
Abstract
Diabetes is a risk factor for breast cancer development and is associated with poor prognosis for breast cancer patients. However, the molecular and biochemical mechanisms underlying the association between diabetes and breast cancer have not been fully elucidated. Here, we investigated estradiol response in MCF-7 breast cancer cells with or without chronic exposure to insulin. We found that insulin priming is necessary and specific for estradiol-induced cancer cell growth, and induces anaplerotic shunting of glucose into macromolecule biosynthesis in the estradiol treated cells. Treatment with ERK or Akt specific inhibitors, U0126 or LY294002, respectively, suppressed estradiol-induced growth. Interestingly, molecular analysis revealed that estradiol treatment markedly increases expression of cyclin A and B, and decreases p21 and p27 in the insulin-primed cells. In addition, estradiol treatment activated metabolic genes in pentose phosphate (PPP) and serine biosynthesis pathways in the insulin-primed cells while insulin priming decreased metabolic gene expression associated with glucose catabolism in the breast cancer cells. Finally, we found that anti-diabetic drug metformin and AMPK ligand AICAR, but not thiazolidinediones (TZDs), specifically suppress the estradiol-induced cellular growth in the insulin-primed cells. These findings suggest that estrogen receptor (ER) activation under chronic hyperinsulinemic condition increases breast cancer growth through the modulation of cell cycle and apoptotic factors and nutrient metabolism, and further provide a mechanistic evidence for the clinical benefit of metformin use for ER-positive breast cancer patients with diabetes.
Abbreviations
| AR | = | androgen receptor |
| CKI | = | cyclin dependent-kinase inhibitor |
| DHT | = | dihydrotestosterone |
| ER | = | estrogen receptor |
| G6PD | = | glucose-6-phosphodehydrogenase |
| GLUT1 | = | glucose transporter 1 |
| HER2 | = | human epidermal growth factor receptor 2 |
| IGF-1 | = | insulin-like growth factor 1 |
| LDHA | = | lactate dehydrogenase A |
| PDK1 | = | pyruvate dehydrogenase kinase 1 |
| PFK | = | phosphofructokinase |
| PFKFB3 | = | 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphate 3 |
| PGD | = | 6-phosphogluconate dehydrogenase |
| PHGDH | = | phosphoglycerate dehydrogenase |
| PKM | = | pyruvate kinase M |
| PPAR γ | = | peroxisome proliferator-activated receptor gamma |
| PPP | = | pentose phosphate pathway |
| PR | = | progesterone receptor |
| PSPH | = | phosphoserine phosphatase |
| RPE | = | ribulose-5-phosphate-3-epimerase |
| RPIA | = | ribulose-5-phosphate isomerase A |
| SHMT | = | serine hydroxymethyltransferase |
| TALDO1 | = | transaldolase 1 |
| TKT | = | transketolase |
| TZDs | = | thiazolidinediones |
Introduction
Breast cancer has become the leading cause of female cancer-related deaths with the most frequent diagnosis worldwide, accounting for 1.38 million newly diagnosed and 0.45 million of cancer deaths in 2008.Citation1 The incidence of this deadly disease is attributed to various biological conditions including reproductive and hormonal factors, postmenopausal hormonal replacement therapy, and diabetes.Citation2,3 Currently, breast cancer prognosis and treatment is guided based on clinico-pathological features such as tumor size, histological grade, lymph node involvement and tumor expression of ER, progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2).Citation4 Tumor expression of ER is the single most studied factor for therapeutic diagnosis of breast cancer, which has led to a better understanding of the role of ER in breast cancer progression and the emergence of therapies that specifically target ER.
Insulin resistance arising from chronic hyperinsulinemia results in Type II diabetes, a metabolic disorder known as a poor prognostic marker for breast cancer and for being associated with an approximately 40% increase in mortality in the first 5 y with breast cancer diagnosis.Citation5 Shanik et al., described that basal insulin levels in patients with insulin resistance are commonly 2-fold higher compared to the non-obese or non-diabetic patients, and the basal hyperinsulinemia is often associated with type 2 diabetes.Citation6 The metabolic disease is also prominently associated with ER-positive tumors in post-menopausal women.Citation7,8 In a recent retrospective study, He et al. demonstrated that insulin use decreased overall breast cancer survival of diabetic patients.Citation9 However, the use of metformin and TZDs to treat type II diabetes showed significant improvement in breast cancer survival in the same cohort study.Citation9 This confirms a clinico-pathological association between diabetes and breast cancer, which includes hyperinsulinemia, hyperglycemia, and chronic inflammation.Citation10 Thus, unearthing the molecular links of breast cancer to diabetes could prove vital for clinical management of breast cancer patients with diabetes.
