
KEYWORDS:
1. Introduction
Around 20,000 protein-coding genes have been predicted from the analysis of the human genome. Many thousands of alternatively spliced isoforms have been predicted from transcriptomic analyses, but only a small fraction of those has been confirmed by mass spectrometry, questioning their existence at protein level, and their functional relevance. During or after translation, protein products are chemically modified, which results in a large proteoform space that varies with time, location, and physiologic or disease conditions. In addition, inter-individual genetic variations can also affect the relative levels of generated proteoforms.
Recent advances in proteomics technologies allow proteins and their modifications to be detected and quantified with high precision. However, many predicted proteins still escape detection, either because they were improperly predicted, their expression is restricted in time and/or space, or their biophysical and chemical properties are not compatible with standard proteomics experiments. Furthermore, the biological role of many of the proteins that have successfully been validated is still unknown.
The HUPO Human Proteome Project (HPP) was launched in 2010 to make proteomics an integral part of multi-omics research in the life sciences and medicine [Citation1]. Its first goal is to ensure that, for every predicted protein-coding gene, at least one protein product has been credibly identified in a biological sample and is functionally characterized. HPP also aims at cataloging genomic variants, splice isoforms and post-translational modifications in order to shed light on the structural and functional diversity of the human proteome.
neXtProt (www.nextprot.org) is a knowledge base on human proteins that integrates high quality data at the genomic, transcriptomic and proteomic levels and provides advanced query tools based on semantic technologies [Citation2]. neXtProt provides for each entry extensive information about the function, structure, localization, post-translational modifications (PTMs), variants, and many other features for the different splice isoforms. Each neXtProt entry has a ‘protein existence’ (PE) score reflecting its experimental validation status. For PE1 entries (‘Evidence at protein level’), at least one protein product has been credibly identified by mass spectrometry or by a direct biochemical assay. PE2-4 entries lack confirmation at the protein level but have evidence at transcript level (PE2), orthologs (PE3), or credible genomic support (PE4). PE5 entries correspond to dubious proteins that are most probably pseudogenes or non-coding RNAs.
Since 2013, neXtProt is the reference knowledgebase for the HPP [Citation3]. The 2020-01-17 release contains 20,350 entries, of which 1,899 (9%) are ‘missing proteins’ (PE2-4) awaiting experimental validation. Over the last 5 years this proportion has steadily decreased thanks to the global efforts of the community to profile human samples with mass spectrometers having increasing precision and sensitivity, and we can reasonably expect that it will continue to do so [Citation4]. Among the validated proteins (PE1), 1,254 are devoid of functional annotation, either predicted or experimental. These proteins, called uPE1, are now prioritized for characterization in the context of the recently launched HPP neXt-CP50 project [Citation5]. Nearly all neXtProt entries have at least one single amino acid variant (SAAV) associated and 78% have at least one PTM associated. Many more PTMs certainly are present in human samples, but their detection remains a challenge [Citation6]. Understanding the impact of SAAVs and PTMs in health and disease is a challenge that can only be solved by integrating data obtained by different omics approaches, from genomics to structural proteomics.
This article summarizes how neXtProt is helping the HPP to identify and fill gaps in our knowledge of the human proteome.
2. What does neXtProt do for the proteomics community?
2.1. neXtProt provides up-to-date lists of ‘missing’ and ‘uncharacterized’ human proteins
According to the HPP guidelines 3.0 [Citation7], a protein is considered validated if at least two non-overlapping peptides of nine amino acids or more have been identified that uniquely map to its sequence. neXtProt integrated more than 2.5 million peptides identified by mass spectrometry from PeptideAtlas [Citation8] and MassIVE [Citation9]. These peptides were mapped to entries using the peptide uniqueness checker [Citation10] and PE2-4 entries were promoted to PE1 in accordance with HPP guidelines. 16ʹ924 entries can now be considered as validated by mass spectrometry according to the HPP guidelines 3.0.
In addition to proteomics data, neXtProt integrates functional annotations from a variety of sources. Some functional annotations are based on the experimental characterization of the protein itself, while others are derived from characterization studies of similar proteins in human or model organisms. The neXtProt team is constantly scrutinizing the literature to find data to update these annotations [Citation11]. Despite this effort, hundreds of proteins still lack any experimental or predicted functional annotation.
