
Abstract
The present studies sought to determine whether the anti-folate pemetrexed (Alimta) and the sphingosine-1-phosphate receptor modulator FTY720 (Fingolimod, Gilenya) interacted to kill tumor cells. FTY720 and pemetrexed interacted in a greater than additive fashion to kill breast, brain and colorectal cancer cells. Loss of p53 function weakly enhanced the toxicity of FTY720 whereas deletion of activated RAS strongly or expression of catalytically inactive AKT facilitated killing. Combined drug exposure reduced the activity of AKT, p70 S6K and mTOR and activated JNK and p38 MAPK. Expression of activated forms of AKT, p70 S6K and mTOR or inhibition of JNK and p38 MAPK suppressed the interaction between FTY720 and pemetrexed. Treatment of cells with FTY720 and pemetrexed increased the numbers of early autophagosomes but not autolysosomes, which correlated with increased LC3II processing and increased p62 levels, suggestive of stalled autophagic flux. Knock down of ATG5 or Beclin1 suppressed autophagosome formation and cell killing. Knock down of ceramide synthase 6 suppressed autophagosome production and cell killing whereas knock down of ceramide synthase 2 enhanced vesicle formation and facilitated death. Collectively our findings argue that pemetrexed and FTY720 could be a novel adjunct modality for breast cancer treatment.
Abbreviations
| ERK | = | extracellular regulated kinase |
| MEK | = | mitogen activated extracellular regulated kinase |
| PI3K | = | phosphatidyl inositol 3 kinase |
| ca | = | constitutively active |
| dn | = | dominant negative |
| PTX | = | pemetrexed |
| PTX | = | pemetrexed |
| ER | = | endoplasmic reticulum |
| mTOR | = | mammalian target of rapamycin |
| MAPK | = | mitogen activated protein kinase |
| PTEN | = | phosphatase and tensin homolog on chromosome 10 |
| ROS | = | reactive oxygen species |
| CMV | = | empty vector plasmid or virus |
| si | = | small interfering |
| SCR | = | scrambled |
| IP | = | immunoprecipitation |
| LASS | = | longevity assurance gene |
| CerS | = | ceramide synthase |
| Ad | = | adenovirus |
| VEH | = | vehicle |
Introduction
Pemetrexed (Alimta®) is an anti-folate drug approved for the treatment of advanced and metastatic non-small cell lung cancer (NSCLC). Pemetrexed was developed as an inhibitor of thymidylate synthase (TS), and the enzyme aminoimidazole-carboxamide ribonucleotide formyl-transferase (AICART) is now known to be a secondary target for pemetrexed.Citation1-4 Inhibition of AICART results in activation of AMP-activated protein kinase (AMPK) and AMPK signaling inhibits mammalian target of rapamycin (mTOR).Citation1,2,5 Inhibition of mTOR stimulates autophagy.Citation6-10
The immuno-modulatory pro-drug FTY720 (Gilenya; Fingolimod) is approved for the treatment of relapsing multiple sclerosis (reviewed in ref. 11). FTY720 reduces peripheral blood lymphocyte levels and blocks the migration of T cells from lymph nodes.Citation12 In cells, FTY720 is phosphorylated predominantly by sphingosine kinase 2, though recent in vivo evidence also argues sphingosine kinase 1 plays a role, and phospho-FTY720 acts as an agonist against sphingosine-1-phosphate receptors (S1P) 1, 4 and 5, and to a lesser extent 3.Citation13-17 By binding to these receptors phospho-FTY720 causes rapid internalization and degradation of the S1P receptors.Citation16 Although FTY720 has been reported to be an inhibitor of ceramide synthase enzymes in vitro, in cells FTY720 elevates ceramide levels.Citation18 In tumor cells FTY720 has been reported to induce autophagy, activate PP2A and to inactivate the ERK1/2 and AKT pathways, with ERK/AKT inactivation causal in drug-induced killing.Citation19-26
Protein phosphatase 2A is a regulator of multiple MAPK pathways. Fulvestrant resistant MCF7 cells i.e. resistant to a pure anti-estrogen, expressed higher levels of the PP2A inhibitor SET/I2PP2A, had lower endogenous PP2A activity, and had elevated basal ERK1/2 activity compared with their estrogen dependent counterparts.Citation6,27 PP2A (and also PP1) can be directly activated by ceramide and SET/I2PP2A complex can be inhibited by increasing levels ceramide. In previous studies inhibition of the de novo ceramide synthase pathway blocked drug-induced ceramide generation, PP2A activation and tumor cell killing.