Several studies have revealed bi-directional signaling pathways between ER and insulin-like growth factor 1 (IGF-1) or insulin of which functional deregulation consequently results in diabetes.Citation11 For example, estrogen positively regulates the expression of members of the IGF family, while insulin/IGF-1 signaling modulates ER activity through post-translational modification of the receptor.Citation12-15 However, these studies have shown acute effect of insulin on estrogen receptor signaling or vice versa. Thus, the study of ER signaling under the condition of chronic hyperinsulinemia is needed to provide a mechanistic insight of diabetic influence on breast cancer pathogenesis.
In this study, we systemically investigated the effect of chronic exposure of insulin or insulin priming on the ER-associated signal transduction and metabolic pathways in ER-positive breast cancer. This allowed us to identify that ER-positive breast cancer might newly acquire metabolic adaptability to anaplerotic glucose utilization for macromolecule biosynthesis that is required for cell proliferation in response to estradiol. Further, we show the differentiated beneficial effects of metformin from TZDs in inhibiting estradiol-induced growth in the insulin-primed breast cancer cells. Overall, this study provides mechanistic as well as metabolic links between diabetes and ER-positive breast cancer, and therapeutic insight into the differentiated use of anti-diabetic drugs for cure of ER-positive breast cancer patients with diabetes.
Results
Insulin priming effect on estradiol-induced breast cancer growth
Several studies have previously shown that insulin or IGF-1 signaling cross-talks with ER in breast cancer.Citation13,16,17 However, little is known about the impact of chronic exposure of insulin on ER signaling, which is the more relevant condition for breast cancer patients with diabetes. Therefore, we here established in vitro hyperinsulinemic breast cancer model in which MCF-7 cells were maintained in supraphysiological concentration of insulin (10 nM) for at least 7 d prior to subsequent experiments. Note that insulin conditioning for 7 d was enough for the insulin priming effect ( bottom). We found that the insulin-primed cells, but not the corresponding unprimed cells, exhibited significant growth response to estradiol treatment that activated the cognate receptor ERα ( and ). Interestingly, the combined treatment of estradiol and insulin showed no growth response in the parental unprimed cells ( top). This suggests that insulin priming process is required for estradiol-induced growth. Consistently, IGF-1 priming showed a similar pattern and extent of estradiol-induced growth response as that of insulin priming ( top). Note that this data was confirmed with multiple, independent batches of insulin priming experiments in MCF-7 cells ( middle). Treatment with 100 pmol of estradiol was enough to achieve maximum cell growth response (), which was striking to us since previous studies used an estradiol concentration of 10 nM to 100 nM to attain a similar magnitude of estradiol-induced growth response as in this study.Citation18,19 This suggests that insulin priming or hyperinsulinemic condition indeed sensitizes breast cancer to physiological estradiol concentration. We further confirmed that the estradiol-induced growth was mediated through ER by treating pure ERα antagonist, ICI182,780 in combination with estradiol ( top). Note that both cell lines showed protein expression of Insulin receptor and ERα while insulin receptor, but not ERα, showed mild increase of expression in the primed cells possibly due to a compensatory increase of the insulin receptor resulting from the chronic insulin exposure in the insulin-primed cells (). Further analysis revealed that both proteins are functional in both cell lines ( and ). The mRNA expression of progesterone receptor (PR), a direct target of ERα, was similarly regulated in both cell lines when treated with estradiol or ICI compound (). These data suggest that additional factor(s), other than ERα activity, may be involved in insulin-primed, estradiol-induced growth response.
Figure 1. Insulin priming effect on estradiol-induced growth of breast cancer MCF-7 cells. (A) Growth response of the insulin-primed and unprimed MCF-7 cells. Relative growth response was assessed using MTT assay as described in materials and methods. MCF-7 cells were treated with 1 nM of estradiol, 10 nM of insulin, 30 nM of IGF-1, 1 μM ICI 182,780 alone or co-treated with estradiol (top). Note that MCF-7-10-42 and MCF-7-1.7-48 represent independent batches of insulin-primed MCF-7 cells (middle). Both primed and unprimed MCF-7 cells were assayed for estradiol-induced growth response in a dose-dependent manner (2nd bottom) Both primed cells (3-day vs. 7-day priming) were assayed for estradiol-induced growth response in a dose-dependent manner (bottom). (B) The expression of ERα and insulin receptor (INSR) were identified using immunoblot assay after treatment of cells with 1 nM of estradiol and/or 10 nM of insulin for 48 h. (C) mRNA expression of PR in response to estradiol in both cell lines. Total RNAs were isolated from cells treated with 1 nM of estradiol alone or in combination with 1 μM of ICI182,780 for 24 h. (D) Functional evaluation of AR for growth response of the insulin primed and unprimed MCF-7 cells; protein expression of AR (Top left) and mRNA expression of an AR target FKBP51 upon DHT treatment (Top right). Growth response for DHT treatment was assessed using MTT assay after treatment with 1 nM DHT alone or in combination with 10 nM insulin or 1 μM bicalutamide (bottom). White bars-unprimed (□); black bars-primed cells (▪). Values are mean ± SEM. *P < 0.05; #P < 0.001.
