamine were dissolved in CH2Cl2 (1 ml). The mixture was cooled to 0 °C with stirring and di-tert-butyl bicarbonate (164 mg, 0.75 mmol) in CH2Cl2 (1 ml) was added dropwise. After stirring at room temperature for 5 min, the solvent was washed with saturated Na2CO3 (3 ml × 2), dried over anhydrous Na2SO4 and concentrated under vacuum to yield 9 as a light yellow oil (165 mg, yield: 97.9%). HRMS (ESI) (m/z): calcd for [C14H19N2O2]+ ([M + H]+): 247.1447, found: 247.1448.
Tert-butyl ((1–(3-methylbenzoyl)-1H-indol-5-yl)methyl)carbamate (10)
To a stirred solution of m-toluic acid (186 mg, 1.37 mmol) in CH2Cl2 (3 ml) at 0 °C was added SOCl2 (3 ml), the mixture was then refluxed for 2 h leading to complete of the reaction. The obtained chloride was concentrated under reduced pressure, dissolved in CH2Cl2 and used in the next step without purification.
To a solution of ((1H-indole-5-methyl) tert-butyl carbamate (169 mg, 0.69 mmol) in CH2Cl2 (3 ml) at 0 °C under N2 atmosphere was added NaH (66 mg, 2.74 mmol), the reaction was then warmed to room temperature and stirred for 30 min, a solution of m-methylbenzoyl chloride in CH2Cl2 was slowly added to the system. After stirring at room temperature for another 2 h, the mixture was carefully poured into ice-water, washed with saturated NaHCO3 (2 ml × 2) and then brine, the separated organic phase was dried with anhydrous Na2SO4, filtered and then concentrated. The crude product was subjected to column chromatography using PE/EtOAc = 4: 1 to afford pure intermediate 10 as an oil (128 mg, yield: 51.3%). HRMS (ESI) (m/z): calcd for [C22H25N2O3]+ ([M + H]+): 365.1865, found: 365.1870.
(5-(Aminomethyl)-1H-indol-1-yl)(m-tolyl)methanone (11)
To a solution of intermediate 10 (79 mg, 0.22 mmol) in CH2Cl2 (2 ml) under an ice-water bath was slowly added trifluoroacetic acid (2 ml) dropwise. The mixture was stirred at room temperature for 0.5 h. Upon completion, the mixture was concentrated and the residue was taken up into 1 N NaOH (5 ml), extracted with CH2Cl2 (5 ml × 2), the organic layers were then combined, washed with brine, and dried over anhydrous Na2SO4 before concentrated. Compound 11 was obtained as colourless oil (453 mg, yield: 91.9%) and used in the next step without purification. HRMS (ESI) (m/z): calcd for [C17H17N2O]+ ([M + H]+):265.1341, found: 265.1342.
2–(4-fluorophenyl)quinoline-4-carboxylic acid (12)
A mixture of 4-fluorobenzaldehyde (173 μL, 1.61 mmol), pyruvic acid (114 μL, 1.61 mmol), and aniline (147 μL, 1.61 mmol) in EtOH (1.5 ml) was refluxed at 80 °C for 3.5 h. Upon completion, the reaction mixture was concentrated, followed by trituration with petroleum ether. The crude product was then recrystallized from EtOH to afford pure product 12 as a light yellow solid (123 mg, yield 28.6%). HRMS (ESI) (m/z): calcd for [C16H11FNO2]+ ([M + H]+): 268.0774, found 268.0770.
2–(4-fluorophenyl)-N-((1–(3-methylbenzoyl)-1H-indol-5-yl)methyl)quinoline-4-carboxamide (ZDS1)
To a solution of intermediate 12 (54 mg, 0.20 mmol), HATU (152 mg, 0.40 mmol) and DIPEA (65 mg, 0.50 mmol) in THF (6 ml), intermediate 11 (53 mg, 0.20 mmol) was added, the reaction was then allowed to proceed for another 12 h. Upon completion of the reaction, the mixture was taken into water (4 ml) and extracted with ethyl acetate (3 ml × 2), the collected organic layers were washed with saturated sodium bicarbonate and then brine. After concentration under vacuum, the crude product was further purified by flash chromatography PE/EtOAc = 4: 1 to yield ZDS1 as a white solid (73 mg, yield: 71.1%) 1H NMR (600 MHz, DMSO-d6) δ = 9.49 (t, J = 5.8 Hz, 1H), 8.40 (dd, J = 8.4, 5.7 Hz, 2H), 8.29 (d, J = 8.5 Hz, 1H), 8.22 (t, J = 3.9 Hz, 2H), 8.13 (d, J = 8.4 Hz, 1H), 7.83 (t, J = 7.6 Hz, 1H), 7.74 (s, 1H), 7.65 (m, 1H), 7.57 (s, 1H), 7.56–7.52 (m, 1H), 7.51–7.46 (m, 3H), 7.44–7.37 (m, 3H), 6.78 (d, J = 3.7 Hz, 1H), 4.73 (d, J = 5.8 Hz, 2H), 2.41 (s, 3H).13C NMR (151 MHz, DMSO-d6) δ 168.8, 167.1, 163.9 (d, J = 247.4 Hz, 1 C), 155.2, 148.3, 143.4, 138.8, 135.2 (d, J = 2.9 Hz, 1 C), 135.1, 135.0, 134.4, 133.2, 131.2, 130.7, 130.1 (d, J = 8.6 Hz, 2 C), 130.0, 129.8, 129.1 (d, J = 4.7 Hz, 2 C), 127.7, 126.6, 125.8, 124.9, 123.8, 120.4, 117.1, 116.4, 116.2 (2 C), 109.0, 43.3, 21.3. HRMS (ESI) (m/z): calcd for [C33H25N3O2]+ [M + H]+: 514.1931, found: 514.1929.
General procedures for preparation of ZDS2
4–(2,5-Dimethyl-1H-pyrrol-1-yl)phenol (13)
A solution of p-aminophenol (5.68 g, 52 mmol), 2,5-hexanedione(5.71 g, 50 mmol) and p-toluenesulfonic acid (50 mg) in toluene (50 ml) was stirred at reflux for 6 h. Upon cooling to room temperature, the mixture was filtered and washed with n-hexane to yield product 13 as a white solid (9.21 g, yield: 98.5%). HRMS (ESI) m/z calcd for [C12H14NO]+ ([M + H]+): 188.1075, found 188.1079.
1–(4-Hydroxyphenyl)-2,5-dimethyl-1H-pyrrole-3-carboxaldehyde (14)
To a 250 ml 3-neck round bottom flask was charged with DMF (45 ml) at 0 °C under N2 atmosphere, phosphorus trichloride (10.83 ml, 128.36 mmol) was added and the reaction was allowed to warm to room temperature slowly over 15 min, intermediate 13 (4.01 g, 21.40 mmol) in DMF (10 ml) was then injected via syringe over 15 min. Upon completion, the reaction was cooled to room temperature and adjusted to pH = 10 by 30% NaOH solution. The resulting solid was filtered, washed with water and dried under vacuum to obtain pure product 14 as a light-yellow solid (3.99 g, yield: 86.6%). HRMS (ESI) m/z calcd for [C13H14NO2]+ ([M + H]+): 216.1025, found: 216.1030.
1–(4-Hydroxyphenyl)-2,5-dimethyl-1H-pyrrole-3-carbaldehyde (15)
A solution of intermediate 14 (1.50 g, 7.25 mmol), K2CO3 (746 mg, 5.34 mmol) and benzyltriethylammonium chloride (1.22 g, 5.34 mmol) in DMF (25 ml) was stirred at room temperature for 0.5 h, methyl 3-bromomethylbenzoate (1.22 g, 5.34 mmol) and KI (89 mg) were added subsequently, the mixture was then stirred at RT overnight. Upon completion, the reaction was quenched by 50 ml water and extracted with ethyl acetate (25 ml × 3). The combined organic phase was successively washed with water and then brine, dried over anhydrous Na2SO4, concentrated, and recrystallized from methanol to give pure product 15 (701 mg, yield: 36.2%) as a white solid. HRMS (ESI) m/z calcd for [C22H22NO4]+ ([M + H]+): 364.1549, found 364.1553.
3-((4–(3-formyl-2,5-dimethyl-1H-pyrrol-1-yl)phenoxy)methyl)benzoic acid (16)
A solution of intermediate 15 (346 mg, 0.956 mmol) and NaOH (152 mg, 1.9 mmol) in MeOH/H2O (1: 1, 12 ml) was heated to 65 °C for 4 h. After consumption of the starting material, the solvent was evaporated, the residue was then acidified using 2 N HCl until pH < 4. The resulting solid was then filtered, washed with water and dried to yield 16 as a white solid (551 mg, yield: 86.9%). HRMS (ESI) m/z calcd for [C21H18NO4]- ([M-H]-): 348.1236 found 348.1227.
3-Aminoquinazolin-4(3H)-one (17)
To a 25 ml round-bottom flask was added o-aminobenzoyl (500 mg, 3.67 mmol), DMF (2 ml) and imidazole hydrochloride (945 mg, 9.18 mmol), the mixture was stirred at 150 °C for 13 h. After the reaction was completed as indicated by TLC, the mixture was poured into ice-water (10 ml) and extracted with ethyl acetate (3 ml × 2). The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and then filtered. After concentrated under reduced pressure, the crude product was then recrystallized from MeOH to give pure product 17 as a light yellow solid (238 mg, yield: 40.2%). HRMS (ESI) m/z calcd for [C8H8N3O]+ ([M + H]+): 162.0667, found 162.0670.
(E)-3-((4–(2,5-dimethyl-3-((4-oxoquinazolin-3(4H)-yl)imino)methyl)-1H-pyrrolidin-1-yl)phenoxy)methyl)benzoic acid (ZDS2)
A solution of intermediate 16 (100 mg, 0.29 mmol), aminoquinazolin-4(3H)-one (46 mg, 0.29 mmol) and p-toluenesulfonic acid (10 mg, 0.057 mmol) in MeOH (4 ml) was stirred at room temperature for 3 h, the resulting solid was then filtered and the filter cake was washed with cold MeOH. After drying under vacuum, compound ZDS2 was obtained as an off-white solid (108 mg, yield: 75.8%).1H NMR (600 MHz, DMSO-d6) δ 13.07 (s,1H), 8.87 (s,1H), 8.43 (s,1H), 8.23–8.19 (m, 1H), 8.08 (s, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.86–7.81 (m, 1H), 7.74 (dd, J = 14.7, 7.9 Hz, 2H), 7.59–7.53 (m, 2H), 7.30 (d, J = 8.8 Hz, 2H), 7.20 (d, J = 8.8 Hz, 2H), 6.40 (s, 1H), 5.26 (s, 2H), 2.12 (s, 3H), 1.98 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.6, 163.3, 158.63, 158.0, 147.5, 146.0, 137.8, 137.1, 134.5, 132.6, 131.6, 131.5, 130.2, 129.6, 129.4, 129.3, 129.0, 127.8, 127.4, 126.9, 122.9, 116.0, 114.5, 104.7, 69.5, 13.0, 11.4. HRMS (ESI) m/z calcd for [C29H25N4O4]+ ([M + H]+): 493.1876, found 493.1871.
General procedures for preparation of ZDS3
Methyl 3-((3,5-dimethyl-1H-pyrazol-1-yl)methyl)benzoate (18)
A mixture of 3,5-dimethylpyrazole (228 mg, 2.37 mmol), sodium tert-butoxide (136 mg, 1.42 mmol) and methyl 3-bromomethylbenzoate (270 mg, 1.12 mmol) in DMF (5 ml) was stirred at 50 °C for 1 h. Upon completion, the mixture was diluted with ethyl acetate (15 ml) and washed successively with saturated NaHCO3 and then brine. After drying over anhydrous Na2SO4 and concentrated, the residue was subjected to flash chromatography to give pure product 18 as a brown solid (214 mg, yield: 74.1%). HRMS (ESI) m/z calcd for [C14H17N2O2]+ ([M + H]+): 245.1290, found 245.1296.
3-((3,5-dimethyl-1H-pyrazol-1-yl)methyl)benzoic acid (19)
A solution of intermediate 18 (214 mg, 0.88 mmol) and NaOH (53 mg, 1.32 mmol) in MeOH/H2O (2: 1, 6 ml) was heated to 70 °C for 5 h before the solvent was removed. The residue was then acidified by 2 N HCl until pH < 5. The obtained slurry was then filtered, washed with water to give pure product 19 as a white solid (73 mg, 36.1%). HRMS (ESI) m/z calcd for [C13H15N2O2]+ ([M + H]+): 231.1134, found 231.1131.
Tert-Butyl 4-(furan-2-formyl)piperazine-1-carboxylate (20)
A solution of 2-furancarboxylic acid (2.00 g, 17.86 mmol), CDI (2.89 g, 17.86 mmol) and TEA (2.97 ml, 21.43 mmol) in CH2Cl2 (20 ml) was stirred at 0 °C, tert-butyl piperazine-1-carboxylate (3.324 g, 17.86 mmol) was added to above system and then stirred at room temperature overnight. Upon completion, water (20 ml) was added and the organic phase was separated, washed successively with 1 N HCl (5 ml) and then brine. After drying over anhydrous Na2SO4, the solvent was removed under reduced pressure to give pure intermediate 20 as a white solid (3.67 g, yield: 73.4%). HRMS (ESI) m/z calcd for [C9H13N2O2]+ ([M + H-BOC]+): 181.0977, found 181.0981.
Furan-2-yl(piperazin-1-yl)methanone (21)
Intermediate 20 (2.00 g, 7.14 mmol) was dissolved in CH2Cl2 (10 ml), followed by the addition of TFA (5 ml). The reaction mixture was stirred at room temperature for 3 h until the complete conversion of the starting material. The volatile was evaporated under reduced pressure and the residue was taken into saturated NaHCO3 (20 ml), extracted with ethyl acetate (10 ml × 2), the combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give pure product 21 as a solid (1.25 g, 97.5%). HRMS (ESI) m/z calcd for [C9H13N2O2]+ ([M + H]+): 181.0977, found 181.0974.