The present studies determined whether manipulation of S1P receptor function and sphingosine-1-phosphate production by FTY720 altered the lethality of pemetrexed. FTY720 enhanced pemetrexed toxicity in a dose-dependent fashion. The drug combination inactivated ERK, AKT and mTOR and increased autophagosome levels, which was regulated by ceramide synthase 6 and ceramide synthase 2. The drug combination of [pemetrexed + FTY720] + sorafenib was highly effective at killing tumor cells.
Materials and Methods
Materials
FTY720 was purchased from Cayman Chemical Inc.., (Ann Arbor, MI, USA). Pemetrexed was purchased from LC Laboratories (Woburn, MA, USA). Sorafenib and 4HPR were purchased from Calbiochem. Trypsin-EDTA, DMEM, RPMI, penicillin-streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies, Grand Island, NY, USA). Validated cells were purchased from the ATCC repository and were not further validated beyond that claimed by ATCC. Reagents and performance of experimental procedures were described in references.Citation6,27,28,40-42
Methods
Culture and in vitro exposure of cells to drugs. All cell lines were cultured at 37 °C (5% (v/v CO2) in vitro using RPMI supplemented with dialyzed 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. Cells growing in “complete” fetal calf serum that contains thymidine were gradually weaned into dialyzed serum lacking thymidine over 2 weeks and were then used for experimental analyses for the following 3 weeks before discarding. Cells were re-isolated in thymidine-less media as required. For short term cell killing assays, immunoblotting studies, cells were plated at a density of 3 × 103 per cm2 (~2 × 105 cells per well of a 12 well plate) and 48h after plating treated with various drugs, as indicated. In vitro pemetrexed et al drug treatments were simultaneous and from 100 mM stock solutions of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study in this manuscript.
In vitro cell treatments, SDS-PAGE and Western blot analysis; transfection of cells with siRNA or with plasmids; microscopy for LC3-GFP-RFP expression. Studies were performed as described in references. 6, 27, 28, 31, 32, 40–42.
Assay for PP2A activity. Cells were plated in 60 mm dishes in triplicate and cultured for 24 h. Cells were treated with vehicle or drugs for 12 h, as indicated. After 12 h drug treatments, cells were washed twice with cold PBS, then harvested in low phosphate lysis buffer. Samples were prepared according to the PP2A Phosphatase Activity Assay Kit protocol (R&D Systems) as described in the protocol. Three separate protein amounts for each condition were immunoprecipiated with a monoclonal antibody against PP2A. After immunoprecipitation, PP2A antibody-coupled sepharose beads were washed and bound protein cleaved using the manufacturer provided reagent at equal volumes for each sample. Samples were then transferred to a 96-well plate and after incubation with the detection substrate, samples were evaluated using a Malachite Green protocol for determination of Absorbance at 595 nm using a Vector 3 plate reader. Triplicate values obtained for each sample type, and 3 variable protein loading amounts, were averaged and plotted as fold-change in PP2A Activity relative to that of the vehicle-treated condition.
Determination of sphingolipid levels Cells were plated at 1 × 106 cells in 60 mm dishes in duplicate every day for 3 days for a total of 6 repeats per condition and cultured for 24 h prior to transfection. Cells were then harvested in 550 µl of cold PBS and 50 µl taken for lysis and protein determination using the Bradford Assay (Bio-Rad). Sphingolipid levels were normalized based on total protein levels for each sample. Cells were processed and subjected to quantitative mass spectrometry to determine the levels of sphingolipid species.Citation27,28
Data analysis. Comparison of the effects of various treatments was performed using one way analysis of variance and a 2 tailed Student's t-test. Statistical examination of in vivo animal survival data utilized log rank statistical analyses between the different treatment groups. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Results
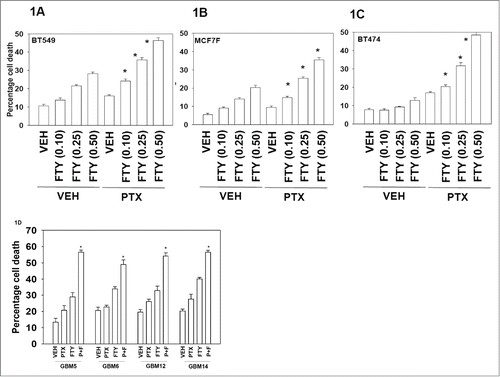
Initial studies examined the dose-dependent interaction on cell viability between FTY720 and pemetrexed. In prior studies we had determined that a pemetrexed concentration in the range of 0.5−1.0 μM exhibited modest short-term toxicity to tumor cells but significantly enhanced sorafenib toxicity.Citation6 In mammary carcinoma cells FTY720 and pemetrexed (PTX) interacted to rapidly kill cells in a dose-dependent and a greater than additive fashion (). Similar data were obtained in primary human glioblastoma cells (). Of note, mammary and GBM cells lacking expression of the oncogene phosphatase and tensin homolog on chromosome 10 (PTEN) were effectively killed as were cells over-expressing other oncogenes such as activated full-length ERBB1, truncated active ERBB1 vIII or ERBB2. These data collectively suggest that a combination of PTX and FTY720 interact to kill diverse types of tumor cells.