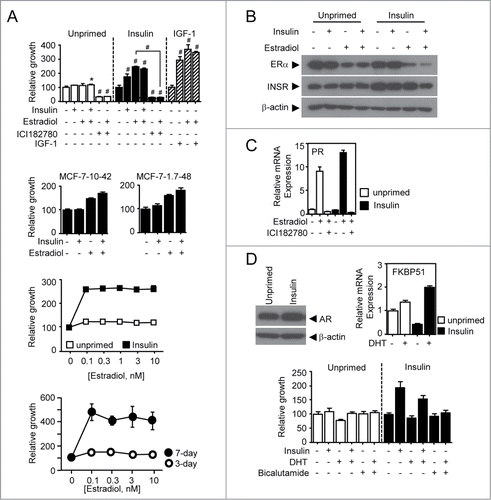
Figure 2. Estradiol-induced signaling depends on PI3K/Akt and ERK signaling. (A) Relative growth response of insulin-primed and unprimed MCF-7 cells. Cells were treated with 10 μM of MEK inhibitor U0126 or 20 μM of PI3K inhibitor LY294,002 in the presence or absence of 1 nM estradiol for 7 d and followed by MTT assay. (B) Immunoblot assay for protein expression in insulin signaling pathway. Total cell lysates were obtained from the cells treated with 1 nM of estradiol in a time-dependent manner and followed by further immunoblot assay for protein expression of interest. Densitometric analysis was performed. Values are mean ± SEM. #P < 0.001.
Since multiple studies have shown a pathological function of androgen receptor (AR) in breast cancer, we examined the functional role of AR in breast cancer growth response in the insulin-primed cells.Citation20-22 AR protein level was higher in the insulin-primed cells compared to the parental cells. AR in both cell lines appeared to be functional as dihydrotestosterone (DHT) treatment induced mRNA expression of FKBP51, a potential AR target ().Citation23 However, both insulin-primed and unprimed MCF-7 cells showed no growth response to treatment of AR agonist DHT, or antagonist bicalutamide (). This indicates that insulin priming is specifically relevant to modulate ER-induced growth response of breast cancer.
Estradiol-induced growth is dependent on Akt and Erk signaling pathways
Since insulin priming is a prerequisite for estradiol-induced breast cancer growth that potentially occurs through ER-mediated non-genomic pathway,Citation24 we next wondered if the estradiol- mediated signal transduction pathways are differently modulated between the insulin-primed and the corresponding unprimed cells. Treatment of the insulin-primed cells with PI3K inhibitor LY294,002 or MEK inhibitor U0126 revealed that the estradiol-induced cellular growth is mediated by PI3K and ERK pathways (), the main downstream pathways of insulin-mediated signaling believed to be involved in cell survival. From the molecular analysis, we observed prompt induction and reduction of ERα phosphorylation in the primed cells when treated with estradiol in a time-dependent manner. However, the unprimed cells sustained the ERα phosphorylation over the same period of estradiol treatment (). Insulin priming appeared to increase the basal level of phosphorylated ERK and total ERK expression while a time-dependent treatment of estradiol showed increased ERK phosphorylation in the unprimed cells but no difference of ERK phosphorylation in the insulin-primed cells (). Both total and phosphorylated-S6 kinase, a downstream target of mTOR, appeared to be more responsive, in the primed cells compared to the unprimed cells when treated with estradiol. However, interestingly, phosphorylation at Thr 308 in Akt protein was markedly decreased in the primed cells but sustained in the parental cells, while phosphorylation at serine 473 residue only changed slightly compared to that at the threonine 308 residue when treated with estradiol in a time-dependent manner. This suggests that insulin priming potentially contributes to the estradiol-induced breast cancer growth by modulating estradiol-insulin signaling crosstalk.
Estradiol modulates the expression of cell cycle and apoptotic factors
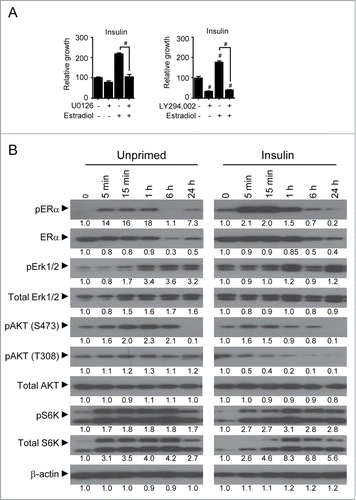
Since the homeostatic balance of cellular factors results in the overall cell growth response for extracellular signaling, we investigated proteins involved in cell cycle progression and apoptosis to determine the mechanism of estradiol-induced growth. From the analysis of cell cycle factors, we found that the basal expression of cyclin A and B, but not cyclin D1, appeared to be decreased in the insulin-primed cells compared to the parental counterpart. However, cyclin A and B were markedly increased with estradiol treatment but were reversed by pure anti-estrogen ICI182,780 compound in the primed cells, suggesting that the resultant protein increase is an ER-dependent. Interestingly, treatment of PI3K inhibitor LY294,002 significantly suppressed overall expression of the cell cyclins, while MEK inhibitor U0126 specifically inhibited cyclin D1 expression. Furthermore, cyclin dependent-kinase inhibitors (CKIs), p21 and p27, showed more decreased expression pattern with estradiol treatment in the primed cells compared to the corresponding cells. An additional interesting finding was that estradiol increased anti-apoptotic Bcl-xL expression in the insulin-primed cells but not in the unprimed one. Estradiol showed no significant difference in the expression of pro-apoptotic BAX and no expression of CKI p16 and anti-apoptotic Bcl-2 in both cell lines ( and data not shown). These data suggest that estradiol drives cell cycle progression by modulating factors in cell cycle and apoptosis in insulin-primed cells, thus resulting in overall cancer growth.