Tert-butyl 4-hydroxypiperidine-1-carboxylate (22)
A solution of 4-hydroxypiperidine (2.00 g, 19.79 mmol), TEA (3.6 ml, 25.66 mmol) and di-tert-butyl dicarbonate anhydride (4.78 g, 25.66 mmol) in CH2Cl2 (20 ml) was stirred at room temperature for 1 h. Upon completion, water was added and the organic phase was separated, the organic phase was washed successively with 10% citric acid solution and then brine. The solvent was then dried over anhydrous Na2SO4, concentrated, and triturated with ether to give crude product 22 as a white solid (3.81 g, yield: 95.6%), which was used in the next step without further purification. HRMS (ESI) m/z calcd for [C10H20NO3]+ ([M + H]+): 202.1443, found 202.1441.
4-((Methylsulfonyl)oxy)piperidine-1-carboxylic acid tert-butyl ester (23)
A solution of N-Boc-4-hydroxypiperidine (2.00 g, 9.95 mmol), TEA (2.08 ml, 14.93 mmol) and methanesulfonyl chloride (0.92 ml, 11.94 mmol) in CH2Cl2 (20 ml) was stirred at room temperature for 2.5 h. Upon completion, saturated sodium bicarbonate solution (10 ml) was added, the organic layers were separated, washed with 10% citric acid solution and then brine. After drying over anhydrous Na2SO4, the solvent was evaporated and the residue was triturated with ether to afford pure product 23 as a white solid (2.72 g, yield: 98.2%). HRMS (ESI) m/z calcd for [C11H22NO5S]+ ([M + H]+): 280.1219, found 280.1222.
Tert-butyl 4–(3-formylphenoxy)piperidine-1-carboxylate (24)
A solution of m-hydroxybenzaldehyde (500 mg, 4.09 mmol), K2CO3 (847 mg, 6.14 mmol) and 1-Boc-4-methanesulfonyloxypiperidine (1.26 g, 4.50 mmol) in DMF (12 ml) was stirred at 90 °C for 48 h. Upon the complete conversion of the starting material, the mixture was poured into 40 ml ice-water, extracted with ethyl acetate (10 ml × 2), the collected organic layers were washed with brine, dried over anhydrous Na2SO4, and then concentrated. The crude product was further purified by column chromatography using 20% ethyl acetate in petroleum ether to give intermediate 24 as a colourless oil (458 mg, yield: 36.67%). HRMS (ESI) m/z calcd for [C12H16NO2]+ ([M + H-BOC]+): 206.1181, found 206.1183.
4–(3-((4-(Furan-2-formyl)piperazin-1-yl)methyl)phenoxy)piperidine-1-carboxylate (25)
A mixture of intermediate 24 (458 mg, 1.50 mmol), 1–(2-furanylcarbonyl)piperazine (676 mg, 3.75 mmol) in THF (5 ml) was stirred at room temperature for 30 min before sodium triacetoxyborohydride (954 mg, 4.50 mmol) was added, the mixture was then heated to 70 °C for 2 h. Upon cooling to room temperature, the reaction was quenched by saturated sodium bicarbonate (5 ml) and extracted with CH2Cl2 (10 ml × 2), the combined organic layers were washed with brine, dried over anhydrous Na2SO4, and then concentrated. The residue was then purified by flash chromatography using PE/EtOAc = 1:1 to give 25 as a brown solid (688 mg, yield: 87.7%). HRMS (ESI) m/z calcd for [C26H36N3O5]+ ([M + H]+): 470.2655, found 470.2660.
Furan-2-yl(4–(3-(piperidin-4-yloxy)benzyl)piperazin-1-yl)methanone (26)
Intermediate 25 (751 mg, 1.60 mmol) was dissolved in CH2Cl2 (10 ml), and trifluoroacetic acid (5 ml) was slowly added under an ice-water bath. The reaction mixture was then stirred at room temperature for 3.5 h. Upon complete of the reaction, the volatiles were evaporated and saturated Na2CO3 (10 ml) solution was added, the aqueous layer was then extracted with CH2Cl2 (10 ml × 2), washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give product 26 as a white solid (559 mg, yield: 94.6%). HRMS (ESI) m/z calcd for [C21H28N3O3]+ ([M + H]+): 370.2131, found 370.2132.
(4–(3-((1–(3-((3,5-dimethyl-1H-pyrazol-1-yl)methyl)benzoyl)piperidin-4-yl)oxy)benzyl)piperazin-1-yl)(furan-2-yl)methanone (ZDS3)
A mixture of 3-((3,5-dimethyl-1H-pyrazol-1-yl)methyl)benzoic acid (338 mg, 1.47 mmol), HATU (698 mg, 1.84 mmol), DIPEA (316 mg, 2.45 mmol) and intermediate 26 (452 mg, 1.22 mmol) in CH2Cl2 (10 ml) was stirred at room temperature overnight. Upon completion, the mixture was quenched by saturated solution of NaHCO3 (10 ml) and transferred to the separatory funnel, the aqueous layer was further extracted with CH2Cl2 (10 ml), the combined extracts were washed by brine, dried over Na2SO4 and concentrated. The crude product was then subjected to flash chromatography with CH2Cl2/MeOH = 99: 1 to give pure product ZDS3 as a white solid (646 mg, yield: 90.8%). 1H NMR (600 MHz, Chloroform-d) δ 7.47–7.45 (m, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.31 (dt, J = 7.6, 1.5 Hz, 1H), 7.24 (t, J = 7.9 Hz, 1H), 7.13–7.04 (m, 2H), 6.98 (d, J = 3.4 Hz, 1H), 6.92 (d, J = 7.4 Hz, 2H), 6.82 (dd, J = 8.5, 1.8 Hz, 1H), 6.47 (dd, J = 3.4, 1.8 Hz, 1H), 5.86 (s, 1H), 5.24 (s, 2H), 3.94–3.73 (m, 6H), 3.59 (s, 1H), 3.52 (s, 2H), 3.31 (s, 1H), 2.51 (s, 4H), 2.23 (s, 3H), 2.16 (s, 3H), 2.08–1.66 (m, 5H). 13C NMR (151 MHz, Chloroform-d) δ 170.05, 159.06, 157.13, 147.86, 143.65, 139.36, 137.96, 136.37, 129.49, 129.00, 127.72, 126.01, 124.86, 122.01, 116.79, 116.32, 114.75, 111.25, 105.73, 71.38, 62.71, 52.07, 44.33, 38.83, 31.10, 30.16, 13.56, 11.13. HRMS (ESI) m/z calcd for [C34H40N5O4]+ ([M + H]+):582.3080, found 582.3083.
General procedures for preparation of ZDS4
4-(1H-pyrazol-1-yl)benzonitrile (27)
A solution of pyrazole (1.00 g, 14.60 mmol) and 4-fluorobenzonitrile (2.14 g, 17.60 mmol) in DMF (20 ml) was heated to 120 °C under N2 atmosphere for 9 h. Upon cooling to room temperature, the mixture was poured into ice-water (100 ml) and extracted with ethyl acetate (20 ml × 2). The combined organic layers were washed with brine, dried over Na2SO4 and then concentrated. The crude product was further crystallised with ethyl acetate to give intermediate 26 as a light yellow solid (1.64 g, yield: 87.7%). HRMS (ESI) m/z calcd for [C10H8N3]+ ([M + H]+): 170.0718, found 170.0725.
4-(1H-pyrazol-1-yl)benzoic acid (28)
A solution of 4-(1H-pyrazol-1-yl)benzonitrile (1 g, 5.92 mmol) and NaOH (355 mg, 8.87 mmol) in MeOH/H2O (3: 2, 15 ml) was heated to 80 °C for 10 h, after consumption of the starting material, the solvent was evaporated. The residue was then acidified using 2 N HCl until pH < 4. The resulting solid was filtered, washed with water and dried under vacuum to yield 28 as a white solid (1.05 g, yield: 94.4%). HRMS (ESI) m/z calcd for [C10H9N2O2]+ ([M + H]+): 189.0664, found 189.0665.
Quinoline-5-carbaldehyde (29)
To a solution of 5-bromoquinoline (2.50 g, 12 mmol) in dry THF (30 ml) at −78 °C under N2 atmosphere, n-BuLi (2.2 M in THF, 6.09 ml, 14.40 mmol) was added. The mixture was then stirred for 1 h before DMF (1.02 ml) was added via syringe. Upon completion of the reaction as indicated by TLC, the mixture was quenched with AcOH (1.5 ml), poured into ice-water, and then extracted with EtOAc (30 ml × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered and then concentrated. The crude product was further subjected to column chromatography using petroleum PE/EtOAc = 4:1 as fluent to afford product 29 as a withe solid (770 mg, yield: 41.2%). HRMS (ESI) m/z calcd for [C10H8NO]+ ([M + H]+): 158.0606, found 158.0610.
3-(((Quinolin-5-ylmethyl)imino)methyl)phenol (30)
A solution of 5-formylquinoline (390 mg, 3.18 mmol), 3-(aminomethyl)phenol (500 mg, 3.18 mmol) and a catalytic amount of AcOH in MeOH (10 ml) was stirred at room temperature for 1 h. Upon completion, the precipitate was filtered to give intermediate 30 as an off-white solid (633 mg, yield: 76.0%). HRMS (ESI) m/z calcd for [C17H15N2O]+ ([M + H]+): 263.1184, found: 263.1188.
3-(((Quinolin-5-ylmethyl)amino)methyl)phenol (31)
A mixture of 3-(((quinolin-5-ylmethyl)amino)methyl)phenol (350 mg, 1.34 mmol) and sodium triacetoxyborohydride (750 mg, 6.68 mmol) in THF (15 ml) was stirred at 70 °C overnight. Upon cooling to room temperature, the reaction was quenched by saturated NaHCO3 (15 ml) and extracted with EtOAc (10 ml × 2), the combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude product was then subjected to flash chromatography with CH2Cl2/MeOH/TEA = 98: 1: 1 as fluent to give product 31 as a brown solid (277 mg, yield: 78.5%). HRMS (ESI) m/z calcd for [C17H17N2O]+ ([M + H]+): 265.1341, found 265.1348.
3-((Methyl(quinolin-5-ylmethyl)amino)methyl)phenol (32)
To a solution of 3-(((quinolin-5-ylmethyl)amino)methyl)phenol (184 mg, 0.69 mmol) in formaldehyde (2 ml) was added formic acid (3 ml), the mixture was heated to 70 °C overnight. Upon completion, the mixture was diluted with EtOAc (5 ml), washed with 1 N NaOH and then brine. After concentration under vacuum, the crude product was purified by flash chromatography with CH2Cl2/MeOH/TEA = 98: 1: 1 to give product 32 as a white solid (152 mg, yield: 78.4%). HRMS (ESI) m/z calcd for [C18H19N2O]+ ([M + H]+): 279.1497, found 279.1496.
Tert-butyl 4–(3-((methyl(quinolin-5-ylmethyl)amino)methyl)phenoxy)piperidine-1-carboxylate (33)
A solution of 3-((methyl(quinolin-5-ylmethyl)amino)methyl)phenol (147 mg, 0.53 mmol), K2CO3 (95 mg, 0.69 mmol) and intermediate 23 (176 mg, 0.63 mmol) in DMF (3 ml) and toluene (3 ml) was stirred at 110 °C overnight. Upon completion, the mixture was poured into ice-water (5 ml) and extracted with EtOAc (5 ml × 2), the collected organic layers were washed with brine, dried over anhydrous Na2SO4, and then concentrated. The crude product was further purified by column chromatography using PE/EtOAc = 4:1 as fluent to give 33 as a white solid (140 mg, yield: 57.7%). HRMS (ESI) m/z calcd for [C28H36N3O3]+ ([M + H]+): 462.2757, found 462.2751.
N-Methyl-N-(3-(piperidin-4-yloxy)benzyl)-1-(quinolin-5-yl)methanamine (34)
To a solution of intermediate 33 (108 mg, 0.234 mmol) in CH2Cl2 (2 ml) was added CF3COOH (2 ml), the mixture was stirred at room temperature for 2 h and the volatiles were evaporated, the residue was then partitioned between water (2 ml) and EtOAc (5 ml), the obtained organic phase was washed with brine, dried over anhydrous Na2SO4 and finally concentrated to give 34 as a colourless oil (56 mg, yield: 71.4%). HRMS (ESI) m/z calcd for [C23H28N3O]+ ([M + H]+): 362.2232, found: 362.2231.
(4-(1H-pyrazol-1-yl)phenyl)(4–(3-(methyl(quinolin-5-yl)amino)methyl)phenoxy)piperidin-1-yl)methanone (ZDS4)
A mixture of 4-(1H-pyrazol-1-yl)benzoic acid (50 mg, 0.27 mmol), HATU (130 mg, 0.33 mmol), DIPEA (60 μL, 0.33 mmol) and N-methyl-N-(3-(piperidin-4-yloxy))-methylamine (80 mg, 0.22 mmol) in CH2Cl2 (4 ml) was stirred at room temperature for 6 h. Upon completion, the mixture was quenched by saturated NaHCO3 (10 ml) and transferred to the separatory funnel, the aqueous layer was further extracted with CH2Cl2 (5 ml × 2), the combined extracts were washed with brine, dried over Na2SO4 and concentrated. The crude product was then subjected to flash chromatography with CH2Cl2/MeOH = 99: 1 to give pure product ZDS4 as a white solid (75 mg, yield: 53.2%). 1H NMR (600 MHz, Chloroform-d) δ 8.92 (dd, J = 4.1, 1.6 Hz, 1H), 8.64 (d, J = 8.5 Hz, 1H), 8.03 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 2.4 Hz, 1H), 7.77–7.75 (m, 1H), 7.74–7.72 (m, 2H), 7.65–7.60 (m, 1H), 7.55–7.52 (m, 2H), 7.50 (d, J = 6.9 Hz, 1H), 7.39 (dd, J = 8.5, 4.2 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.91 (d, J = 7.6 Hz, 1H), 6.87 (s, 1H), 6.80 (dd, J = 8.1, 2.1 Hz, 1H), 6.49 (t, J = 2.1 Hz, 1H), 4.56–4.51 (m, 1H), 3.91 (s, 2H), 3.88–3.36 (m, 6H), 2.20 (s, 3H), 2.03–1.74 (m, 4H). 13C NMR (151 MHz, Chloroform-d) δ = 169.60, 157.02, 150.08, 148.74, 141.58, 140.98, 133.72, 133.53, 129.39, 128.66, 128.43, 127.90, 127.65, 126.75, 121.86, 120.54, 118.88, 116.50, 114.62, 108.14, 71.24, 62.01, 60.06, 42.41, 38.88, 29.68. HRMS (ESI) m/z calcd for [C33H34N5O2]+ ([M + H]+): 532.2713, found: 532.2721.