Figure 1. Pemetrexed and FTY720 interact to kill tumor cells. (A–C) BT474, BT549 and MCF7F breast cancer cells were treated with Vehicle (VEH) or pemetrexed (PTX, 0.5 μM) and increasing concentrations of FTY720 (FTY, 0.10−0.5 μM). Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than corresponding value in VEH treated cells. (D) Glioblastoma cells (GBM5/6/12/14) were treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than corresponding value in VEH treated cells.
We next examined the roles of RAS signaling and p53 in the response of tumor cells to FTY720, alone and in combination with pemetrexed, using genetically manipulated colon cancer cells.Citation31-33 Deletion of p53 did not significantly enhance or decrease the lethality of FTY720 in HCT116 cells (). In contrast, deletion of the single allele of K-RAS D13 significantly enhanced FTY720 lethality. In HCT116 cells deleted for the single allele of K-RAS D13 and expressing H-RAS V12, FTY720 toxicity was reduced compared to that in wild type HCT116 cells expressing K-RAS D13. Using HCT116 cells expressing H-RAS V12 isoforms with additional point mutations that specifically activate downstream signaling pathways we found that expression of a mutant H-RAS V12 which only activates PI3K (V12/40) resulted in a reduced protective effect when compared to wild type H-RAS V12. Point mutants that only activate RAF-1 (V12/35) or RAL GDS (V12/37) were less protective than either wild type H-RAS V12 or the H-RAS V12 mutant which only activates PI3K (V12/40). This data set would tend to argue that PI3K/AKT signaling, and to a lesser extent RAF-1/ERK signaling, antagonize FTY720 lethality as a single agent in HCT116 cells.
Figure 2. RAS-dependent PI3K signaling plays a protective role against FTY720 toxicity. (A) HCT116 colon cancer cells (Wild type cells expressing K-RAS D13; p53 null −/− cells; null −/− cells with K-RAS D13 deleted; null cells expressing H-RAS V12; null cells expressing H-RAS V12-35 that activates RAF-1; null cells expressing H-RAS V12-40 that activates PI3K) were treated with vehicle (DMSO), or FTY720 (2.0 μM). Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3 +/− SEM). *P < 0.05 greater than corresponding value in HCT116 −/− H-RAS V12 cells; **P < 0.05 greater than corresponding value in HCT116 −/− H-RAS V12 / 40 cells; ***P < 0.05 greater than corresponding value in HCT116 WT cells. (B) HCT116 cells were treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM). *P < 0.05 greater than corresponding value in HCT116 −/− H-RAS V12 cells; **P < 0.05 greater than corresponding value in HCT116 −/− H-RAS V12 = 40 cells; ***P < 0.05 greater than corresponding value in HCT116 WT cells.
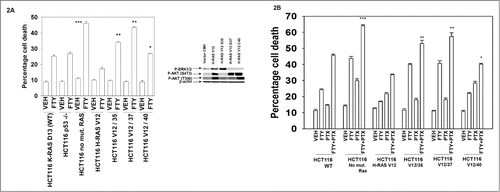
We then determined the impact of altered oncogenic RAS signaling on the sensitivity of HCT116 cells to treatment with FTY720 in combination with pemetrexed. As was observed for treatment with FTY720 alone in , expression of wild type H-RAS V12 or H-RAS V12/40 that activates PI3K were killed by pemetrexed and FTY720 treatment to a lesser extent than the mutants that only activate RAF-1 (V12/35) or RAL GDS (V12/37) (). Deletion of the single allele of K-RAS D13 significantly enhanced the toxicity of FTY720 and pemetrexed combination treatment compared to wild type HCT116 cells. These findings again point to PI3K signaling as being an important pathway in mediating control of pemetrexed and FTY720 toxicity.
Based on our data in , we next determined changes in the activities of recognized signaling pathways downstream of RAS and PI3K, after drug combination treatment. Within 6 h, drug combination treatment had very modestly activated the ERK pathway, modestly inhibited AKT signaling and inactivated p70 S6K (). By 12 h following drug exposure the activities of AKT, p70 S6K and mTOR were significantly reduced. These findings again are suggestive that PI3K signaling may mediate control of pemetrexed and FTY720 toxicity in colon cancer cells.