Figure 3. Growth response of the insulin-primed cells upon estradiol-mediated regulation of cell cycle factors. Immunoblot assay was performed for the expression of cell cycle factors involving cyclins (A, B, D1), cyclin-dependent kinase inhibitor (p21, p27) and apoptosis (Bcl-xL, BAX). Total cell lysates were obtained from both primed and unprimed cells after 24 h treatment with 1 nM estradiol alone or together with 1 μM of ICI182,780, 100 μM of LY294,002, 10 μM of U0126, or 3 mM of metformin. Densitometric analysis was performed.
Estradiol modulates genes involved in glucose metabolic flux and serine biosynthesis
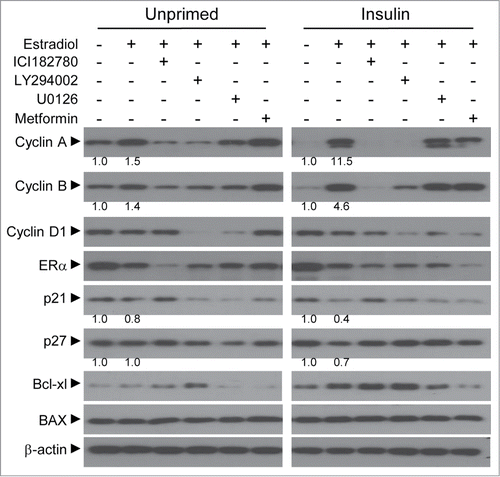
Recent studies of cancer metabolism have proposed that tumors develop mechanisms for metabolic shunting of nutrients into macromolecule biosynthesis pathways including proteins, fatty acids, and nucleotides in order to sustain a high proliferative index.Citation25,26 To comply with the cellular needs, cancer cells employ a strategy to increase glycolytic flux, which in turn allows shunting of carbon into other biosynthetic pathways, i.e. serine biosynthesis and pentose phosphate pathways.Citation26 To determine if estradiol-induced growth is accompanied by an increase in glucose metabolism, we profiled the mRNA expression of 17 genes involved in metabolic flux of glucose using QPCR (). Insulin priming decreased the expression of 6 genes involved in glycolysis. Included in this group are muscle isoform of phosphofructokinase (PFKM), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), pyruvate kinase (PKM 1 and 2), pyruvate dehydrogenase kinase 1 (PDK1), and lactate dehydrogenase A (LDHA). Although insulin priming decreased overall catabolism of glucose, interestingly, estradiol treatment induced the expression of glucose transporter 1 (GLUT1) and PKM2 in insulin-primed cells (). It is known that GLUT1 is a key glucose transporter molecule involved in glucose uptake and PKM2 is an inactive isoform of pyruvate kinase in cancer.Citation27,28 This further drove us to investigate if the uptaken glucose is shunted away from the glycolytic pathway into other intermediate pathways. Interestingly, we found that genes involved in serine biosynthesis were markedly induced in response to estradiol only in the insulin-primed cells (). Moreover, this induction was reversed by ICI 182,780 treatment, suggesting that the induction is an ER-dependent. These genes include phosphoglycerate dehydrogenase (PHGDH) and phosphoserine phosphatase (PSPH). Furthermore, mitochondrial serine hydroxymethyltransferase 2 (SHMT2) also increased in response to estradiol (). This result further prompted us to rationalize that the cells might accompany anaplerotic shunting pathway to replenish the potential deficit of 3-carbon carbohydrate due to serine biosynthesis. Indeed, a subset of genes was induced to direct glucose-6-phosphate into glyceraldehyde-3-phosphate in the insulin-primed cells when treated with estradiol. These genes involve glucose-6-phosphodehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (PGD), ribulose-5-phosphate-3-epimerase (RPE), ribulose-5-phosphate isomerase A (RPIA), transaldolase 1 (TALDO 1), and transketolase (TKT) (). Along with the molecular change in cells, these data suggest that insulin priming process might contribute to shunting of glucose carbon into anabolic pathways to comply with cellular biosynthetic needs.