General procedures for preparation of ZDS5
Methyl 4–(4-methylpiperazin-1-yl)-3-nitrobenzoate (35)
A mixture of methyl 4-fluoro-3-nitrobenzoate (1.00 g, 5.03 mmol), N-methyl piperazine (654 mg, 6.53 mmol) and N, N-diisopropylethylamine (1.14 ml, 6.53 mmol) in DMF (10 ml) was stirred at room temperature for 0.5 h. Upon completion, the mixture was poured into ice-water (5 ml) and extracted with ethyl acetate (30 ml × 3), the collected organic layers were washed with brine, dried over anhydrous Na2SO4, and then concentrated. 35 was obtained as an orange oil (1.38 g, yield: 91.9%) which was used in the next step without further purification. HRMS (ESI) m/z calcd for [C11H18N3O4]+ ([M + H]+): 280.1297, found: 280.1287.
Methyl 3-amino-4–(4-methylpiperazin-1-yl)benzoate (36)
To a solution of intermediate 35 (1.442 g, 5.12 mmol) in EtOAc (10 ml) was added Tin(II) chloride dihydrate (3.49 g, 15.50 mmol), the reaction was then warmed to 80 °C and stirred overnight. Upon completion of the reaction, the mixture was taken into saturated NaHCO3 solution (15 ml) and extracted with EtOAc (10 ml × 2), the collected organic layers were washed with brine and then dried over Na2SO4. After concentration under vacuum, 36 was obtained as a brown solid (907 mg, yield: 71.1%). HRMS (ESI) m/z calcd for [C13H20N3O2]+ ([M + H]+): 250.1556, found: 250.1559.
Methyl 3–(3-chlorobenzamido)-4–(4-methylpiperazin-1-yl)benzoate (37)
To a solution of 3-chlorobenzoic acid (754 mg, 4.82 mmol) in CH2Cl2 (10 ml) at 0 °C was added thionyl chloride (3 ml) dropwise, the mixture was then warmed to reflux for 2 h leading to complete reaction. The obtained chloride was concentrated under reduced pressure, dissolved in CH2Cl2 (3 ml) and used in the next step without purification.
To a mixture of intermediate 36 (800 mg, 3.21 mmol), TEA (1.34 ml, 9.64 mmol) in CH2Cl2 (8 ml) at 0 °C was added the above solution of chloride dropwise. After stirring at room temperature for another 1 h, the reaction was poured into ice-water (5 ml), washed with saturated NaHCO3 solution (8 ml × 2) and then brine. After drying over anhydrous Na2SO4, the crude product was triturated with a small amount of EtOAc to afford 37 as a white solid (870 mg, yield: 69.9%). HRMS (ESI) m/z calcd for [C20H23ClN3O2]+ ([M + H]+): 388.1428, found: 388.1426.
3–(3-chlorobenzamido)-4–(4-methylpiperazin-1-yl)benzoic acid (38)
To a mixture of intermediate 37 (656 mg, 1.70 mmol) in MeOH/THF (3: 2, 10 ml) was added a solution of NaOH (203 mg, 5.10 mmol) in water (4 ml), the mixture was then heated to 50 °C for 4 h before the solvent was removed. The residue was acidified by 1 N HCl until pH < 5. The obtained slurry was then filtered, washed with water to give pure product 38 as a white solid (551 mg, yield: 86.9%). HRMS (ESI) m/z calcd for [C19H21ClN3O3]+ ([M + H]+): 374.1271, found: 374.1277.
Tert-butyl 2–(2-hydroxyethyl)piperidine-1-carboxylate (39)
A solution of 4-hydroxyethylpiperidine (2.00 g, 19.79 mmol), TEA (3.60 ml, 25.66 mmol) and di-tert-butyl dicarbonate (4.78 g, 25.66 mmol) in CH2Cl2 (20 ml) was stirred at room temperature for 1 h. Upon completion, the mixture was partitioned between water and CH2Cl2. The pooled extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum to give crude product 39 as a colourless oil (3.32 g, yield: 93.6%). HRMS (ESI) m/z calcd for [C12H24NO3]+ ([M + H]+): 230.1756, found: 230.1754.
Tert-butyl 2–(2-(4-nitrophenoxy)ethyl)piperidine-1-carboxylate (40)
A solution of 1,4-trans-4-aminocyclohexanol (1.01 g, 4.37 mmol) in DMF (10 ml) was added NaH (229 mg, 9.54 mmol) at 0 °C under nitrogen atmosphere. The mixture was then warmed to room temperature and stirred for 0.5 h before addition of 4-fluoronitrobenzene (1.74 g, 6.36 mmol), the mixture was then heated to 80 °C for 12 h until the consumption of starting material. After cooling to room temperature, the mixture was carefully poured into ice-water, extracted with ethyl acetate (10 ml × 3), washed successively with saturated NaHCO3, and then brine. After drying over anhydrous Na2SO4 and concentrated, the crude product was then subjected to flash chromatography using PE/EtOAc = 9: 1 to give 40 as a yellow solid (1.876 g, yield: 84.3%). HRMS (ESI) m/z calcd for [C18H26N2O5Na]+ ([M + Na]+): 373.1739, found: 373.1745.
2–(2-(4-nitrophenoxy)ethyl)piperidine (41)
Intermediate 40 (800 mg, 2.28 mmol) was dissolved in CH2Cl2 (10 ml), followed by the addition of TFA (4 ml). The mixture was stirred at room temperature for 3 h until the consumption of the starting material. The volatiles were evaporated under reduced pressure and the residue was taken into 20 ml saturated NaHCO3, extracted with ethyl acetate (10 ml × 2), the combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The resulting residue was then subjected to flash chromatography using 10% MeOH in dichloromethane to give 41 as a yellow solid (515 mg, yield: 90.3%). HRMS (ESI) m/z calcd for [C13H19N2O3]+ ([M + H]+): 251.1396, found: 251.1402.
1-ethyl-2–(2-(4-nitrophenoxy)ethyl)piperidine (42)
To a solution of intermediate 41 (500 mg, 2.00 mmol) in DMF (10 ml) was added NaH (144 mg, 6 mmol) at 0 °C under N2 atmosphere. The reaction was then warmed to room temperature and stirred for 30 min before the addition of iodoethane (240 μL), the mixture was further stirred at ambient temperature for another 2 h. Upon completion of the reaction, the mixture was diluted with ethyl acetate, washed successively with water, and then brine. Further drying over anhydrous Na2SO4 and concentration under vacuum afforded 42 as a yellow solid (340 mg, yield: 64.9%). HRMS (ESI) m/z calcd for [C15H23N2O3]+ ([M + H]+): 279.1709, found: 279.1715.
4–(2-(1-ethylpiperidin-2-yl)ethoxy)aniline (43)
To a solution of intermediate 42 (354 mg, 1.35 mmol) in EtOAc (8 ml) was added Tin(II) chloride dihydrate (1.52 g, 6.75 mmol), the mixture was then warmed to 80 °C and stirred overnight. Upon cooling to room temperature, the mixture was taken into saturated NaHCO3 solution (15 ml) and extracted with EtOAc (15 ml × 2), the collected organic layers were washed with brine and concentrated under vacuum. The crude product was then subjected to flash chromatography using 10% MeOH in dichloromethane to give pure product 43 as a brown solid (181 mg, yield: 53.8%). HRMS (ESI) m/z calcd for [C15H25N2O]+ ([M + H]+):249.1967, found 249.1970.
2–(3-chlorobenzamido)-N-(4–(2-(1-ethylpiperidin-2-yl)ethoxy)phenyl)-4–(4-methylpiperazin-1-yl)benzamide (ZDS5)
A mixture of intermediate 38 (337 mg, 0.87 mmol), intermediate 43 (180 mg, 0.73 mg), HATU (414 mg, 1.09 mmol) and DIPEA (141 mg, 1.09 mmol) in DMF (6 ml) was stirred at room temperature for 4 h. Upon completion, the mixture was quenched by saturated NaHCO3 (20 ml) and transferred to the separatory funnel, the aqueous layer was further extracted with EtOAc (10 ml × 2), the pooled extracts were washed with brine, dried over Na2SO4 and concentrated. The crude product was then subjected to flash chromatography with CH2Cl2/MeOH = 99: 1 to give pure product ZDS5 as a white solid (283 mg, yield: 64.6%). 1H NMR (600 MHz, DMSO-d6) δ 10.07 (s, 1H), 9.78 (s, 1H), 8.38 (s, 1H), 8.02 (s, 1H), 7.95 (d, J = 7.1 Hz, 1H), 7.81 (d, J = 8.1 Hz, 1H), 7.71 (d, J = 7.5 Hz, 1H), 7.69–7.59 (m, 3H), 7.29 (d, J = 8.1 Hz, 1H), 6.91 (d, J = 8.1 Hz, 2H), 3.99 (m, 2H), 3.54–3.26 (m, 3H), 3.02–2.92 (m, 4H), 2.81–2.62 (m, 2H), 2.49 (s, 3H), 2.31–2.16 (m, 4H), 2.03–1.75 (m, 2H), 1.70–1.16 (m, 6H), 0.96 (t, J = 6.7, 3H). 13C NMR (151 MHz, DMSO-d6) δ = 164.9, 163.9, 155.3, 148.6, 136.9, 134.0, 132.7, 132.1, 131.6, 131.2, 130.2, 127.7, 126.4, 125.6, 124.5, 122.4, 120.0, 114.7, 65.5, 56.4, 55.5, 51.2, 50.4, 46.8, 46.3, 30.0, 29.8, 25.2, 23.2, 11.4. HRMS (ESI) m/z calcd for [C34H43ClN5O3]+ ([M + H]+): 604.3054, found 604.3057.
General procedures for preparation of ZDS6
4-methyl-3-nitrobenzaldehyde (44)
A mixture of 70% nitric acid and concentrated sulphuric acid (1: 3, 10 ml) was added dropwise to 4-methylbenzaldehyde (3 g, 25 mmol) on an ice-water bath with vigorous stirring. The mixture was stirred for another 2 h at ambient temperature and carefully poured into 150 ml ice-water. The resulting solid was collected via filtration, washed with water, recrystallized from hexane-chloroform to afford 4-methyl-3-nitrobenzaldehyde 44 as a yellow solid (2.83 g, yield: 68.6%). HRMS (ESI) m/z calcd for [C8H8NO3]+ ([M + H]+): 166.0504, found: 166.0508.
5-(1H-benzo[d]imidazol-2-yl)-2-methylaniline (45)
To a 100 ml round-bottom flask was charged with nitrobenzaldehyde (1.00 g, 6.06 mmol), sodium metabisulfite (3.46 g, 18.18 mmol) and DMF (10 ml), the mixture was then warmed to reflux for 1 h. Upon cooling to room temperature, a solution of o-phenylenediamine (654 mg, 6.06 mmol) in DMF (3 ml) was added, the mixture was further refluxed for another 10 h until the reaction was completed. Upon cooling, the mixture was poured into ice-water (100 ml), the resulting precipitate was filtered, washed with cold ethyl acetate, and then ether. The product was dried under vacuum to give pure 45 as a light yellow solid (361 mg, yield: 24.2%). HRMS (ESI) m/z calcd for [C14H14N3]+ ([M + H]+):224.1188, found 224.1183.
Methyl 4-((4-chlorobenzyl)oxy)-3-methoxybenzoate (46)
A solution of methyl 4-hydroxy-3-methoxybenzoate (1.00 g, 5.49 mmol) and 1-(bromomethyl)-4-chlorobenzene (1.35 g, 6.59 mmol) in toluene (16 ml) was treated with K2CO3 (1.288 g, 9.33 mmol). The resulting mixture was warmed to 120 °C and stirred for 7 h. Upon completion, was added (5 ml water) and the organic phase was separated, washed with brine and concentrated, the crude product was recrystallized from EtOAc to give 46 as a white solid (1.05 g, yield: 62.5%). HRMS (ESI) m/z calcd for [C16H16ClO4]+ ([M + H]+): 307.0737, found 307.0745.
2-((4-chlorobenzyl)oxy)-3-methoxybenzoic acid (47)
To a solution of intermediate 46 (415 mg, 1.36 mmol) in MeOH/THF (1:1, 10 ml) was added a solution of NaOH (217 mg, 5.42 mmol) in water (3 ml). The reaction mixture was heated to 50 °C for 3 h until the consumption of the starting material. The volatiles were removed under reduced pressure, and the residue was acidified to pH < 4 by 2 N HCl. The slurry was then stirred at room temperature for 30 min and filtered, washed with water and dried to give 47 as a white solid (328 mg, yield: 82.8%). HRMS (ESI) m/z calcd for [C15H14ClO4]+ ([M + H]+): 293.0581, found 293.0589.
N-(5-(1H-benzo[d]imidazol-2-yl)-2-methylphenyl)-4-((4-chlorobenzyl)oxy)-3-methoxybenzamide (ZDS6)
To a solution of intermediate 47 (158 mg, 0.536 mmol) in CH2Cl2 (3 ml) was added thionyl chloride (50 µL) at 0 °C under ice-water bath, the mixture was then warmed to reflux for 2 h, Upon completion, the volatiles were removed and the residue was redissolved in CH2Cl2 (2 ml), which was directly used in the next step without further purification. Then, to a 3-neck flask was charged with intermediate 45 (100 mg, 0.45 mmol), CH2Cl2 (3 ml), and NaH (16 mg, 0.67 mmol) under N2 atmosphere, the above obtained acyl chloride was added slowly to the system. The mixture was stirred at room temperature for 2 h before quenched by saturated NaHCO3. The organic phase was further washed with brine and dried over anhydrous Na2SO4 before concentrated. The crude residue was subsequently purified using silica gel chromatography (CH2Cl2/MeOH = 99: 1) to obtain ZDS6 as a white solid (162 mg, yield: 73.1%). 1H NMR (600 MHz, DMSO-d6) δ 12.90 (s, 1H), 9.95 (s, 1H), 8.18 (d, J = 1.5 Hz, 1H), 7.98 (dd, J = 7.9, 1.5 Hz, 1H), 7.68–7.63 (m, 3H), 7.53–7.48 (m, 5H), 7.46 (d, J = 8.0 Hz, 1H), 7.23–7.17 (m, 3H), 5.21 (s, 2H), 3.88 (s, 3H), 2.31 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ = 165.3, 151.4, 150.9, 149.2, 137.6, 136.4, 136.3, 133.0, 131.4, 130.1, 129.0, 128.7, 127.4, 125.4, 124.3, 122.9, 121.3, 119.2, 113.1, 111.7, 69.5, 56.2, 18.4. HRMS (ESI) m/z calcd for [C29H25ClN3O3]+ ([M + H]+): 498.1584, found 498.1577.