Figure 3. Regulation of PI3K/AKT/mTOR signaling by FTY720 and pemetrexed treatment. (A) BT474 cells were treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination. Six and 12h after drug treatment cells were isolated and lysates subjected to SDS PAGE followed by immunoblotting against the indicated phospho-proteins. (B) BT474 cells were transfected with either: empty vector plasmid CMV or with plasmids to express activated forms of AKT and MEK or express dominant negative forms of AKT and MEK. Twenty four h after transfection cells were treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM). *P < 0.05 less than corresponding value in CMV transfected cells; #p < 0.05 greater than corresponding value in CMV transfected cells. (C) BT474 cells were transfected with either: empty vector plasmid CMV or with plasmids to express activated forms of p70 S6K and mTOR or express a dominant negative form of p38α. As indicated cells were pre-treated with the JNK inhibitory peptide (JNK-IP, 10 μM). Twenty four h after transfection cells were treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM). *P < 0.05 less than corresponding value in CMV cells.
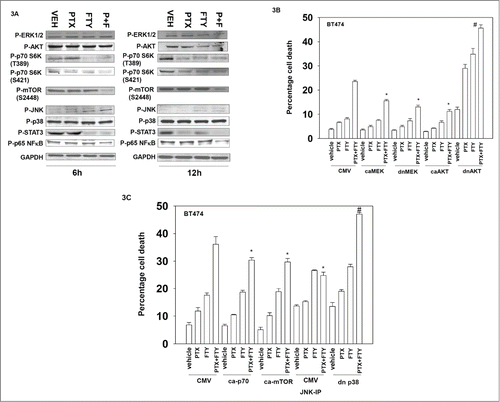
Expression of activated forms of MEK and AKT protected tumor cells from pemetrexed and FTY720 killing (). In a manner similar to that previously observed in our studies combining pemetrexed and multi-kinase inhibitor sorafenib, expression of dominant negative MEK unexpectedly also reduced drug combination toxicity.Citation6 Expression of dominant negative AKT enhanced killing under all treatment conditions. Expression of activated p70 S6K or activated mTOR reduced drug combination toxicity, as did expression of dominant negative p38 MAPK or inhibition of JNK pathway signaling (). Although p38 MAPK phosphorylation was not altered by drug treatment, PTX+FTY treatment decreased the phosphorylation and activity of the chaperone HSP27 (data not shown). These data sets again argue that signaling by the PI3K/AKT pathway plays a major role in regulating pemetrexed and FTY720 lethality.
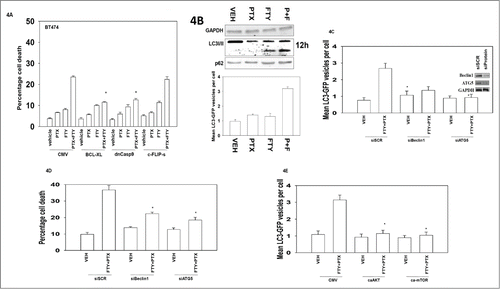
The pathways mediating cell killing were then explored. Inhibition of mitochondrial dysfunction and the intrinsic apoptosis pathway reduced drug combination lethality whereas inhibition of the extrinsic pathway was ineffective at protection (). In our prior studies combining the multi-kinase inhibitor sorafenib and pemetrexed we observed the 2 drugs interacted to promote formation of autophagosomes visualized by GFP+ punctae and also autolysosomes visualized by Lysotracker red and RFP+ punctae.Citation6 FTY720 and pemetrexed interacted to cause autophagosome formation that correlated with: increased LC3II processing and with a modest increase in p62 levels; increased levels of GFP+ punctae but no significant increase in RFP+ punctae levels or Lysotracker red staining (, data not shown). Knock down of the autophagy regulatory proteins Beclin1 or ATG5 suppressed the increase in the levels of autophagosomes after drug exposure and blocked cell killing by FTY720 and pemetrexed (). Signaling by the PI3K/AKT/mTOR pathway is known to suppress autophagy and expression of activated forms of AKT or of mTOR prevented autophagosome formation ().Citation36,37 Thus our data in strongly argue the inhibitory effect of [pemetrexed + FTY720] on the PI3K pathway results in reduced mTOR activity which facilitates a toxic form of autophagosome vesicle formation to proceed, but that based on the increase in p62 levels and lack of an increase in RFP+ vesicles, we conclude that killing was proceeding after stalled autophagy.