Figure 4. Estradiol modulation of gene expression involved in glucose metabolism. The QPCR assay was performed for metabolic genes in insulin-primed and unprimed cells. Total RNAs were isolated from cells treated with 1 nM of estradiol alone or in combination with 1 μM of ICI182,780 for 24 h. (A) Genes associated with glycolytic pathway. (B) Genes involved in serine biosynthesis from 3-phosphoglycerate. (C) Genes for pentose phosphate pathway for anaplerotic reaction to replenish glyceraldehyde-3-phosphate. (D) Schematic diagram of glycolysis and the associated biosynthetic pathways. Continuous bold arrows show proposed flow of carbon in insulin-primed cells after treatment with estradiol. White bars-unprimed (□); black bars-primed cells (▪). Values are mean ± SEM. *P < 0.05; ΔP < 0.01; #P < 0.001.
Metformin but not TZDs inhibits estradiol-induced growth in MCF-7 cells
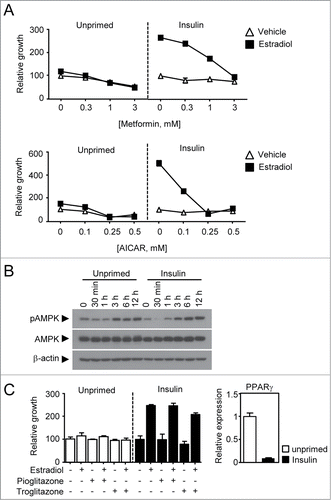
Having established that insulin priming confers breast cancer cells the ability to shunt glucose metabolism into serine biosynthesis, we next wondered if treatment of the primed cancer cells with anti-diabetic drugs, i.e., metformin and TZDs, could be therapeutically effective. This idea is supported by a previous study in which metformin and TZDs have been associated with better prognosis, but insulin with poor outcome, in breast cancer patients.Citation5 Indeed, the biguanide compound similarly inhibited growth response of the unprimed cells with or without estradiol treatment in a dose-dependent manner. However, the insulin-primed cells exhibited dramatic growth inhibitory response to the metformin treatment in the presence of estradiol but no response without estradiol (). Consistent with this, metformin markedly decreased the expression of ERα and cyclin D1 (). It is an interesting to note that metformin treatment increased AMPK phosphorylation in a time-dependent manner but exhibited no difference between the primed and unprimed cells (). As metformin has several mechanisms of action such as adenylate cyclase activationCitation29 or inhibition of complex I of the mitochondrial respiratory chain that causes a decrease in ATP/ADP ratio consequently activating AMPK signaling pathway,Citation30,31 we tested if AMPK activation inhibits the estradiol-induced growth. Thus, insulin-primed and unprimed cells were treated with AMPK specific activator, AICAR alone or in combination with estradiol. In a very similar manner to metformin, AICAR was able to inhibit the growth of both primed and unprimed cells (). We next tested alternative anti-diabetic drugs, TZDs, for their potential anti-tumorigenic activity in diabetic breast cancer. The TZDs, i.e. pioglitazone and troglitazone, are well-known ligands for nuclear receptor peroxisome proliferator-activated receptor gamma (PPAR γ). To our surprise, we found that the TZDs showed no growth inhibition in both unprimed and primed cancers with PPAR γ expression (). Note that the chemical activity of TZDs used in this study was confirmed by luciferase assay (data not shown). These data suggests that AMPK activation, but not PPAR γ, is critical for inhibiting the estradiol-induced growth of ER-positive diabetic breast cancer.
Figure 5. Therapeutic evaluation of anti-diabetic drugs in the insulin-primed breast cancer. (A) Growth response of both primed and unprimed breast cancer cells to metformin and AICAR. Cells were treated with metformin or AICAR in a dose-dependent manner in the presence or absence of 1 nM estradiol. (B) Immunoblot assay for phosphorylated-AMPK and total AMPK. Cells were treated with 5 mM of metformin in a time-dependent manner for immune blot assay of AMPK phosphorylation. (C) Growth response of both primed and unprimed breast cancer cells to TZDs. Cells were treated with 3 μM of pioglitazone or troglitazone in the presence or absence of 1 nM estradiol. The mRNA expression of PPAR γ were measured in both unprimed(□, cycle time = □24) and insulin primed (▪, cycle time = □28) MCF-7 cells.
Discussion
Epidemiological studies have previously demonstrated that patients with diabetes are at higher risk of developing many types of cancer including breast cancer.Citation32 In ER-positive breast cancer, insulin or IGF-1 signaling is known to crosstalk with ER for breast cancer growth.Citation12-14 However, most mechanistic studies involve acute insulin treatment, rather than employing chronic insulin exposure which is more relevant to type II diabetes, and which should be used to determine impact of diabetes on breast cancer growth. This rationale led us to establish in vitro chronic hyperinsulinemic model to investigate insulin priming effect on estradiol signaling in ER-positive breast cancer. In the present study, to demonstrate that the insulin priming introduces some cellular and metabolic changes in ER-positive breast cancer cells, we executed several independent molecular and biochemical approaches; 1) to interrogate if the chronic exposure of insulin sensitizes breast cancer responsiveness to estradiol treatment; 2) to investigate metabolic changes of breast cancer cells in response to estradiol upon insulin priming; 3) to assess the difference of therapeutic response to anti-diabetic drugs between insulin-primed and unprimed breast cancer cells.