General procedures for preparation of ZDS7
Methyl 4-acetyl-5-oxohexanoate (intermediate 48)
To a mixture of pentane-2,4-dione (5.13 ml, 50 mmol) and MeONa (14 mg, 2.50 mmol) in 50 ml round-bottom flask was added methyl acrylate (4.95 ml, 55 mmol) dropwise over 15 min. The system was heated to 100 °C and maintained for 3 h until the reaction was completed. Upon cooling, the mixture was partitioned between water and ethyl acetate, the obtained organic phase was washed with brine, dried over Na2SO4 and concentrated under reduced pressure to give 48 as a colourless liquid (8.24 g, yield: 88.6%). HRMS (ESI) m/z calcd for [C9H15O4]+ ([M + H]+): 187.0970, found 187.0980.
Methyl 3–(5,7-dimethyl-2-phenylpyrazolo[1,5-a]pyrimidin-6-yl)propanoate (intermediate 49)
Methyl 4-acetyl-5-oxo-caproate (2.00 g, 10.75 mmol) and 3-amino-5-phenylpyrazole (1.14 g, 7.20 mmol) were dissolved in EtOH/AcOH (2:1, 30 ml) and stirred at 80 °C overnight under nitrogen atmosphere. Upon completion, the solvent was evaporated and the residue was partitioned between saturated NaHCO3 (15 ml) and EtOAc (15 ml), the aqueous phase was extracted once more with ethyl acetate (15 ml), the combined organic layers were washed with brine, dried over Na2SO4 and concentrated. The crude product was subjected to flash chromatography (PE/EtOAc = 5: 1) to obtain 49 as a white solid (1.64 g, 73.6%). HRMS (ESI) m/z calcd for [C18H20N3O2]+ ([M + H]+): 310.1556, found 310.1551.
3–(5,7-dimethyl-2-phenylpyrazolo[1,5-a]pyrimidin-6-yl)propanoic acid (intermediate 50)
A solution of intermediate 49 (1 g, 3.24 mmol) and NaOH (640 mg, 16.18 mmol) in MeOH/H2O (1:1, 10 ml) was heated to 50 °C for 3 h. Upon completion, the volatiles were evaporated and the residue was acidified with 2 N HCl, the obtained solid was filtered, washed with water and dried under vacuum to give 50 as a white solid (780 mg, yield of 81.4%). HRMS (ESI) m/z calcd for [C17H16N3O2]+ ([M + H]+):294.1243, found 294.1248.
Bis(2-chloroethyl)amine hydrochloride (intermediate 51)
A solution of diethanolamine (5 ml, 43.35 mmol) and SOCl2 (13 ml, 66.71 mmol) in CH2Cl2 (15 ml) was heated to reflux for 0.5 h. Upon cooling, the precipitate obtained was filtered, washed with CH2Cl2 and then ether to give 51 as a white solid (7.06 g, yield: 90.4%).
1–(3-Chlorophenyl)piperazine hydrochloride (intermediate 52)
A mixture of 2-chloroaniline (1.53 g, 12.01 mmol), bis(2-chloroethyl)amine hydrochloride (2.12 g, 12.00 mmol) and diethylene glycol monomethyl ether (5 ml) was heated to 150 °C for 12 h. Upon cooling to 0 °C, MeOH (10 ml) was added to precipitate the product, the obtained solid was filtered and washed with ether (10 ml) to give m-chloro-arylpiperazine hydrochloride (1.98 g, yield of 71.4%) as a white solid, which was used in the next reaction without further purification. HRMS (ESI) m/z calcd for [C10H14ClN2]+ ([M + H]+): 197.0846, found 197.0839.
2–(3-(4–(3-chlorophenyl)piperazin-1-yl)propyl)isoindoline-1,3-dione (intermediate 53)
To a 100 ml round-bottom flask was charged with m-chloro-arylpiperazine hydrochloride (1.91 g, 8.20 mmol), acetonitrile (20 ml) and NaOH (820 mg, 20.5 mmol) under nitrogen atmosphere. After stirring at room temperature for 30 min, N-(3-bromopropyl) phenylenediamine (2 g, 7.46 mmol) was added and the mixture was then heated to reflux for 14 h. Upon completion, the mixture was poured into ice-water and extracted with ethyl acetate (15 ml × 2), the combined organic layers were washed with brine, dried over Na2SO4 and concentrated, the crude product was recrystallized from MeOH to give pure 53 as a white solid (1.54 g, yield of 53.8%). HRMS (ESI) m/z calcd for [C21H23ClN3O3]+ ([M + H]+):384.1479, found 384.1471.
(4–(3-chlorophenyl)piperazin-1-yl)propan-1-amine (intermediate 54)
To a solution of intermediate 53 (1.20 g, 3.13 mmol) in EtOH (6 ml) was added 80% hydrazine hydrate (3 ml), the mixture was heated to reflux for 2 h. Upon completion of the reaction, the solvent was evaporated and water (10 ml) was added, the aqueous layer was extracted with EtOAc (10 ml × 2), the combined organic layers were washed with brine, dried over Na2SO4 and concentrated. 54 was obtained as a colourless oil (573 mg, yield of 72.4%) which was used in the next reaction without further purification. HRMS (ESI) m/z calcd for [C13H21ClN3]+ ([M + H]+):254.1424, found 254.1427.
N-(3–(4-(3-chlorophenyl)piperazin-1-yl)propyl)-3–(5,7-dimethyl-2-phenylpyrazolo[1,5-a]pyrimidin-6-yl)propanamide(ZDS7)
A solution of 3–(5,7-dimethyl-2-phenylpyrazolo[1,5-a]pyrimidin-6-yl)propanoic acid (643 mg, 2.18 mmol), HATU (1.04 g, 2.73 mmol), DIPEA (470 mg, 3.64 mmol) and 3–(4-(3-chlorophenyl)piperazin-1-yl)propan-1-amine (460 mg, 1.86 mmol) in CH2Cl2 (15 ml) was stirred at room temperature overnight. Upon completion, the mixture was washed with saturated NaHCO3 (5 ml) and then brine. After drying over anhydrous Na2SO4, the solvent was evaporated and the crude product was subjected to flash chromatography with CH2Cl2/MeOH = 99: 1 as fluent to obtain ZDS7 (716 mg, yield: 74.2%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 8.04 (d, J = 7.3 Hz, 2H), 7.89 (s, 1H), 7.48 (t, J = 7.6 Hz, 2H), 7.40 (t, J = 7.3 Hz, 1H), 7.18 (t, J = 8.1 Hz, 1H), 7.02 (s, 1H), 6.84 (s, 1H), 6.77 (d, J = 7.3 Hz, 2H), 3.13–2.99 (m, 6H), 2.96 (t, J = 7.4 Hz, 2H), 2.78 (s, 3H), 2.57 (s, 3H), 2.36 (t, J = 7.4 Hz, 4H), 2.33–2.06 (m, 4H), 1.48 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ = 171.43, 158.70, 154.01, 152.49, 147.97, 143.29, 134.26, 133.40, 130.81, 129.23, 129.14, 126.45, 118.45, 114.87, 113.98, 92.27, 55.55, 55.39, 52.71, 47.80, 37.20, 35.61, 26.52, 24.41, 23.72, 13.73. HRMS (ESI) m/z calcd for [C30H35ClN6O]+ ([M + H]+):531.2639, found 531.2644.
General procedures for preparation of ZDS8
1–(2-chlorobenzyl)-4–(4-nitrophenyl)piperazine (intermediate 55)
To a solution of 1–(4-nitrophenyl)piperazine (1.50 g, 7.25 mmol) and 2-chlorobenzaldehyde (0.85 g, 6.04 mmol) in THF (35 ml) was added sodium triacetoxyborohydride (3.841 g, 18.12 mmol) portion-wise with stirring. The reaction was then heated to 70 °C for 2 h. Upon completion, the mixture was quenched by 1 N NaOH (15 ml) and extracted with EtOAc (15 ml), the organic phase was washed with brine and then concentrated. The crude residue was recrystallized from PE/EtOAc to give pure 55 as a light yellow solid (1.62 g, yield: 80.3%). HRMS (ESI) m/z calcd for [C17H19ClN3O2]+ ([M + H]+): 332.1166, found: 332.1157.
4–(4-(2-chlorobenzyl)piperazin-1-yl)aniline (intermediate 56)
To a 100 ml round-bottom flask was charged with Tin(II) chloride dihydrate (5.23 g, 23.18 mmol), intermediate 55 (1.61 g, 7.73 mmol) and EtOAc (25 ml), the mixture was stirred at 80 °C for 17 h until the completion of the reaction. The mixture was neutralised with saturated Na2CO3, filtered through celite and washed with EtOAc. The organic phase was separated, washed with brine, dried over Na2SO4 and concentrated. The crude residue was purified via silica gel chromatography (PE/EA = 4: 1) to give pure 56 as a yellow solid (1.53 g, yield: 65.7%). HRMS (ESI) m/z calcd for [C17H21ClN3]+ ([M + H]+): 302.1424, found: 302.1425.
5–(2,5-dichlorophenyl)furan-2-carboxylic acid (Intermediate 57)
To a mixture of (2,5-dichlorophenyl)boronic acid (3.36 g, 17.60 mmol), methyl 5-bromofuran-2-carboxylate (3.00 g, 14.60 mmol), Na2CO3 (3.10 g, 29.2 mmol) and Pd(PPh3)4 (845 mg, 0.73 mmol) was added ethylene glycol dimethyl ether (30 ml) and water (15 ml) under nitrogen atmosphere. The resulting mixture was then stirred at 100 °C for 24 h. Upon completion, the mixture was acidified with 1 N HCl and extracted with ethyl acetate (20 ml × 3). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated. The crude product was subjected to flash chromatography to give 57 as a white solid (2.75 g, yield of 73.6%). HRMS (ESI) m/z calcd for [C11H7Cl2O3]+ ([M + H]+): 256.9772, found: 256.9779.
5–(2,5-dichlorophenyl)furan-2-carbonyl isothiocyanate (intermediate 58)
Intermediate 57 (1.00 g, 3.91 mmol) was dissolved in CH2Cl2 (6 ml) under an ice-water bath, followed by the addition of SOCl2 (3 ml) and a catalytic amount of DMF. After stirring at room temperature for 3 h, the volatiles were removed under reduced pressure. The resulting product was further dissolved in dry acetone (5 ml) and then added dropwise to the solution of potassium thiocyanate (379 mg, 3.91 mmol) in acetone (5 ml) at 0 °C. The mixture was then stirred at room temperature for another 0.5 h before filtered. 58 was obtained as a yellow solid (732 mg, yield: 63.1%) and used in the next step without further purification. HRMS (ESI) m/z calcd for [C12H6Cl2NO2S]+ ([M + H]+): 297.9496, found: 297.9501.
N-((4–(4-(2-chlorobenzyl)piperazin-1-yl)phenyl)carbamothioyl)-5–(2,5-dichlorophenyl)furan-2-carboxamide (ZDS8)
To a stirred solution of intermediate 56 (713 mg, 2.37 mmol) in dry acetone (15 ml) was added intermediate 58 (642 mg, 2.16 mmol), after stirring at room temperature for 30 min, the mixture was filtered, washed with a small amount of cold acetone to give pure ZDS8 as a yellow solid (1.21 g, yield: 82.8%). 1H NMR (600 MHz, DMSO-d6) δ 12.26 (s, 1H), 11.59 (s, 1H), 8.33 (d, J = 1.9 Hz, 1H), 7.82 (d, J = 3.9 Hz, 1H), 7.66 (d, J = 8.6 Hz, 1H), 7.57–7.49 (m, 4H), 7.47–7.43 (m, 2H), 7.33 (dt, J = 29.1, 7.5 Hz, 2H), 6.97 (d, J = 8.6 Hz, 2H), 3.63 (s, 2H), 3.18 (t, J = 5.0 Hz, 4H), 2.59 (m, J = 5.0 Hz, 4H). 13C NMR (151 MHz, DMSO-d6) δ = 178.4, 157.7, 152.2, 149.7, 145.3, 135.9, 133.8, 133.1, 132.9, 131.4, 130.6, 129.8, 129.6, 129.1, 129.1, 128.8, 127.5, 125.5, 120.8, 115.4, 114.7, 59.0, 53.0, 48.6. HRMS (ESI) m/z calcd for [C29H26Cl3N4O2S]+ ([M + H]+):599.0842, found: 599.0847.
General procedure for preparation of ZDS9
1,4-bis(((S)-oxiran-2-yl)methoxy)benzene (intermediate 59)
A mixture of hydroquinone (0.36 g, 3.24 mmol), (S)-2-(chloromethyl)oxirane (3.00 g, 32.43 mmol) and benzyl triethylammonium chloride (74 mg, 0.34 mmol) was warmed to 90 °C for 12 h under nitrogen atmosphere. A solution of 30% NaOH (3.5 ml) was added to the mixture subsequently, the mixture was stirred for another 4 h until the completion of the reaction. The mixture was then partitioned between water and CH2Cl2. The separated organic phase was washed with brine, dried over Na2SO4 and concentrated. The crude residue was purified by silica gel chromatography (PE/EA = 4: 1) to give pure 59 as a white solid (496 mg, yield: 24.9%). HRMS (ESI) m/z calcd for [C12H15O4]+ ([M + H]+): 223.0970, found: 223.0973.