Figure 4. The regulation of FTY720 and pemetrexed toxicity by apoptosis pathways and autophagy. (A) BT474 cells were infected with recombinant adenoviruses to express empty vector (CMV); BCL−XL; dominant negative caspase 9; the caspase 8 inhibitor c-FLIP-s (50 moi). Twenty four h after infection cells were treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM). *P < 0.05 less than corresponding value in CMV cells. (B) Lower graph: BT474 cells were transfected with a plasmid to express LC3-GFP. Twenty four h after transfection cells were treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination. Cells were microscopically examined after 6h and the number of GFP+ intense staining punctae counted (n = 3 +/− SEM); Upper blots: BT474 cells were treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination. Cells were isolated after 12h and the expression of LC3I/II and p62 determined. (C) BT474 cells were transfected with a plasmid to express LC3-GFP and in parallel transfected with scrambled siRNA control molecule (siSCR) or siRNA molecules to knock down expression of ATG5 or Beclin1. Twenty four h after transfection cells were treated with Vehicle (VEH) or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. Cells were microscopically examined after 6h and the number of GFP+ intense staining punctae counted (n = 3 +/− SEM). (D) BT474 cells were transfected with scrambled siRNA control molecule (siSCR) or siRNA molecules to knock down expression of ATG5 or Beclin1. Twenty four h after transfection cells were treated with Vehicle (VEH) or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM). *P < 0.05 less than corresponding value in siSCR cells. (E) BT474 cells were transfected with a plasmid to express LC3-GFP and in parallel transfected with either: empty vector plasmid CMV or with plasmids to express activated forms of AKT or mTOR. Twenty four h after transfection cells were treated with Vehicle (VEH) or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. Cells were microscopically examined after 6h and the number of GFP+ intense staining punctae counted (n = 3 +/− SEM) *P < 0.05 less than corresponding value in CMV cells.
Because it has been suggested that FTY720 might affect ceramide biosynthesis, we next examined effects on sphingolipids that have been implicated autophagy and cell death.Citation38 Pemetrexed as a single agent increased the levels of multiple ceramide and dihydro-ceramide species (). Remarkably, FTY720 markedly increased C22:0, C24:1 and C24:0 dihydro-ceramide species (). These drugs only had small effects on the levels of sphingosine and dihydro-sphingosine and their phosphorylated derivatives (). Treatment with fumonisin B1, a pan ceramide synthase inhibitor, although greatly increasing sphingosine and dihydro-sphingosine levels, drastically reduced elevations of ceramide and dihydro-ceramide induced by the combination of FTY720 and pemetrexed suggesting that altered dihydro-ceramide and ceramide levels rather than changes in sphingoid base levels contribute to its killing effect. Treatment of cells with fumonisin B1 or knock down of ceramide synthase 6 suppressed the induction of autophagosome production and of cell killing by pemetrexed and FTY720 (). Knock down of ceramide synthase 6 (CerS6) which regulates C16 dihydro-ceramide levels reduced the levels of drug-induced autophagosomes and reduced drug combination toxicity (). This is of note as although pemetrexed increased C16:0 ceramide / dihydro-ceramide levels, [pemetrexed + FTY720] treatment reduced C16:0 ceramide levels and only weakly increased C16:0 dihydro-ceramide levels ().