To this end, we first established insulin-primed breast cancer MCF-7 cells that have been maintained in 10 nM insulin, a supra-physiological concentration that was intended to safely set hyperinsulinemic condition in this study. This provided a unique system for the molecular and biochemical dissection of estradiol-induced breast cancer growth. Note that blood insulin levels fluctuate, with normal levels ranging from 0.057 to 0.079 nM between meals and can rise up to 0.43 nM after meals.Citation33 In addition, a previous study reported that basal insulin levels are more than 2-fold higher in type II diabetic patients compared to the non-obese or non-diabetic patients.Citation6 We found that the insulin priming dramatically sensitizes breast cancer growth to 100pmol of estradiol, a concentration that is 100–1000 fold lower compared to the one utilized in previous studies ().Citation18,19 It is worthwhile to note that the Kd value of ERα to the estradiol is approximately 100 pmol. Intriguingly, given that both primed and unprimed cell lines showed comparable levels of functional ERα expression and pure anti-estrogen ICI 182,780 mediated-growth inhibition, ERα is necessary but not sufficient for estradiol-induced growth of the insulin-primed breast cancer cells (). Interestingly, treatment of both agonistic and antagonistic AR ligands showed no growth response in both breast cancer cells (). This suggests that insulin priming introduces an additional factor that is specifically required for estradiol-induced growth.
Secondly, from the molecular analysis of the primed cells, various cellular factors and a subset of metabolic genes were identified to differentiate the primed cells from the unprimed counterpart for estradiol responsiveness. Estradiol treatment of the insulin-primed cells modulated the cellular homeostasis of cell cyclins, CKIs, and anti-apoptotic factors resulting in overall cell proliferation (). In addition, the female hormone induced expression of metabolic genes involving PPP for anaplerosis of glyceraldehde-3-phosphate and serine biosynthesis pathway for further macromolecule production including other amino acids as well as nucleotides. This finding suggests that the insulin priming process confers estradiol the capability to regulate cell cycle progression and metabolic shunting in cancer cells in order to meet the anabolic needs for cell proliferation. Further, estradiol induced the expression of PKM2 and GLUT1, critical factors for regulating glucose metabolism, in the insulin primed MCF-7 cells. It might be interesting to see if these genes are directly involved in estradiol-dependent breast cancer growth. Likewise, LDHA and PDK1 expressions were increased upon estradiol treatment, which is expected to increase lactate production in the insulin-primed cells. Thus, chronic insulin exposure may prime ER-positive breast cancer cells to preferentially breakdown glucose to lactate even in the presence of oxygen, the so-called Warburg effect which is a common feature in cancer cells, when treated with estradiol. Along with the biosynthetic shunting, it will be intriguing to investigate other critical cancer metabolic hallmarks, i.e., bioenergetics and redox control, in this system.
Lastly, one notable finding from this study is that metformin, but not TZDs, inhibited estradiol-induced growth of the insulin primed cells. This is partly consistent with the previous study where the use of both anti-diabetic drugs, i.e. metformin and TZDs, improved the overall survival of diabetic breast cancer patients.Citation9 Even with the same anti-diabetic outcome, these 2 groups of drugs employ different mechanisms to achieve the resulting hypoglycemia which could account for the difference in inhibition of estradiol-induced growth. While metformin inhibits complex I of the respiratory chain which leads to activation of AMPK eventually increasing cellular utilization of glucose,Citation30,31 TZDs mainly work to improve insulin sensitivity by counteracting TNF-α-induced insulin resistance through restoration of insulin receptor and IRS-1 phosphorylation in adipocytes.Citation34 This mechanistic difference would explain the different anti-cancer effect of both drugs especially in the insulin-primed ER-positive breast cancer cells. It would be interesting to see if these anti-diabetic drugs would present differentiated benefits for prognosis of the diabetic breast cancer patients according to ER status.
Collectively, our study explicitly presents mechanistic crosstalk of insulin and ER signaling and shows biochemically the anaplerotic shunting of glucose utilization and further that metformin, but not TZD, inhibit cancer growth in the insulin-primed ER-positive breast cancer. Our work provides a new insight into differentiated clinical use of anti-diabetic drugs for ER associated breast cancer.
Materials and Methods
Cell culture
Estrogen receptor-positive breast cancer cell line, MCF-7, was cultured in RPMI supplemented with 5% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin. To establish the insulin-primed breast cancer cell model, MCF-7 cells were maintained in a supra-physiological insulin concentration of 10 nM or Insulin-like growth factor-1 (IGF-1) of 30 nM for at least 7days before carrying out further experiments.