(2S,2’S)-3,3’-(1,4-phenylenebis(oxy))bis(1–(4-(3-chlorophenyl)piperazin-1-yl)propan-2-ol) (ZDS9)
To a solution of intermediate 59 (100 mg, 0.41 mmol) and 1–(3-chlorophenyl) piperazine hydrochloride (228 mg, 0.49 mmol) in EtOH (2 ml) at room temperature was added DIPEA (156 mg, 1.21 mmol) dropwise. The mixture was heated to 80 °C for 1h until the completion of the reaction. Upon cooling, the precipitate was filtered and washed by EtOH to obtain ZDS9 as a white solid (206 mg, yield of 81.7%). 1H NMR (600 MHz, DMSO-d6) δ = 7.19 (t, J = 8.1 Hz, 2H), 6.92 (t, J = 2.3 Hz, 2H), 6.87–6.89 (m, 6H), 6.77 (d, J = 7.8 Hz, 2H), 4.87 (s, 2H), 3.99–3.96 (m, 2H), 3.93–3.90 (m, 2H), 3.82 (dd, J = 9.7, 6.1 Hz, 2H), 3.15 (t, J = 5.0 Hz, 8H), 2.64–2.53 (m, 8H), 2.52–2.45 (m, 2H), 2.41 (dd, J = 12.8, 6.6 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ = 153.3, 152.7, 134.3, 130.8, 118.4, 115.9, 114.9, 114.0, 72.0, 67.2, 61.5, 53.7, 48.2. HRMS (ESI) m/z calcd for [C32H41Cl2N4O4]+ ([M + H]+): 615.2505, found: 615.2508.
General procedures for preparation of ZDS10
Tert-butyl-4–(2,2-dimethyl-4,6-dioxo-1,3-dioxane-5-carbonyl)piperidine-1-carboxylate(intermediate 60)
A mixture of 2,2-dimethyl-1,3-dioxane-4,6-dione (5.01 g, 34.71 mmol), 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (8.75 g, 38.16 mmol) and DMAP (6.36 g, 52.05 mmol) in CH2Cl2 (300 ml) was added DCC (9.31 g, 45.11 mmol) portionwise at 0 °C under an ice-water bath. The bath was removed and mixture was then stirred at room temperature for 12 h. The slurry formed was filtered and the filtrate was washed successively with 5% citric acid solution, saturated sodium bicarbonate and then brine. The organic phase was dried with anhydrous Na2SO4 and concentrated under reduced pressure to obtain 60 as a light-yellow solid (10.68 g, yield: 86.7%). HRMS (ESI) (m/z): calcd for [C17H26NO7]+ ([M + H]+): 356.1709, found: 356.1704.
Tert-butyl 4–(3-ethoxy-3-oxopropanoyl)piperidine-1-carboxylate (intermediate 61)
To a 100 ml round-bottom flask was added intermediate 60 (5.02 g, 14.81 mmol) and EtOH (25 ml), the mixture was heated to reflux for 20 h until the completion of the reaction. The solvent was evaporated to give 61 as a yellow oil (4.12 g, yield: 97.9%), which was used in the next step without further purification. HRMS (ESI) (m/z): calcd for [C15H26NO5]+ ([M + H]+): 300.1811, found 300.1821.
Tert-Butyl 4–(3-ethoxy-2-(ethoxycarbonyl)acryloyl)piperidine-1-carboxylate (intermediate 62)
A mixture of intermediate 61 (6.67 g, 22.50 mmol), acetic anhydride (4.18 ml, 67.50 mmol) and triethyl orthoformate (11.16 ml, 44.50 mmol) was stirred at 100 °C for 48 h. The volatiles were removed under reduced pressure and the residue was partitioned between water and EtOAc (80 ml). The organic phase was then separated, washed with brine, dried over Na2SO4 and concentrated. The crude product was purified by flash chromatography (CH2Cl2/MeOH = 99: 1) to give pure 62 as yellow oil (6.57 g, yield: 82.2%). HRMS (ESI) (m/z): calcd for [C18H30NO6]+ ([M + H]+): 356.2068, found 356.2067.
Tert-Butyl 4–(4-(ethoxycarbonyl)-1-(p-tolyl)-1H-pyrazol-5-yl)piperidine-1-carboxylate (intermediate 63)
To a mixture of p-methylphenylhydrazine hydrochloride (161 mg, 1.01 mmol) and sodium bicarbonate (85 mg, 1.01 mmol) in EtOH (10 ml) was added intermediate 62 (300 mg, 0.85 mmol), the mixture was then warmed to 100 °C and stirred for 10 h. Upon completion, the mixture was concentrated under reduced pressure, diluted with EtOAc, washed with 10% citric acid, and then brine. The separated organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give 63 as an oil (0.33 g, yield: 77.8%), which was used in the next step without purification. HRMS (ESI) (m/z): calcd for [C23H32N3O4]+ ([M + H]+): 414.2393, found 414.2398.
5–(1-(Tert-butoxycarbonyl)piperidin-4-yl)-1-(p-tolyl)-1H-pyrazole-4-carboxylic acid (intermediate 64)
A solution of intermediate 63 (300 mg, 0.75 mmol) in MeOH (3 ml) was added a solution of NaOH (68 mg, 2.25 mmol) in water (3 ml), the mixture was then heated to 80 °C for 2 h. Upon cooling to room temperature, the volatiles were removed and the residue was acidified with 2 N HCl, the obtained solid was filtered, washed with ether, dried under vacuum to obtain 64 as a white solid (249 mg, yield: 86.4%). HRMS (ESI) (m/z): calcd for [C21H28N3O4]+ ([M + H]+): 386.2080, found: 386.2084.
Tert-butyl 4–(2-methoxybenzyl)piperazine-1-carboxylate (intermediate 65)
To a mixture of 2-methoxybenzaldehyde (4.00 g, 29.38 mmol), tert-butyl piperazine-1-carboxylate (6.00 g, 32.32 mmol) in THF (56 ml) was stirred at room temperature for 30 min before sodium triacetoxyborohydride (18.38 g, 88.15 mmol) was added, the mixture was then heated to 70 °C for 5 h before quenched by water. The aqueous layer was then extracted with EtOAc (10 ml × 2), the combined extracts were washed with brine, dried over Na2SO4 and concentrated. The crude product was recrystallized from MeOH to give 65 as a light yellow solid (6.91 g, yield: 76.5%). HRMS (ESI) (m/z): calcd for [C17H27N2O3]+ ([M + H]+): 307.2022, found: 307.2024.
1–(2-methoxybenzyl)piperazine (intermediate 66)
To a solution of tert-butyl 4–(2-methoxybenzyl)piperazine-1-carboxylate (1.00 g, 3.26 mmol) in CH2Cl2 (8 ml) was added CF3COOH (4 ml) dropwise. The reaction was stirred at room temperature for 2.5 h until the completion of the reaction. The volatiles were removed and the residue was partitioned between saturated Na2CO3 and EtOAc, the organic layer was separated, washed with brine, dried over Na2SO4 and concentrated. After trituration with cold acetone, intermediate 66 was obtained as a white solid (52 mg, yield: 75.4%) and used in the next step without further purification. HRMS (ESI) (m/z): calcd for [C12H19N2O]+ ([M + H]+): 207.1497, found: 207.1492.
2–(3-(4–(2-methoxybenzyl)piperazin-1-yl)propyl)isoindoline-1,3-dione (intermediate 67)
A mixture of 1–(2-methoxybenzyl)piperazine (483 mg, 2.34 mmol), Cs2CO3 (2.78 g, 8.53 mmol) and N-(3-bromopropyl) phenylenediamine (571 mg, 2.13 mmol) in MeCN (10 ml) was stirred at 80 °C for 2 h. Upon completion, the solvent was removed and the residue was partitioned between water and EtOAc, the separated organic phase was washed with brine, dried over Na2SO4 and concentrated. The crude product was then subjected to flash chromatography with PE/EA = 4: 1 to give 67 as a light yellow solid (696 mg, yield: 75.6%). HRMS (ESI) (m/z): calcd for [C23H28N3O3]+ ([M + H]+): 394.2131, found: 394.2135.
3–(4-(2-methoxybenzyl)piperazin-1-yl)propan-1-amine (intermediate 68)
To a solution of 2–(3-(4–(2-methoxybenzyl)piperazin-1-yl)propyl)isoindoline-1,3-dione (500 mg, 1.27 mmol) in EtOH (8 ml) was added 80% hydrazine hydrate (203 μL, 6.36 mmol), the mixture was heated to reflux for 3 h. Upon completion, the solid was decanted and the filtrate was concentrated, dissolved in EtOAc (5 ml), washed with brine and then dried over Na2SO4. After concentration under vacuum, the 68 was obtained as a yellow solid which was used in the next step without further purification (218 mg, yield: 65.3%). HRMS (ESI) (m/z): calcd for [C15H26N3O]+ ([M + H]+): 264.2076, found: 264.2071.
Tert-butyl 4–(4-((3–(4-(2-methoxybenzyl)piperazin-1-yl)propyl)carbamoyl)-1-(p-tolyl)-1H-pyrazol-5-yl)piperidine-1-carboxylate (ZDS10)
A mixture of intermediate 64 (1.66 g, 4.32 mmol), HATU (3.28 g, 8.63 mmol), DIPEA (1.67 g, 12.95 mmol) and 3–(4-(2-methoxybenzyl)piperazin-1-yl)propan-1-amine (1.14 g, 4.32 mmol) in THF (20 ml) was stirred at room temperature for 8 h. Upon completion, the mixture was poured into water (20 ml) and extracted with EtOAc (10 ml × 2), the combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was then subjected to flash chromatography with DCM/MeOH = 99: 1 as fluent to give compound ZDS10 as a white solid (2.14 g, yield: 78.7%). 1H NMR (600 MHz, DMSO-d6) δ 8.33 (s, 1H), 8.06 (s, 1H), 7.35 (d, J = 7.9 Hz, 2H), 7.34–7.28 (m, 2H), 7.27 (d, J = 8.0 Hz, 3H), 7.02 (d, J = 8.0 Hz, 1H), 6.95 (t, J = 7.2 Hz, 1H), 3.92 (s, 2H), 3.79 (s, 3H), 3.69–3.56 (m, 1H), 3.44–3.31 (m, 6H), 3.06 (q, J = 7.3 Hz, 2H), 3.00–2.84 (m, 4H), 2.40 (s, 3H), 2.21–2.11 (m, 2H), 1.92–1.77 (m, 2H), 1.51 (d, J = 11.2 Hz, 2H), 1.38 (s, 9H), 1.20 (t, J = 7.3 Hz, 2H), 1.09 (t, J = 7.0 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 163.4, 162.1, 158.6, 158.0, 155.7, 154.3, 147.8, 140.1, 139.2, 137.3, 130.2, 126.9, 120.7, 115.8, 111.5, 79.0, 65.4, 55.88, 51.4, 50.2, 45.8, 34.9, 29.1, 28.6, 21.2, 15.6, 8.9. HRMS (ESI) (m/z): calcd for [C36H51N6O4]+ ([M + H]+): 631.3972, found: 631.3971.
General procedures for preparation of ZDS11
Ethyl 2-formylthiazole-4-carboxylate (intermediate 69)
To a stirred solution of ethyl 2-methylthiazole-4-carboxylate (3.00 g, 17.52 mmol) in glacial acetic acid (30 ml), selenium oxide (9.72 g, 87.6 mmol) was added, the reaction was then heated to 120 °C for 24 h. Upon cooling to room temperature, the mixture was filtered and the solvent were removed under reduced pressure. The residue was neutralised with saturated NaHCO3 and extracted with CH2Cl2 (20 ml × 2). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. Further purification by flash chromatography on silica gel with DCM/MeOH = 99: 1 afforded 69 as a yellow oil (1.69 g, yield: 52.4%). HRMS (ESI) (m/z): calcd for [C7H8NO3S]+ ([M + H]+): 186.0225, found: 186.0229.
1–(3-(Trifluoromethyl)phenyl)piperazine (intermediate 70)
A mixture of 3-(trifluoromethyl)aniline (1.50 g, 9.31 mmol), bis(2-chloroethyl)amine hydrochloride (1.68 g, 9.31 mmol) and diethylene glycol monomethyl ether (2.5 ml) was heated to 150 °C for 12 h. Upon cooling to room temperature, ether (20 ml) was added, the precipitate was filtered and dried to give 70 as a white solid (1.06 g, yield: 71.4%), which can be used in the next reaction without further purification. HRMS (ESI) (m/z): calcd for [C11H14F3N2]+ ([M + H]+): 231.1109, found: 231.1114.
N-(4-(benzyloxy)benzyl)-1–(4-fluorophenyl)methanamine (intermediate 71)
A mixture of 4-(benzyloxy)benzaldehyde (2.00 g, 9.43 mmol), (4-fluorophenyl)methanamine (1.18 g, 9.43 mmol) in THF (40 ml) was stirred at room temperature for 30 min before sodium triacetoxyborohydride (3.99 g, 18.86 mmol) was added, the mixture was then heated to 70 °C for 3h. Upon completion, the mixture was carefully quenched by saturated Na2CO3 and then extracted with EtOAc (10 ml × 2). The pooled extracts were washed with brine, dried over anhydrous Na2SO4, concentrated and then triturated with ether to give 71 as a white solid (2.80 g, yield: 92.3%). HRMS (ESI) (m/z): calcd for [C21H21FNO]+ ([M + H]+): 322.1607, found: 322.1610.
Ethyl 2-(((4-(benzyloxy)benzyl)(4-fluorobenzyl)amino)methyl)thiazole-4-carboxylate (intermediate 72)
A solution of N-(4-(benzyloxy)benzyl)-1–(4-fluorophenyl)methanamine (1.74 g, 5.40 mmol), ethyl 2-formylthiazole-4-carboxylate (1 g, 5.41 mmol) in THF (30 ml) was stirred at room temperature for 30 min before sodium triacetoxyborohydride (2.86 g, 13.52 mmol) was added, the mixture was then heated to 70 °C for 4.5 h until the completion of the reaction. The mixture was carefully quenched by saturated Na2CO3 and then extracted with EtOAc (10 ml × 2). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrate under reduced pressure, the crude product was purified by flash chromatography with PE/EA = 4: 1 to give 72 as a white solid (2.20 g, yield: 83.2%). HRMS (ESI) (m/z): calcd for [C28H28FN2O3S]+ ([M + H]+): 491.1805, found: 491.1812.