Figure 5. Dihydro-ceramide generation plays a central role in the toxicity of FTY720 and pemetrexed. (A-D) BT474 cells were incubated with vehicle or the ceramide synthase inhibitor fumonisin B1 (FB1, 25 μM) as indicated. Cells were then treated with Vehicle (VEH), pemetrexed (PTX, 0.5 μM) FTY720 (FTY, 0.25 μM) or the drugs in combination, as indicated. Cells were isolated after 12h and the levels of ceramides, dihydro-ceramides and other sphingolipids under each condition determined by mass spectrometry (n = 2, 6 independent samples total +/− SEM). *P < 0.05 greater than corresponding value in VEH cells; **P < 0.05 greater than corresponding value in pemetrexed treated cells; % p < 0.05 greater increase over vehicle than in corresponding value in cells treated with FB1. (E) BT474 were transfected with a plasmid to express LC3-GFP-RFP and in parallel transfected with scrambled siRNA control molecule (siSCR) or an siRNA molecule to knock down expression of CerS6 / LASS6 (ceramide synthase 6). Twenty four h after transfection cells were treated with the ceramide synthase inhibitor fumonisin B1 (FB1, 25 μM) then Vehicle (VEH) or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. Cells were microscopically examined after 6h and the number of GFP+ and RFP+ intense staining punctae counted (n = 3 +/− SEM). *P < 0.05 less than corresponding value in siSCR+VEH cells. (F) BT474 were transfected with a scrambled siRNA control molecule (siSCR) or an siRNA molecule to knock down expression of CerS6 / LASS6 (ceramide synthase 6). Twenty four h after transfection cells were treated the ceramide synthase inhibitor fumonisin B1 (FB1, 25 μM) then with Vehicle (VEH) or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM). *P < 0.05 less than corresponding value in siSCR+VEH cells. (G) BT474 were transfected with a plasmid to express LC3-GFP and in parallel transfected with scrambled siRNA control molecule (siSCR) or siRNA molecules to knock down expression of CerS2 / LASS2 and CerS3 / LASS3 (ceramide synthases 2 and 3). Twenty four h after transfection cells were treated with Vehicle (VEH) or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. Cells were microscopically examined after 6h and the number of GFP+ intense staining punctae counted (n = 3 +/− SEM). * P < 0.05 less than corresponding value in siSCR+VEH cells. (H) BT474 were transfected with a scrambled siRNA control molecule (siSCR) or siRNA molecules to knock down expression of LASS2 and LASS3 (ceramide synthases 2 and 3). Twenty four h after transfection cells were treated with Vehicle (VEH) or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM). *P < 0.05 less than corresponding value in siSCR+VEH cells.
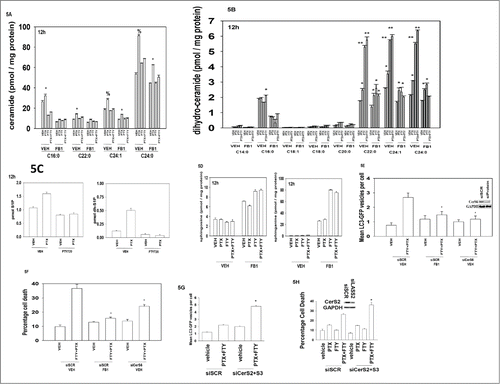
In contrast to findings with CerS6, knock down of ceramide synthases 2 and 3 (CerS2, CerS3) which regulate C22-C24 dihydro-ceramide levels enhanced the levels of drug-induced autophagosomes and enhanced drug combination toxicity (). This would suggest that ceramides and dihydro-ceramides of different chain lengths differentially alter the cellular response to pemetrexed and FTY720 treatment, i.e., because siCerS6, but not siCerS2, decreases lethality it could be argued that basal levels of C16 ceramide and dihydro-ceramide plays a more important role than increased levels of C22-C24 ceramides and dihydro-cermides in modulating cell viability.
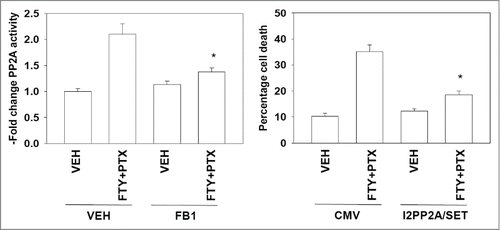
Moreover, based on data in , and in agreement with previous reports that binding of ceramide to inhibitor 2 of PP2A (I2PP2A) activates PP2A and causes cell death (see reference 27, and references therein), we found that FTY720 and pemetrexed activated protein phosphatase 2A (PP2A), an effect that was blocked by incubation of cells with the ceramide synthase inhibitor fumonisin B1; expression of a mutant I2PP2A/SET that does not bind ceramide suppressed killing by pemetrexed and FTY720 (), supporting the involvement of PP2A in cell killing.
Figure 6. Pemetrexed and FTY720 activate PP2A in a ceramide-dependent fashion. Left graph: BT474 were pre-treated with vehicle or fumonisin B1 (FB1, 25 μM), then with vehicle or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. After 12h cells were isolated and the activity of PP2A determined (n = 3 +/− SEM). Right graph: BT474 cells were transfected with an empty vector plasmid (CMV) or a plasmid to express I2PP2A/SET (VIK-SSS). Twenty four h after transfection cells were treated with Vehicle (VEH) or with pemetrexed (PTX, 0.5 μM) and FTY720 (FTY, 0.25 μM) in combination. Twenty four h after drug treatment cells were isolated and viability determined by trypan blue exclusion assay (n = 3, +/− SEM).