MTT assay
Three thousand MCF-7 cells, both unprimed and insulin-primed, were seeded per well in 96-well plates in phenol red-free media supplemented with 5% charcoal-stripped serum (Sigma Aldrich, F6765). The following day, the cells were treated with 17β-estradiol, insulin, pure anti-estrogen ICI182,780, MEK inhibitor U0126, PI3K inhibitor LY294,002, metformin, or AICAR. MTT assay was carried out as previously described.Citation35
Quantitative real-time polymerase chain reaction
Total RNA was isolated from cell lines using TRIzol reagent (Invitrogen, BRL15596-026) according to the manufacturer's instructions. Total RNA was reverse-transcribed into cDNA (cDNA) using ReverTra Ace® qPCR RT Master Mix with gDNA Remover (Toyobo Co. Ltd, FSQ-301) according to the manufacturer's instructions. The mRNA expression for genes of interest was profiled using qPCR in an ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). Data analysis was performed using the delta-delta Ct method with 18S as the reference gene.Citation36
Western blot
Total cell lysates was obtained from cells using PRO-PREPTM Protein Extraction Solution (Intron Biotechnology, 17081) according to the manufacturer's instructions. Immunoblot assay was employed for protein expression analysis. Primary antibodies include cyclin D1 (2926), cyclin B (4135), ERα (8644), ERα-pSer118 (2511), AR (5153), Akt-pSer473 (4060), Akt-pThr308 (9275), Akt (9272), p70 S6 Kinase-pThr421/Ser424 (9204), p70 S6 Kinase (9202), p44/42 Erk1/2-pThr202/Tyr204 (9101) and p44/42 Erk1/2 (9102) from Cell Signaling Technologies; cyclin A (sc-239) was purchased from Santa Cruz Biotechnology while β-actin (ab6276) and IRβ (ab69508) were purchased from Abcam.
Statistical analysis
All results were expressed as mean ± SEM and they were analyzed using GraphPad Prism 5.03 (GraphPad Software Inc.). Data was analyzed using one-way ANOVA with Tukey's post hoc test. Results with p-value <0.05 were considered to be statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was financially supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A1A05005075 to Y.J.).
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: Cancer J Clin 2011; 61:69-90; PMID:21296855
- Hulka BS, Moorman PG. Breast cancer: hormones and other risk factors. Maturitas 2001; 38:103-13; discussion 13-6; PMID:11311599; http://dx.doi.org/10.1016/S0378-5122(00)00196-1
- Kaplan MA, Pekkolay Z, Kucukoner M, Inal A, Urakci Z, Ertugrul H, Akdogan R, Firat U, Yildiz I, Isikdogan A. Type 2 diabetes mellitus and prognosis in early stage breast cancer women. Med Oncol 2012; 29:1576-80; PMID:22083554; http://dx.doi.org/10.1007/s12032-011-0109-4
- Cadoo KA, Fornier MN, Morris PG. Biological subtypes of breast cancer: current concepts and implications for recurrence patterns. Q J Nucl Med Mol Imag: Off Pub Ital Assoc Nucl Med 2013; 57:312-21; PMID:24322788
- Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, Hux JE. The impact of diabetes on survival following breast cancer. Br Cancer Res Treat 2008; 109:389-95; PMID:17659440; http://dx.doi.org/10.1007/s10549-007-9654-0
- Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 2008; 31 Suppl 2:S262-8; PMID:18227495; http://dx.doi.org/10.2337/dc08-s264
- Michels KB, Solomon CG, Hu FB, Rosner BA, Hankinson SE, Colditz GA, Manson JE, Nurses' Health Study. Type 2 diabetes and subsequent incidence of breast cancer in the Nurses' Health Study. Diabetes Care 2003; 26:1752-8; PMID:12766105; http://dx.doi.org/10.2337/diacare.26.6.1752
- Gunter MJ, Hoover DR, Yu H, Wassertheil-Smoller S, Rohan TE, Manson JE, Li J, Ho GY, Xue X, Anderson GL, et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J Natl Cancer Inst 2009; 101:48-60; PMID:19116382; http://dx.doi.org/10.1093/jnci/djn415
- He X, Esteva FJ, Ensor J, Hortobagyi GN, Lee MH, Yeung SC. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann Oncol: Off J Eur Soc Med Oncol/ESMO 2012; 23:1771-80; PMID:22112968; http://dx.doi.org/10.1093/annonc/mdr534
- Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report. CA: Cancer J Clin 2010; 60:207-21; PMID:20554718
- Sachdev D, Yee D. The IGF system and breast cancer. Endocr-relat Cancer 2001; 8:197-209; PMID:11566611; http://dx.doi.org/10.1677/erc.0.0080197
- Yee D, Lee AV. Crosstalk between the insulin-like growth factors and estrogens in breast cancer. J Mammary Gland Biol 2000; 5:107-15; PMID:10791773; http://dx.doi.org/10.1023/A:1009575518338