2-(((4-(Benzyloxy)benzyl)(4-fluorobenzyl)amino)methyl)thiazole-4-carboxylic acid (intermediate 73)
A solution of intermediate 72 (992 mg, 2.02 mmol) and NaOH (324 mg, 8.09 mmol) in MeOH/H2O (3: 1, 12 ml) was stirred at 50 °C for 3 h. Upon completion, the volatiles were removed and the residue was acidified with 2 N HCl, the obtained solid was filtered, washed with water and then acetone to give 73 as a white solid (688 mg, yield: 73.5%). HRMS (ESI) (m/z): calcd for [C26H24FN2O3S]+ ([M + H]+): 463.1492, found: 463.1490.
(2-(((4-(Benzyloxy)benzyl)(4-fluorobenzyl)amino)methyl)thiazol-4-yl)(4–(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (ZDS11)
A mixture of intermediate 73 (526 mg, 1.14 mmol), HATU (649 mg, 1.71 mmol), DIPEA (440 mg, 12.95 mmol) and 1–(3-(trifluoromethyl)phenyl)piperazine (262 mg, 1.14 mmol) in DMF (5 ml) was stirred at room temperature for 5 h. Upon completion, the mixture was then poured into ice-water (30 ml) and extracted with EtOAc (10 ml × 2). The combined extracts were washed with brine, dried over Na2SO4, and concentrated. The crude product was then subjected to flash chromatography with PE/EA = 4: 1 to give ZDS11 as a white solid (585 mg, yield: 72.2%). 1H NMR (600 MHz, Chloroform-d) δ 7.92 (s, 1H), 7.42 (d, J = 7.4 Hz, 2H), 7.39–7.37 (m, 3H), 7.36 (d, J = 1.7 Hz, 1H), 7.35–7.30 (m, 3H), 7.25 (s, 1H), 7.14–7.10 (m, 2H), 7.07 (d, J = 8.8 Hz, 1H), 7.03 (t, J = 8.5 Hz, 2H), 6.96 (d, J = 8.5 Hz, 2H), 5.05 (s, 2H), 4.12–3.83 (m, 6H), 3.62 (d, J = 14.7 Hz, 4H), 3.37–3.17 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 171.3, 162.6, 161.9 (d, J = 242.8 Hz, 1 C), 158.2, 151.5, 149.9, 137.6, 135.0 (d, J = 2.8 Hz, 1 C), 130.9 (d, J = 8.2 Hz, 2 C), 130.7, 130.6, 130.6, 130.4, 130.3, 128.9 (2 C), 128.3, 128.2 (2 C), 126.3, 125.4, 123.6, 119.6, 115.7 (d, J = 21.3 Hz, 2 C), 115.2, 111.9, 111.8, 69.7, 65.4, 57.06, 57.1, 56.8, 56.8, 54.3, 38.8, 15.7. HRMS (ESI) (m/z): calcd for [C37H35F4N4O2S]+ ([M + H]+): 675.2417, found: 675.2414.
PD-1/PD-L1 binding assay
The ability of newly compounds in impeding PD-1/PD-L1 binding was studied in a well-established HTRF assay.Citation18 The PD-1/PD-L1 binding assay kit (Cat. No. 64PD1PEH) was purchased from Cisbio and the experiments were performed according to the manufacturer’s guidelines. Briefly, the compounds were dissolved in DMSO and transferred to 384-well plate, the appropriate concentration of compounds was preincubated with Tag-PD-L1 followed by Tag-PD-1 solution in detection buffer. The mixture was incubated at room temperature for 60 min. Then detection buffer including anti-Tag1-Eu and anti-Tag2-XL665 was added to assay plate, sealed and incubate at room temperature for 60 min. Fluorescent emissions from RFU 665 nm and RFU 615 nm were recorded by using Envision.
Western blot
Murine melanoma B16F10 (CL-0319) was purchased from Procell Life Science& Technology Co., Ltd. (Wuhan, China). B16F10 cells were cultured in Roswell Park Memorial Institute-1640 medium (Sangon Biotech, Shanghai) supplemented with 10% foetal bovine serum (FBS, Excell Bio, Shanghai) and 1% penicillin/streptomycin (PS, MacGene, Beijing). The cells were maintained in a 37 °C incubator with a humidified atmosphere containing 5% CO2. For western blot analysis, B16F10 cells were seeded in six-well plates and treated with DMSO (control), ZDS20 (3 μM, 15 μM and 27 μM) for 36 h, respectively. Protein lysates were obtained using RIPA buffer supplemented with PMSF and a Phosphatase A inhibitor. The protein concentration was determined using the BCA Protein Assay Kit (P0010, Beyotime). The proteins were then separated using SDS-PAGE and subsequently transferred to a PVDF membrane. Following the transfer, the membrane was blocked with 5% non-fat milk powder for 1 h at room temperature. Primary antibodies were then incubated overnight at 4 °C, followed by three washes with TBST. Subsequently, secondary antibodies were applied for 2 h, followed by an additional three washes with TBST. Finally, protein bands were visualised and obtained using a Chemiluminescence Imaging System (VILBER-VILBER FUSION FX6.EDGE). The antibodies used were PD-L1 (Proteintech, 66248) and Vinculin (Abcam, ab129002). The secondary antibodies used were goat anti-rabbit IgG H&L (HRP) (Abcam, ab205718) and Anti-mouse IgG, HRP-linked Antibody (Cell Signalling Technology, 7076).
ADMET and physicochemical properties prediction
ADMET-related in silico models have provided a preliminary screening of ADMET properties before compounds being further investigated. To reduce the risk of late-stage attrition, the ADME properties of compounds were predicted by Discovery Studio software. Furthermore, the toxicity and physicochemical properties of drugs were also predicted through a dedicated online website (ADMETlab 2.0 (scbdd.com)).
Results and discussion
Virtual screening
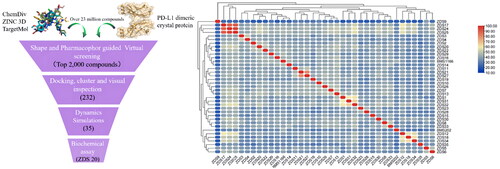
The workflow for our virtual screening is summarised in . To predict ligands those potentially disrupt PD-1/PD-L1 binding, a ligand-based 3D molecular similarity method was utilised. Molecules fitting the query shape are expected to be likely active as PD-L1 inhibitors. The shape query was performed by screening chemical databases derived from ZINC15, Topscience, and ChemDiv with BMS1166 as the query molecule. To additionally increase the chance of finding potential PD-L1 hits, the resultant molecules were subsequently subjected to pharmacophore-based virtual screening. A well-defined pharmacophore proposed by Lin’s group was utilised. The pharmacophore model consisted of five main features: one hydrogen bond donor, one positive ionisable point, and three hydrophobic points. A total of 9,115 molecules matched the pharmacophore model were returned during this process. To further provide elementary predictions of the binding modes and affinities between PD-1/PD-L1 and the ligands, the data set obtained was further subjected to docking-based virtual screening using Autodock Vina. The bioactive conformation of of BMS1166 in complex with PD-1/PD-L1 (PDB: 6R3K) was used as a template. 2,000 top-ranking ligands were retrieved as candidates and clustered by structural similarities using ChemMine Tools (http://chemmine.ucr.edu/) to reduce their structural redundancy. To narrow down the number of compounds, each pose with the lowest score was carefully checked manually by visual inspection to remove nonbinders. Finally, 232 compounds were taken forward for molecular dynamics simulations to monitor the ligand behaviour in PD-1/PD-L1 binding site. At last, 35 compounds with binding energy lower than −50 kcal/mol were retained and chosen as representative molecules for further study. Besides, molecular fingerprints based on RDKit were generated and the molecular similarity was calculated (). It can be observed that these candidate hits have significant structural differences when compared to the reference compound BMS202 and BMS1166. The final hits were either obtained from commercial resources or synthesised in our lab.
Figure 3. (A) Schematic representation of virtual screening protocol leading to identify potent PD-L1 inhibitors. (B) Heatmap presentation of topological similarities of the 35 hits to the reported PD-L1 inhibitors.
Chemical synthesis
Preparation of ZDS1
The synthetic route for ZDS1 is illustrated as below . Firstly, 4-fluorobenzaldehyde was cyclized with pyruvic acid and aniline in EtOH to afford the quinoline-4-carboxylic acid derivative 12. Secondly, protection of 5-(aminomethyl) indole with (Boc)2O followed by acylation with m-methylbenzoyl chloride afforded intermediate 10, which was further deprotected with CF3COOH in CH2Cl2 and condensed with 12 in the presence of HATU and DIPEA to yield the target compound ZDS1.
Preparation of ZDS2
To start with, 2-aminobenzohydrazide was cyclized with imidazole hydrochloride in DMF to afford important intermediate 17. Subsequently, p-aminophenol was reacted with 2,5-hexanedione with a catalytic amount of p-TsOH to afford intermediate 13. Further Vilsmeier reaction afforded the aldehyde 14, which was then substituted by methyl 3-bromomethylbenzoate and hydrolysed to the carboxylic acid 16. Final condensation of 16 with 17 in MeOH afforded the target compound ZDS2 .
Preparation of ZDS3
The starting material 3-bromomethylbenzoate was reacted with 3,5-dimethylpyrazole in the presence of t-BuOK to provide 18, which was subsequently hydrolysed to the corresponding acid 19 by NaOH aq at room temperature. On the other hand, 2-furancarboxylic acid was condensed with tert-butyl piperazine-1-carboxylate in the presence of CDI to give 20, further deprotection with CF3COOH in CH2Cl2 furnished intermediate 21. 4-hydroxypiperidine was protected with (Boc)2O to provide 22, which was then reacted with methanesulfonyl chloride to give 23. Nucleophilic substitution by m-hydroxybenzaldehyde and further reductive amination with 21 furnished intermediate 25, which was then deprotected to give 26. The target compound ZDS3 was finally obtained by condensation of 26 with 19 in the presence of HATU and DIPEA .
Preparation of ZDS4
The preparation of ZDS4 is outlined in Scheme 4. 4-fluorobenzonitrile was reacted with pyrazole in DMF under N2 atmosphere to provide 27, which was then converted to the corresponding carboxylic acid 28 in the presence of NaOH (aq) in EtOH. 5-bromoquinoline can be easily converted to quinoline-5-carbaldehyde in the presence of n-BuLi and DMF, which was then condensed with 3-(aminomethyl)phenol and reduced by sodium triacetoxyborohydride to afford 31. Further methylation and nucleophilic substitution with 23 furnished 33. Subsequent N-deprotection by CF3COOH and condensation with 28 in the presence of HATU and DIPEA provided the final product ZDS4.
Scheme 1. Reagents and conditions: (i) aniline, pyruvic acid, EtOH, 80 °C, 3.5 h; (ii) TEA, (Boc)2O, DCM, 0 °C to rt, 0.5 h; (iii) a). m-toluic acid, SOCl2, DCM, reflux, 2 h; b). acyl chloride, NaH, DCM, 0 °C to rt, 0.5 h; (iv) TFA, DCM, 0 °C to rt, 0.5 h; (v) 12, HATU, DIPEA, THF, rt, overnight.
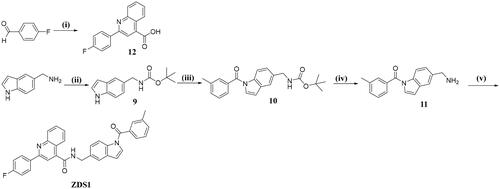
Scheme 2. Reagents and conditions: (i) DMF, imidazole hydrochloride, 150 °C, 13 h; (ii) p-TsOH, toluene, hexane-2,5-dione, reflux, 6 h; (iii) POCl3, DMF, 0 – 100 °C, 3 h; (iv) 3-bromomethylbenzoate, K2CO3, DMF, TEBA, rt, overnight; (v) NaOH, MeOH/H2O, 65 °C, 4 h; (vi) 17, TsOH, MeOH, rt, 3 h.
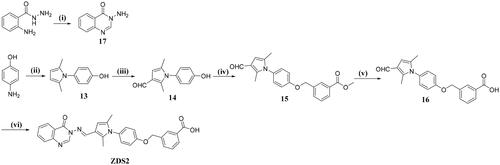
Scheme 3. Reagents and conditions: (i) 3,5-dimethylpyrazole, K2CO3, DMF, 50 °C, 1 h; (ii) NaOH, MeOH/H2O, 65 °C, 2 h; (iii) tert-butyl piperazine-1-carboxylate, CDI, DCM, TEA, overnight; (iv) TFA, DCM, 0 °C to rt, 3 h; (v) (Boc)2O, TEA, DCM, 0 °C to rt, 1 h; (vi) methanesulfonyl chloride, TEA, DCM, 0 °C to rt, 2.5 h; (vii) 3-hydroxybenzaldehyde, K2CO3, DMF, 90 °C, 48 h; (viii) 21, STAB, THF, 70 °C, 3 h; (ix) TFA, DCM, 0 °C to rt, 2 h; (x) 19, HATU, DIPEA, DCM, overnight.
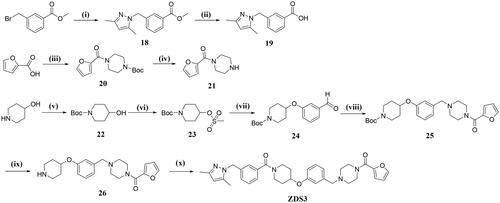
Scheme 4. Reagents and conditions: (vii) pyrazole, DMF, N2, 120 °C, 9 h; (ii) NaOH, 80 °C, EtOH/H2O, 6 h; (iii) BuLi, DMF, -78 °C, 1 h; (iv) 3-(aminomethyl)phenol, AcOH, MeOH, rt, 1 h; (v) NaBH4, THF, overnight; (vi) CH2O, HCOOH, 70 °C, 8 h; (vii) 23, K2CO3, toluene, 110 °C, overnight; (viii) TFA, DCM, 0 °C to rt, 2 h; (ix) 28, HATU, DIPEA, DCM, rt, 6 h.