As noted in , pemetrexed increases the levels of multiple ceramide species and FTY720 treatment increases dihydro-ceramide levels and increased dihydro-ceramide levels, particularly C16:0 dihydro-ceramide, were responsible for cell killing. Based on our pre-clinical studies, the multi-kinase inhibitor sorafenib is presently under phase I clinical evaluation, combined with pemetrexed, at VCU/MCVH (NCT01450384).Citation6 One reason for this drug interaction is that sorafenib strongly down-regulates ERK1/2 signaling and to a lesser extent AKT signaling, as well as causing an endoplasmic reticulum stress response with reduced expression of protective proteins, e.g. MCL−1, and increasing the levels of toxic autophagosomes.Citation6,27 Preliminary findings from our trial combining pemetrexed and sorafenib have been very positive for breast cancer patients in particular and will be reported in the late spring of 2015 at the ASCO meeting (Poklepovic and Dent, unpublished observations). Thus we next determined whether sorafenib, pemetrexed and FTY720 interact to suppress the growth of mammary carcinoma cells in vitro. As we have reported previously, treatment of cells with sorafenib increased the levels of multiple dihydro-ceramide species and also very modestly the levels of sphingosine-1-phosphate; FTY720 as a sphingosine kinase inhibitor blocked the increase in sphingosine-1-phosphate levels (data not shown). Sorafenib at low concentrations well below the plasma C max of 21 μM significantly enhanced [pemetrexed + FTY720] lethality within 24 h in HER2+ and triple negative PTEN mutant breast cancer cell lines ().
Figure 7. FTY720 enhances the anti-tumor effects of [sorafenib + pemetrexed] treatment. BT474 and BT549 cells were incubated with vehicle or sorafenib (SOR, 1−3 μM) as indicated. Cells were also treated as indicated with vehicle or FTY720 (50 nM). Cells were isolated 24h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM).
![Figure 7. FTY720 enhances the anti-tumor effects of [sorafenib + pemetrexed] treatment. BT474 and BT549 cells were incubated with vehicle or sorafenib (SOR, 1−3 μM) as indicated. Cells were also treated as indicated with vehicle or FTY720 (50 nM). Cells were isolated 24h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM).](/cms/asset/632168ec-de39-4672-9b2b-3c057e4d0057/kcbt_a_1026509_f0007_b.gif)
Discussion
The present studies demonstrate that the drug FTY720, in a dose-dependent and apparent greater than additive fashion enhanced tumor cell killing induced by pemetrexed. Cell killing by the FTY720 and pemetrexed drug combination required mitochondrial dysfunction and increased levels of autophagosome formation. The activity of signaling downstream of RAS was initially examined to prospectively define which signal transduction pathways played roles in regulating FTY720 and FTY720 and pemetrexed toxicity. Deletion of mutant active K-RAS D13 resulted in cells that were killed more readily by FTY720 and FTY720 and pemetrexed. Expression of H-RAS V12 suppressed FTY720 and FTY720 and pemetrexed toxicity to a greater extent than K-RAS D13 which correlated with enhanced basal AKT activity in these cells. Using point mutants of H-RAS V12 we found that signaling downstream of PI3K was more protective that signaling through RAF-1 or RAL GDS. We discovered that the drug combination reduced signaling through the PI3K/AKT/p70S6K/mTOR pathway and expression of activated forms of AKT, p70 S6K or mTOR suppressed both the induction of autophagy and the increase in cell killing. As we noted previously for the combination of sorafenib and pemetrexed expression of activated MEK1 or dominant negative MEK1 protected cells from FTY720 and pemetrexed lethality.
Our previous results have demonstrated in colitis/colon cancer models that FTY720 interferes with sphingosine-1-phosphate/sphingosine receptor 1 feed-forward signaling to NFκB and STAT3.Citation17 In our studies we noted that pemetrexed and FTY720 treatment reduced p65 NFκB and STAT3 phosphorylation at activating sites. These data argue that our drug combination causes inactivation of not only the AKT pathway but also of parallel cell growth/survival/invasion pathways including p65 NFκB and STAT3.
In several prior studies using a variety of different drug combinations we have observed ceramide synthase –dependent increases in ceramide levels (see references 39–42, and references therein). In these studies increased ceramide levels played a role in activation of the death receptor CD95 as well as the induction of autophagy. In other studies however we have observed increased ceramide levels that did not associate with CD95 activation but enhanced a toxic form of autophagy.Citation28 The present data demonstrated that pemetrexed and FTY720 interacted to increase dihydro-ceramide levels, and that increased autophagy and cell killing were CerS6/C16 dihydro-ceramide dependent; as judged using the caspase 8 inhibitor c-FLIP-s, enhanced killing was not due to death receptor signaling.