- Mawson A, Lai A, Carroll JS, Sergio CM, Mitchell CJ, Sarcevic B. Estrogen and insulin/IGF-1 cooperatively stimulate cell cycle progression in MCF-7 breast cancer cells through differential regulation of c-Myc and cyclin D1. Mol Cell Endocrinol 2005; 229:161-73; PMID:15607540; http://dx.doi.org/10.1016/j.mce.2004.08.002
- Belfiore A, Frasca F. IGF and insulin receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia 2008; 13:381-406; PMID:19016312; http://dx.doi.org/10.1007/s10911-008-9099-z
- Bartella V, De Marco P, Malaguarnera R, Belfiore A, Maggiolini M. New advances on the functional cross-talk between insulin-like growth factor-I and estrogen signaling in cancer. Cell Signalling 2012; 24:1515-21; PMID:22481093; http://dx.doi.org/10.1016/j.cellsig.2012.03.012
- Hawsawi Y, El-Gendy R, Twelves C, Speirs V, Beattie J. Insulin-like growth factor - oestradiol crosstalk and mammary gland tumourigenesis. Biochimica et Biophysica Acta 2013; 1836:345-53; PMID:24189571
- Fox EM, Miller TW, Balko JM, Kuba MG, Sanchez V, Smith RA, Liu S, González-Angulo AM, Mills GB, Ye F, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res 2011; 71:6773-84; PMID:21908557; http://dx.doi.org/10.1158/0008-5472.CAN-11-1295
- Madak-Erdogan Z, Lupien M, Stossi F, Brown M, Katzenellenbogen BS. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol Cell Biol 2011; 31:226-36; PMID:20956553; http://dx.doi.org/10.1128/MCB.00821-10
- Marquez DC, Pietras RJ. Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cancer cells. Oncogene 2001; 20:5420-30; PMID:11571639; http://dx.doi.org/10.1038/sj.onc.1204729
- Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, Rimm DL, Liu XS, Brown M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell 2011; 20:119-31; PMID:21741601; http://dx.doi.org/10.1016/j.ccr.2011.05.026
- Peters AA, Buchanan G, Ricciardelli C, Bianco-Miotto T, Centenera MM, Harris JM, Jindal S, Segara D, Jia L, Moore NL, et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res 2009; 69:6131-40; PMID:19638585; http://dx.doi.org/10.1158/0008-5472.CAN-09-0452
- Doane AS, Danso M, Lal P, Donaton M, Zhang L, Hudis C, Gerald WL. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006; 25:3994-4008; PMID:16491124; http://dx.doi.org/10.1038/sj.onc.1209415
- Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem 2012; 287:19736-49; PMID:22532567; http://dx.doi.org/10.1074/jbc.M112.352930
- Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol 2005; 19:833-42; PMID:15695368; http://dx.doi.org/10.1210/me.2004-0486
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11:85-95; PMID:21258394; http://dx.doi.org/10.1038/nrc2981
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029-33; PMID:19460998; http://dx.doi.org/10.1126/science.1160809
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008; 452:181-6; PMID:18337815; http://dx.doi.org/10.1038/nature06667
- Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005; 202:654-62; PMID:15389572; http://dx.doi.org/10.1002/jcp.20166
- Miller RA, Chu QW, Xie JX, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013; 494:256-60; PMID:23292513; http://dx.doi.org/10.1038/nature11808
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci 2012; 122:253-70; PMID:22117616; http://dx.doi.org/10.1042/CS20110386
- Klip A, Leiter LA. Cellular mechanism of action of metformin. Diabetes Care 1990; 13:696-704; PMID:2162756; http://dx.doi.org/10.2337/diacare.13.6.696
- Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr-Relat Cancer 2009; 16:1103-23; PMID:19620249; http://dx.doi.org/10.1677/ERC-09-0087
- Iwase H, Kobayashi M, Nakajima M, Takatori T. The ratio of insulin to C-peptide can be used to make a forensic diagnosis of exogenous insulin overdosage. Forensic Sci Int 2001; 115:123-7; PMID:11056282; http://dx.doi.org/10.1016/S0379-0738(00)00298-X
- Diaz-Delfin J, Morales M, Caelles C. Hypoglycemic action of thiazolidinediones/peroxisome proliferator-activated receptor gamma by inhibition of the c-Jun NH2-terminal kinase pathway. Diabetes 2007; 56:1865-71; PMID:17416798; http://dx.doi.org/10.2337/db06-1293
- Jeong Y, Xie Y, Lee W, Bookout AL, Girard L, Raso G, Behrens C, Wistuba II, Gadzar AF, Minna JD, et al. Research resource: Diagnostic and therapeutic potential of nuclear receptor expression in lung cancer. Mol Endocrinol 2012; 26:1443-54; PMID:22700587; http://dx.doi.org/10.1210/me.2011-1382
- Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Current Protocols in Molecular Biology, edited by Frederick M Ausubel [et al] 2006; Chapter 15:Unit 15 8. PMID:18265376; doi:10.1002/0471142727.mb1508s73