The list of missing (PE2-4) proteins and that of proteins lacking functional annotation from the 2020-01-17 release will serve as baselines for studies performed by HUPO HPP teams in 2020. These lists can be obtained using the SPARQL queries NXQ_00204 (https://www.nextprot.org/proteins/search?mode=advanced&queryId=NXQ_00204) and NXQ_00022 (https://www.nextprot.org/proteins/search?mode=advanced&queryId=NXQ_00022), respectively.
2.2. neXtProt provides extensive information about the proteoforms found in human healthy and disease samples
neXtProt currently reports additional splice isoforms for half the human proteome (10ʹ535 entries). For 916 of these entries, it was shown that the different splice isoforms have a different subcellular location or function. The list of these 916 entries can be obtained using the SPARQL query NXQ_00218 (https://www.nextprot.org/proteins/search?mode=advanced&queryId=NXQ_00218).
neXtProt currently reports 190,888 PTM sites. The complete list of PTM sites, with mappings to isoforms and data provenance, can be retrieved in csv, json or xml format by running the following SPARQL query in the neXtProt SNORQL interface at https://snorql.nextprot.org/:
select distinct ?iso ?pos str(?txt) ?source where {
?entry :isoform ?iso.
?iso :ptm ?ptm.
?ptm :start ?pos .
?ptm :evidence/:assignedBy ?source .
?ptm rdfs:comment ?txt .
}
Approximately half of the sites are phosphorylation sites from PeptideAtlas; the others are imported from UniProt or GlyConnect or were manually annotated from publications. Among the 27,764 sites that were manually annotated by the neXtProt team, 16,759 are ubiquitin-like conjugation sites, 3,647 are glycosylation sites, 3,354 are phosphorylation sites, 2,468 are acetylation sites and 1,174 are methylation sites.
neXtProt reports more than nine million single amino acid variations from UniProtKB, dbSNP, COSMIC, the genome Aggregation Database (gnomAD), and manually annotated from the literature. Of these, 19% are variations found in cancer samples, and 43% are polymorphisms with frequency data.
As for past releases, PTM and variant information included in the 2020-01-17 release are made available in various formats, including the HUPO PSI PEFF [Citation12] that allows protein identification software to consider PTMs and variants in the search space when analyzing spectra. The peptide uniqueness checker already takes variants into account when mapping peptides to entries [Citation9].
2.3. neXtProt helps the community plan targeted mass spectrometry experiments to obtain evidence for ‘missing proteins’
neXtProt provides tissue expression profiles for each entry at the protein and RNA levels, with updated data from the Human Protein Atlas (HPA) [Citation13]. This information helps narrow down the samples to be analyzed when looking for missing proteins. Furthermore, the protein digestion tool (www.nextprot.org/tools/protein-digestion) can be used to define the best proteolysis conditions to find unique peptides complying with HPP guidelines.
2.4. neXtProt helps the community find the function of uncharacterized proteins
Based on deep data mining, the neXtProt team recently proposed functional hypotheses for 34 uncharacterized proteins [Citation11,Citation14], which are awaiting experimental confirmation. We are extending this strategy with a more extensive phylogenetic profiling approach.
neXtProt already links to the I-TASSER/COFACTOR tool that predicts GO terms using structural information [Citation15]. We would be happy to link to other functional prediction tools provided by the community.
2.5. neXtProt helps answer complex questions on human proteins by integrating different types of data
neXtProt uses an RDF data model and community-approved standards for all its annotations, ensuring good interoperability with other life science resources. It offers a SPARQL endpoint and associated search tools allowing complex queries to be performed, not only on neXtProt data, but also on data from semantically compatible resources. Nearly 200 pre-built queries are provided on https://snorql.nextprot.org/. Many of them were written in response to specific user requests. Among those queries, 17 make use of proteomics data, 24 of PTM data, 28 of subcellular location data, 19 of variant data, 25 of functional annotations, and 13 of expression data. For example, the query NXQ_00099 retrieves 3,498 secreted proteins that have at least one PTM in a position of a variant. The query NXQ_00251 retrieves 3,044 entries that have at least one proteotypic peptide of nine amino acids or more that does not map on the canonical isoform but rather on an alternatively spliced isoform. We encourage HPP users to use these queries, customize them according to their own questions and contact us should they need any help.