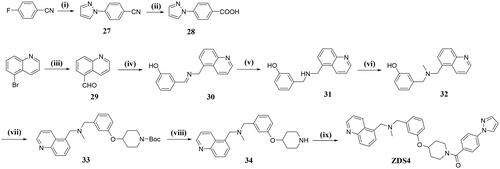
Scheme 5. Reagents and conditions: (i) 1-methylpiperazine, DIPEA, DMF, rt, 0.5 h; (ii) SnCl2.2H2O, EA, 80 °C, 8 h; (iii) Step1: 3-chlorobenzoic acid, SOCl2, DCM, reflux, 2 h; Step2: TEA, DCM, 0 °C to rt; (iv) NaOH, MeOH/THF/H2O, 50 °C, 4 h; (v) TEA, (Boc)2O, DCM, 0 °C to rt, 7 h; (vi) 1-fluoro-4-nitrobenzene, NaH, DMF, Ar2, 80 °C, 12 h; (vii) TFA, DCM, rt; (viii) NaH, iodoethane, DMF, 0 °C to rt; (ix) SnCl2.2H2O, EA, 80 °C, 6 h; (x) 38, HATU, DIPEA, THF, rt, 4 h.
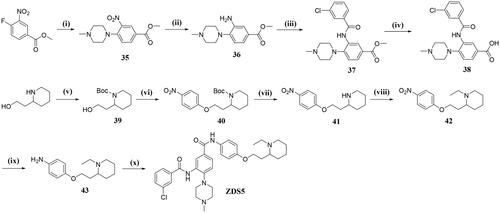
Preparation of ZDS5
The starting material 4-fluoro-3-nitrobenzoate was reacted with N-methyl piperazine in the presence of DIPEA in DMF to give intermediate 35, which was further reduced by SnCl2 in EtOH to provide 36. Further acylation with 3-chlorobenzoyl chloride afforded 37, which was then hydrolysed to the corresponding carboxylate acid 38. N-selective protection of 2-hydroxyethylpiperidine with (Boc)2O provided 39, which underwent O-substitution, N-deprotection, N-ethylation, and nitroreduction to give piperidine derivative 43. Final condensation of 38 with 43 in the presence of HATU and DIPEA provided the target compound ZDS5 .
Preparation of ZDS6
By starting with 4-methylbenzaldehyde, 45 can be easily synthesised via nitration, in situ reduction and cyclisation. On the other hand, 4-hydroxy-3-methoxybenzoate was substituted by 1-(bromomethyl)-4-chlorobenzene and then hydrolysed to the corresponding acid, which was finally condensed with 45 to give the target compound ZDS6 .
Scheme 6. Reagents and conditions: (i) HNO3/H2SO4 = 1:3, 0 °C to rt, 1 h; (ii) o-phenylenediamine, Na2S2O4, DMF, 150 °C, 10 h; (iii) 1-(bromomethyl)-4-chlorobenzene, toluene, K2CO3, 120 °C, 7 h; (iv) NaOH, MeOH/THF/H2O, 50 °C, 3 h; (v) Step1: SOCl2, DCM, reflux, 2 h; Step2: 45, NaH, DCM, 0 °C to rt.
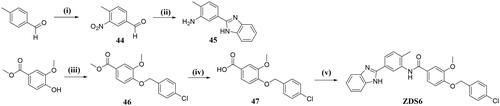
Preparation of ZDS7
Pentane-2,4-dione was reacted with methyl acrylate in the presence of MeONa to afford 48, which was further cyclized with 3-amino-5-phenylpyrazole in EtOH/AcOH under N2 atmosphere to furnish pyrazolo[1,5-a]pyrimidine derivative 49. Subsequent hydrolysis in NaOH (aq) provided the carboxylic acid 50. Chlorination of diethanolamine by SOCl2 afforded bis(2-chloroethyl)amine hydrochloride 51, which was further reacted with 2-chloroaniline to give 52. N-substitution of 52 and further deprotection furnished 54, which was finally condensed with 50 to obtain the final product ZDS7 ().
Scheme 7. Reagents and conditions:(i) methyl acrylate, MeONa, 100 °C, 3 h; (ii) 3-amino-5-phenylpyrazole, EtOH/AcOH, N2, 80 °C, overnight; (iii) NaOH, EtOH/H2O, reflux, 3 h; (iv) SOCl2, DCM, rt to reflux, 0.5 h; (v) 2-chloroaniline, 150 °C, 12 h; (vi) N-(3-bromopropyl) phenylenediamine, K2CO3, CH3CN, Ar2, reflux, 14 h; (vii) 80% N2H4-H2O, EtOH, reflux, 2 h; (viii) 50, HATU, DIPEA, DCM, rt, overnight.
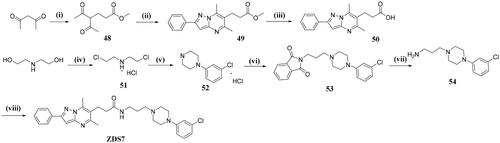
Preparation of ZDS8
Taking 1–(4-nitrophenyl)piperazine as starting material, intermediate 56 can be easily obtained by N-substitution and SnCl2 reduction. Suzuki coupling of (2,5-dichlorophenyl)boronic acid with methyl 5-bromofuran-2-carboxylate provided 57, which was converted to isothiocyanate 58 by chlorination and substitution by potassium thiocyanate. Final reaction of 58 with amine 56 furnished the target compound ZDS8 .
Scheme 8. Reagents and conditions: (i) 2-chlorobenzaldehyde, STAB, THF, 70 °C, 1 h; (ii) SnCl2.2H2O, EA, 70 °C; 17 h; (iii) methyl 5-bromofuran-2-carboxylate, Na2CO3, Pd(PPh3)4, DME/H2O, 100 °C, 2 d; (iv) Step1: SOCl2, DCM, DMF, rt; Step2: KSCN, acetone, 0 °C to rt, 0.5 h; (v) acetone, rt, 5 min;
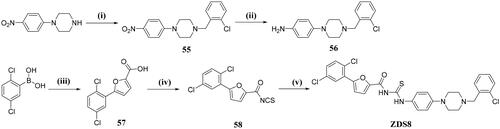
Preparation of ZDS9
Hydroquinone was substituted with (S)-2-(chloromethyl)oxirane under basic condition to provide 59, which was then reacted with 1–(3-chlorophenyl) piperazine hydrochloride in the presence of DIPEA in EtOH to obtain the symmetric compound ZDS9 .
Scheme 9. Reagents and conditions: (i) (S)-2-(chloromethyl)oxirane, NaOH, benzyl triethylammonium chloride, 90 °C, 12 h; (ii) 1–(3-chlorophenyl) piperazine hydrochloride, DIPEA, EtOH, 80 °C, 1 h;
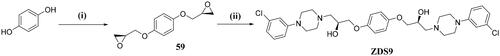
Preparation of ZDS10
N-Boc-piperidine-4-carboxylic acid was condensed with 2,2-dimethyl-1,3-dioxane-4,6-dione in the presence of DCC and DMAP to give 60, which was subsequently converted into 61 by heating in EtOH. Further reaction with triethyl orthoformate in the presence of Ac2O afforded 62, which was then cyclized with p-methylphenylhydrazine to give pyrazole derivative 63. The carboxylate acid 64 was obtained by hydrolysis of 63 by NaOH (aq) in quantitative yield. The intermediate 68 was prepared via a similar route with 54. The reductive amination of 2-methoxybenzaldehyde with tert-butyl piperazine-1-carboxylate provided 65, which underwent deprotection and substitution by N-(3-bromopropyl) phenylenediamine to give 67. Further hydrazinolysis of 67 provided 68, which was finally condensed with 64 in the presence of HATU and DIPEA to give the target compound ZDS10 ().
Scheme 10. Reagents and conditions:(i) 2,2-dimethyl-1,3-dioxane-4,6-dione, DMAP, DCC, 0 °C to rt, 13 h; (ii) EtOH, 80 °C, 20 h; (iii) triethyl orthoformate, Ac2O, 100 °C, 48 h; (iv) p-methylphenylhydrazine hydrochloride, EtOH, NaHCO3, 100 °C, 9 h; (v) NaOH, EtOH/H2O, 80 °C, 2 h; (vi) tert-butyl piperazine-1-carboxylate, STAB, THF, 70 °C, 5 h; (vii) TFA, DCM, 0° C to rt, 2 h; (viii) N-(3-bromopropyl) phenylenediamine, K2CO3, MeCN, 80 °C, 2 h; (ix) 80% N2H4·H2O, EtOH, reflux, 2 h; (x) 64, HATU, DIPEA, THF, rt, 8 h.
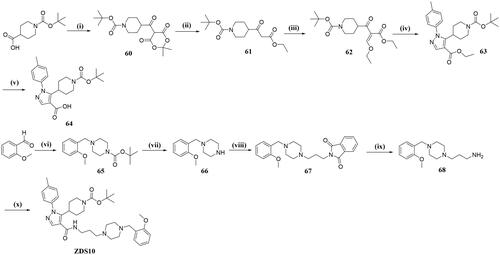
Preparation of ZDS11
Two separate intermediates 69 and 70 were synthesised via oxidation of 2-methylthiazole-4-carboxylate with SeO2 and cyclisation of 3-(trifluoromethyl)aniline with bis(2-chloroethyl)amine hydrochloride, respectively. By starting with 4-(benzyloxy)benzaldehyde, a two-step reductive amination with (4-fluorophenyl)methanamine and then 69 provided thiazole derivative 72, which was easily hydrolysed to carboxylic acid 73. Finally, ZDS11 can be easily obtained via the condensation of 73 with 70 in the presence of HATU and DIPEA .
Scheme 11. Reagents and conditions: (i) SeO2, AcOH, 120 °C, 24 h; (ii) bis(2-chloroethyl)amine hydrochloride, 150 °C, 12 h; (iii) (4-fluorophenyl)methanamin, STAB, THF, 70 °C, 3 h; (iv) 69, STAB, THF, 70 °C, 4.5 h; (v) NaOH, EtOH/H2O, 3 h; (vi) 70, HATU, DIPEA, DMF, rt, 4.5 h.
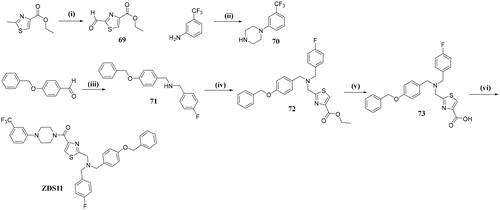
In vitro PD-/PD-L1 inhibition assay
Based on virtual screening results, a total of 35 compounds were selected for further experimental validation. Preliminary screening was performed at 30 μM concentration using a well-defined HTRF assay. The ability of these compounds to impede the PD-1/PD-L1 binding was evaluated. As shown in , 10 out of 35 compounds showed inhibition higher than 50%. Among them, seven compounds with higher inhibition rate were selected for further IC50 determination with BMS202 as positive control. As shown in , the inhibition of these predicted compounds was further confirmed, five out of seven compounds showed singe-digital micromolar IC50 values. Although they were not as potent as BMS202 (IC50 = 142.5 nM), ZDS20 carrying novel scaffold deserves to be further investigated.
Figure 4. Inhibitory activities of 35 compounds against PD-1/PD-L1 binding at 30 μM.
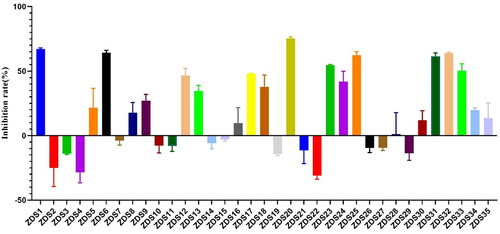
Figure 5. (A) PD-1/PD-L1 blockade effect of seven selected compounds. BMS202 was chosen as a positive control. (B) Structures of seven predicted compounds and BMS202.
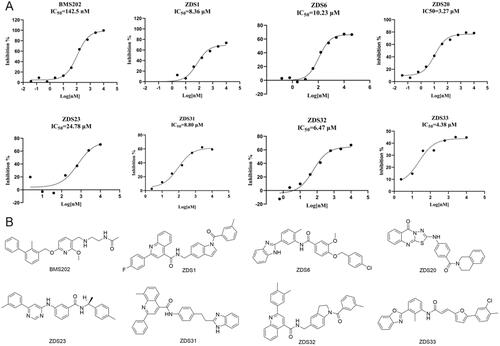
Western blot
To determine whether postinternalization degradation occurs following treatment with ZDS20, B16F10 cells expressing human PD-L1 were re-treated with ZDS20 at various concentrations for 36 h. After cell lysing and immunoblotting, the expression of PD-L1 was investigated. As shown in , the Western blots revealed that ZDS20 can promote PD-L1 degradation and that the level of expression of PD-L1 decreased in a dose-dependent manner.
Figure 6. Degradation of PD-L1 in B16F10 cells treated with ZDS20 at different concentrations (3, 15, 27 μM) for 36 h (left). Quantification of the results (right). ***p < 0.001 and ****p < 0.0001, vs control group.
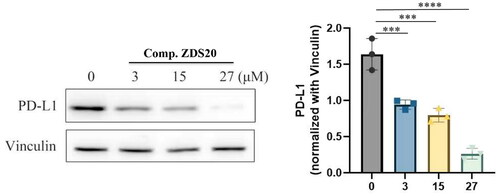
Binding modes analysis
To further validate the method and the postulated binding poses, we redocked BMS1166 to the PD-l/PD-L1 crystal structure using the AutoDock Vina program. The root-mean-square deviation (RMSD) between the docked pose and the X-ray structure was 0.14 Å, thus demonstrating the accuracy of our molecular scheme (see Figure S1). Due to its desirable activity in disrupting PD-1/PD-L1 binding, ZDS20 was then subjected to a binding mode analysis. As depicted in , the C2 symmetry of PD-L1 dimer was broken upon binding with the small molecule ZDS20. The tetrahydroisoquinoline-2-carbonyl)phenyl core of ZDS20 was embedded into the hydrophobic cavity formed by the two PD-L1 monomers, forming hydrophobic interactions with hotspot residue Ile54, Tyr56, Met115, and Ala121 on A chain and Met115, Ala121, and Tyr123 on B chain, the central benzene ring formed a π-π stacking interaction with the Tyr56 on A chain at an ideal angle and a perpendicular T shaped π stacking interaction with the Tyr123 on B chain. Additionally, the thiazolidine-quinazolin-5-one core unit of ZDS20, located in the near-solvent region, formed two important hydrogen bonds with the Tyr56 on A chain, Tyr123 on B chain as well as two cation-π interactions with the Lys124 on B chain. Notably, the backbone of ZDS20 undergoes a diversity of Pi-stacking, hydrophobic, and hydrogen bond interactions with the hotspot residue Tyr on both chains, which contribute to the binding affinity of compound to the dimeric PD-L1, resulting in its desirable inhibitory activity. Overall, ZDS20 breaks the framework restriction of biphenyl nucleus as a hydrophobic group and provides a novel structure to generate various types of interaction forces.
Figure 7. (A) Binding mode of ZDS20 with PD-L1 dimer (PDB 6R3K). (B) The overlay of ZDS20 with BMS202 in PD-L1 dimer. Images of A and B were generated by PyMol. (C) Detailed hydrophobic interaction of ZDS20 with the dimeric PD-L1 protein. Image was generated by LigPlus. (D) Binding map contacts of ZDS20 with PD-L1 protein. Image was generated by Discovery Studio.
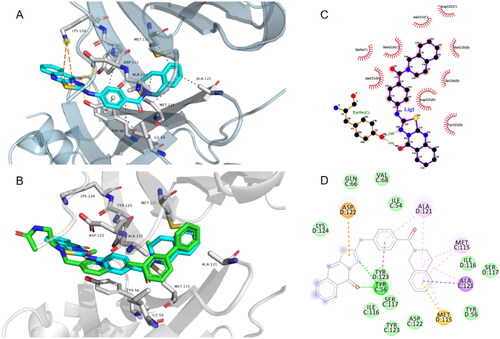
Stability of the PD-1/PD-L1 inhibitor complexes
RMSD is used to describe the difference between the simulated system and the initial conformation during the motion process, while RMSF can be used to describe the degree of atomic motion freedom. These two methods are commonly used to provide insights into the dynamic evolution of the simulation system. As shown in , BMS202 and the receptor protein reach equilibrium at 12 ns and fluctuated within the range of 0.5–2 Å. On the other hand, ZDS20 reached equilibrium at 10 ns and maintained a stable fluctuation within the range of 0.5–1.5 Å throughout the simulation (see also ), indicating that its binding to PD-L1 is more stable than that of BMS202. Meanwhile, it was observed that the complex of ZDS20 and PD-L1 kept a low RMSF value and the RMSF trends of BMS202 and ZDS20 were similar, thereby indicating that the complexes maintained similar stability and consistent state of motion during the simulation process. The state of motion of the complex is shown in . Three replicas RMSD of BMS202 and ZDS20 are shown in Figure S2 and S3, and the movement process of ZDS20 in 100 ns is shown in Figure S4.
Figure 8. Analysis of classical MD trajectories of inhibitor-PD-L1 dimer complexes. (A) RMSD plot of PD-L1-inhibitor complex. (B) RMSF plot of PD-L1-inhibitor complex. (C) Radius of gyration plot of PD-L1-inhibitor complex. (D) Solvent Accessible Surface Area (SASA) plot of PD-L1-inhibitor complex. Images were generated by Xmgrace software.
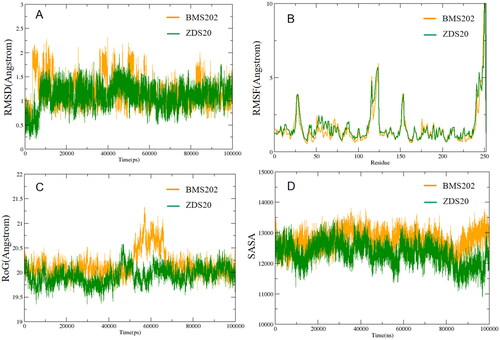
Figure 9. Conformation changes of ZDS20 with PD-L1 dimer during 100 ns simulation time.
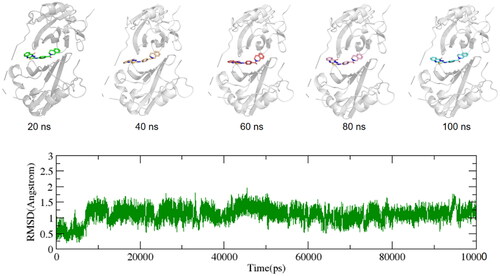
The radii of gyration (ROG) indicates how the atoms are distributed in a particular axis, and serves as a measure of the proximity between molecules.Citation19 As shown in , the radius of gyration of the complex in the ZDS20-PD-L1 system is lower than that of BMS202, indicating that ZDS20 makes the complex system compact and stable. Additionally, the solvent accessible surface area (SASA) of a protein is widely recognised as a critical determinant in protein folding and stability investigations.Citation20 It works as a significant role in the structure-function interplay of proteins. Examining the compactness treatment of backbone atoms can be assessed by the SASA parameter. As shown in the , the SASA value of the ZDS20 complex system is lower than that of the BMS202 system, indicating less compactness of the latter.
Binding free energy Calculation
To further probe the binding affinity between ZDS20 and PD-L1, the MM-GBSA method was utilised to calculate the binding free energy (ΔGbind) based on the equilibrium stage of the complex, the positive control BMS202 was co-investigated. The binding free energies of the three times molecular dynamics simulations were shown in . From the perspective of binding free energy, ZDS20 displayed equivalent binding free energies to BMS202, with values of −52.04 and −50.62 kcal/mol, respectively. Moreover, the ΔEvdw, ΔEele, ΔGgb and ΔGsurf of the two compounds were almost identical. In terms of the energy contribution, the major favourable contributor to ligand binding was the van der Waals term, and the values of the ΔEvdw for ZDS20 (-59.50 kcal/mol) were equal to that of BMS202 (-60.58 kcal/mol), suggesting that both ZDS20 and BMS202 fitted well into the hydrophobic cavity created by the PD-L1 dimer. Besides, the entropy values of BMS202 and ZDS20 were also evaluated, with an average entropy value of −23.57 and −19.02 Kcal/mol for these two compounds, respectively. The ΔETranslational and ΔERotational values of the two compounds are almost equivalent, and the difference in entropy values mainly comes from the ΔEVibrational term. BMS202 shows more negative TΔS value (see supporting information Table S2), the final calculated binding energies of BMS202 and ZDS20 are −27.05 and −33.02 Kcal/mol, respectively.
Table 1. Predicted binding free energy contributions of compound BMS202 and ZDS20 to the PD-L1 by MM/PBSA method.
To further investigate the contribution of hotspot residues in PD-L1-inhibitor complex, the energetic decomposition of amino acid residues of the PD-L1 complexes was performed. As shown in , residues with high absolute values of energy contributions were selected, this include Ile54, Tyr56, Val76, Met115, Ala121, and TYR123 for APD-L1, as well as Ile54, Met115, Ile116, Ala121, and Tyr123 for BPD-L1. Obviously, compound ZDS20 and BMS202 maintained consistent energy contribution values at four hotspot residues, namely Ile54, Tyr56, Val76, and Met115 for APD-L1. However, there were also significant energy differences observed at six residues, including Ala121 and Tyr123 for APD-L1, and Ile54, Met115, Ile116, Ala121, and Tyr123 for BPD-L1. This phenomenon illustrated that novel framework of tetrahydroisoquinoline and thiazolidinedione-quinazoline-5-ketone in ZDS20 played a different role in energy contribution to the PD-L1 complex than that of BMS202, which may explain the difference in the inhibitory activities of the two inhibitors.
Figure 10. Total energy contribution for each residue of compound ZDS20 and BMS202.
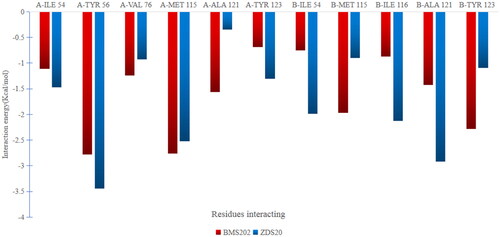
ADMET and physicochemical properties prediction
Reasonable ADMET properties are important for compound efficacy and safety as therapeutics. Therefore, predicted ADMET properties have been of great interest to medicinal chemist in recent years.Citation21 In this study, ZDS20 with excellent binding affinity and desirable in vitro inhibitory activity was selected for ADMET and physiochemical properties prediction. As shown in , all the physicochemical property parameters of ZDS20 were acceptable and completely satisfied the Lipinski rule. Compound ZDS20 also exhibited high plasma protein binding, moderate BBB penetration, high permeability in Caco-2 cells, low clearance and desirable oral availability. For drug-drug interaction, compound ZDS20 showed potency to inhibit CYP3A4. Furthermore, the predicted toxicity profile revealed that ZDS20 showed no risk in hERG potassium channel inhibition, mutagenic response, respiratory sensitisation, and irritation for eye (), irrespective of risk in carcinogencity and DILI (drug induced liver injury). Overall, the results suggest that compound ZDS20 covers acceptable drug-like properties.
Figure 11. (A) Plot of t-PSA versus AlogP for three compounds showing the 95% and 99% confidence limit ellipses corresponding to the BBB and intestinal absorption models. (B) Predicted physicochemical properties of ZDS20.33.
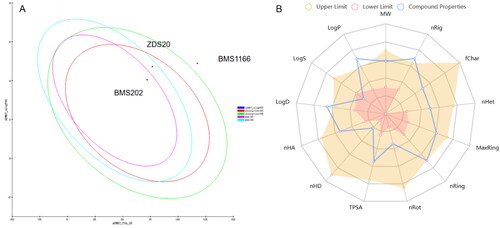
Table 2. Predicted ADMET properties of ZDS20 by ADMET 2.0.
Conclusion
In this study, by performing shape, pharmacophore and docking-based virtual screening strategies, a diversity range scaffolds of PD-L1 inhibitors were identified from ChemDiv, ZINC and Topscience databases. Subsequently, molecular dynamics simulations were performed to screen compounds and 35 hits were finally discovered. In vitro HTRF assay confirmed that 7 out of 35 hits were capable of disrupting PD-1/PD-L1 recognition. Among them, ZDS20 exhibited an IC50 value of 3.23 μM, was the most potent compounds that we obtained. The docking results showed that ZDS20 was highly stacked with BMS202 and formed three hydrogen bonds with Tyr56 of the A-chain, Tyr123 and Asp122 of the B-chain. Most importantly, ZDS20 still maintained a π-π stacking interaction with Tyr56 of the A-chain. Molecular dynamics simulations indicated that ZDS20 exhibited a consistent change in motion trend with BMS202. Moreover, the results of residue binding free energy decomposition may explain the difference in activity between ZDS20 and BMS202. Further ADMET evaluation also indicated that ZDS20 possessed desirable physicochemical properties and safety profiles. Overall, ZDS20 can be regarded as a potentially innovative lead compound for developing novel small molecule inhibitors disrupting PD-1/PD-L1 binding, which are ongoing in our laboratory.
Author contributions
Tingting Wu and Hu Cheng synthesised the compounds, and wrote the manuscript. Lijie Sima and Weiwei Ouyang conducted in vitro activity evaluation and data analysis. Zhongyuan Wang conducted in the ADMET calculations. Jianta Wang assisted in the synthesis of compounds. Dongsheng Zhao and Yunlei Hou conducted the shape, pharmacophore screening, molecular docking, and dynamics simulations. Chujiao Hu guided the design and implementation of the project. Weike Liao designed and guided the entire project and edited the manuscript. All authors approved the final version of the manuscript.
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Additional information
Funding
References
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):1–23.
- He X, Xu CQ. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30(8):660–669.
- Dömling A, Holak TA. Programmed death-1: therapeutic success after more than 100 years of cancer immunotherapy. Angew Chem Int Ed Engl. 2014;53(9):2286–2288.
- Mould DR, Meibohm B. Drug development of therapeutic monoclonal antibodies. BioDrugs. 2016;30(4):275–293.
- Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, Shabafrouz K, Ribi C, Cairoli A, Guex-Crosier Y, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563–580.
- Guzik K, Zak KM, Grudnik P, Magiera K, Musielak B, Törner R, Skalniak L, Dömling A, Dubin G, Holak TA, et al. Small-molecule inhibitors of the programmed cell death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) interaction via transiently induced protein states and dimerization of PD-L1. J Med Chem. 2017;60(13):5857–5867.
- Guzik K, Tomala M, Muszak D, et al. Development of the Inhibitors that Target the PD-1/PD-L1 Interaction-A Brief look at progress on small molecules. Peptides and macrocycles. Molecules. 2019;24:2071–2101.
- Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, Holak TA. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. 2015;23(12):2341–2348.
- Zhong Y, Li X, Yao H, Lin K. The characteristics of PD-L1 inhibitors, from peptides to small molecules. Molecules. 2019;24(10):1940–1957.
- Spitzer R, Jain AN. Surflex-Dock: Docking benchmarks and real-world application. J Comput Aided Mol Des. 2012;26(6):687–699.
- Wang J, Piotr C, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules?. J Comput Chem. 2000;21(12):1049–1074.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791.
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3(1):33–47.
- Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr., Onufriev A, Simmerling C, Wang B, Woods RJ, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688.
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174.
- Nayar D, Agarwal M, Chakravarty C. Comparison of tetrahedral order, liquid state anomalies, and hydration behavior of mTIP3P and TIP4P water models. J Chem Theory Comput. 2011;7(10):3354–3367.
- Wang E, Sun H, Wang J, Wang Z, Liu H, Zhang JZH, Hou T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem Rev. 2019;119(16):9478–9508.
- OuYang Y, Gao J, Zhao L, Lu J, Zhong H, Tang H, Jin S, Yue L, Li Y, Guo W, et al. Design, Synthesis, and Evaluation of o-(Biphenyl-3-ylmethoxy)nitrophenyl Derivatives as PD-1/PD-L1 Inhibitors with Potent Anticancer Efficacy In Vivo. J Med Chem. 2021;64(11):7646–7666.
- Zhao S-S, Wang Y-J, Tang L, Guo B, Wang L, Zhang J-Q, Yang S-G. Identifying novel selective PPO inhibitors through structure-based virtual screening and bio-evaluation. RSC Adv. 2023;13(16):10873–10883.
- Ali S, Hassan M, Islam A, Ahmad F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. CPPS. 2014;15(5):456–476.
- Dong J, Wang N-N, Yao Z-J, Zhang L, Cheng Y, Ouyang D, Lu A-P, Cao D-S. ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform. 2018;10(1):29.