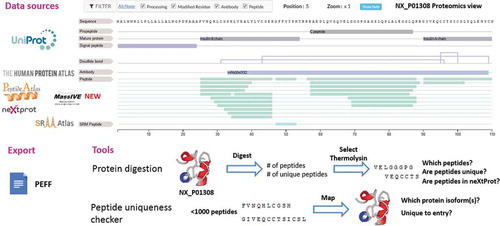
Figure 1. neXtProt curates and integrates data from various sources, displays it on the proteomics view, exports it in different formats including the HUPO-PSI PEFF format, and offers tools to prepare and interpret proteomics experiments.
3. Conclusion and perspectives
As summarized in the , neXtProt curates and integrates data from various sources including the key HPP resources (HPA, PeptideAtlas, and MassIVE), displays it in its proteomics view, exports it in different formats including the HUPO-PSI PEFF format, and offers tools to prepare and interpret proteomics experiments. Proteomics technologies are rapidly evolving, and quantitative proteomics methods are now mature enough for us to integrate the data. In the near future, we plan to add quantitative proteomics data to our expression view, so that it can be directly compared to the transcriptomics data and to the antibody-based data.
Beyond the integration and display of proteomic data, neXtProt provides advanced search functionalities that put this data into a broader context and facilitate the generation of functional hypotheses. In order to facilitate the adoption of these functionalities by the scientific community, we plan to create public notebooks combining documentation, visualization, data analytics, and code that can be shared, modified and published.
As the HUPO HPP project turns ten and faces new challenges, we celebrate being part of this initiative and welcome suggestions on how to improve our services and continue to fit the needs of this vibrant community.
4. Expert opinion
The quantity and quality of the proteomics data generated across the world is providing a better picture of the diversity of the proteins found in human samples. The HUPO Human Proteome Project launched in 2010 promotes the standardization, access and reuse of this data, in order to make proteomics an integral part of multi-omics research. By acting as the reference knowledgebase for this project, neXtProt helps the community to identify and fill gaps in our knowledge of the human proteome.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Legrain P, Aebersold R, Archakov A, et al. The human proteome project: current state and future direction. Mol Cell Proteomics. 2011. DOI:10.1074/mcp.O111.009993.
- Zahn-Zabal M, Michel P-A, Gateau A, et al. The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res. 2019;48(D1):D328–D334..
- Lane L, Bairoch A, Beavis RC, et al. Metrics for the human proteome project 2013–2014 and strategies for finding missing proteins. J Proteome Res. 2014;13:15–20.
- Omenn GS, Lane L, Overall CM, et al. Progress on identifying and characterizing the human proteome: 2019 metrics from the HUPO human proteome project. J Proteome Res. 2019;18:4098–4107.
- Paik Y-K, Lane L, Kawamura T, et al. Launching the C-HPP pilot project for functional characterization of identified proteins with no known function. J Proteome Res. 2018;17:4042–4050. .
- Doll S, Burlingame AL. Mass spectrometry-based detection and assignment of protein posttranslational modifications. ACS Chem Biol. 2015;10:63–71.
- Deutsch EW, Lane L, Overall CM, et al. Human proteome project mass spectrometry data interpretation guidelines 3.0. J Proteome Res. 2019;18:4108–4116.
- Deutsch EW, Sun Z, Campbell D, et al. State of the human proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through the AtlasProphet. J Proteome Res. 2015;14:3461–3473.
- Wang M, Wang J, Carver J, et al. Assembling the community-scale discoverable human proteome. Cell Syst. 2018;7:412–421.e5.
- Schaeffer M, Gateau A, Teixeira D, et al. The neXtProt peptide uniqueness checker: a tool for the proteomics community. Bioinformatics. 2017;33:3471–3472.
- Duek P, Gateau A, Bairoch A, et al. Exploring the uncharacterized human proteome using neXtProt. J Proteome Res. 2018;17:4211–4226.
- Binz P-A, Shofstahl J, Vizcaíno JA, et al. Proteomics standards initiative extended FASTA format. J Proteome Res. 2019;18:2686–2692.
- Thul PJ, Lindskog C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018;27:233–244.
- Duek P, Lane L. Worming into the uncharacterized human proteome. J Proteome Res. 2019;18:4143–4153.
- Zhang C, Lane L, Omenn GS, et al. Blinded testing of function annotation for uPE1 proteins by I-TASSER/COFACTOR pipeline using the 2018–2019 additions to neXtProt and the CAFA3 challenge. J Proteome Res. 2019;18:4154–4166.