In our prior studies combining pemetrexed and sorafenib we found that the drug combination activated protein phosphatase 2A (PP2A), and inhibition of the de novo ceramide synthase pathway blocked drug-induced ceramide generation, PP2A activation and tumor cell killing.Citation6,27 Recent studies have argued that FTY720 directly binds to the inhibitor of PP2A, I2PP2A/SET, leading to PP2A activation and tumor cell killing through a RIP-1 dependent mechanism.Citation43 However, we found that knock down of RIP-1 did not alter cell killing by pemetrexed and FTY720 (unpublished observations). We found that FTY720 and pemetrexed treatment activated PP2A and inhibition of PP2A using a mutant form of I2PP2A/SET that does not bind ceramide protected cells from FTY720 and pemetrexed toxicity. Moreover, reduction of elevated levels of ceramide and dihydro-ceramide with fumonisin B1 reduced PP2A activity. Hence, our results support the notion that elevation of ceramide and dihydro-ceramide, rather than FTY720 itself, is responsible for the increased activity of PP2A. FTY720 as a single agent has been reported to have anti-tumor effects in multiple systems through modulation of ceramide and S1P levels, and our data argue that its combination with pemetrexed also relies on altering sphingolipid metabolism (e.g., see reference 17, and references therein).
The generation of ceramide has been linked to autophagy, promoting mitophagy.Citation44 Inhibition of de novo ceramide synthesis blocked the induction of autophagy by pemetrexed and sorafenib as well as pemetrexed and FTY720.Citation6 This data would therefore argue that pemetrexed also stimulates cell killing through ceramide-dependent / autophagy-dependent pathways.
Prior pre-clinical studies from our group have demonstrated that pemetrexed and the multi-kinase inhibitor sorafenib interact in a synergistic manner to kill a wide variety of tumor cell types.Citation6,27 In our initial studies we found that sorafenib interacted in a greater than additive fashion with pemetrexed to increase autophagy that lead to activation of the intrinsic apoptosis pathway, and to kill a diverse array of tumor cell types.Citation6 Tumor cell types that displayed high levels of cell killing after combination treatment showed elevated levels of AKT, p70 S6K, and/or phosphorylated mTOR, in addition to class III receptor tyrosine kinases such as platelet-derived growth factor receptor β and VEGF receptors, known in vivo targets of sorafenib. Thus our new data combining FTY720 and pemetrexed appears to follow a similar biologic pattern, and also kills tumor cells. As sorafenib + pemetrexed therapy will shortly be moving into a phase II trial in recurrent mammary carcinoma, the present studies argue that a new phase I trial combining pemetrexed + sorafenib + FTY720 could yield useful information and further enhance anti-tumor effects of the [pemetrexed + sorafenib] diad in patients.
In conclusion, the present studies demonstrate that pemetrexed lethality can be enhanced by a clinically relevant drug which increases intracellular ceramide and dihydro-ceramide levels. It will be of interest to determine in patients receiving pemetrexed and sorafenib whether FTY720 can prolong tumor free survival or reduce the growth of established tumors, as was observed in our prior and the present manuscripts.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Thanks to Mrs. Grizzard for her support to the Dent lab and to Dr. H.F. Young and the Betts family fund for support in the purchase of the Hermes Wiscan instrument. We gratefully acknowledge the assistance of the VCU lipidomics / Metabolomics core in performing our ceramide and S1P analyses, which is supported in part by funding from the NIH-NCI Massey Cancer Center support grant CA016059. PD is the holder of the Universal Inc.. Chair in Signal Transduction Research.
Funding
Support for the present study was funded from PHS grants from the National Institutes of Health [R01-CA141704, R01-CA150214, R01-DK52825, R01-CA61774].
References
- Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009; 69: 5467-74; PMID:19549896; http://dx.doi.org/10.1158/0008-5472.CAN-08-4979
- Rothbart SB, Racanelli AC, Moran RG. Pemetrexed indirectly activates the metabolic kinase AMPK in human carcinomas. Cancer Res. 2010; 70: 10299-10309; PMID:21159649; http://dx.doi.org/10.1158/0008-5472.CAN-10-1873
- Jarmuła A. Antifolate Inhibitors of Thymidylate Synthase as Anticancer Drugs. Mini Rev Med Chem. 2010; 13: 1211-22; http://dx.doi.org/10.2174/13895575110091211
- Fleeman N, Bagust A, McLeod C, Greenhalgh J, Boland A, Dundar Y, Dickson R, Tudur Smith C, Davis H, Green J, et al. Pemetrexed for the first-line treatment of locally advanced or metastatic non-small cell lung cancer. Health Technol Assess. 2010; 14 S1:47-53
- Paglin S, Lee NY, Nakar C, Fitzgerald M, Plotkin J, Deuel B, Hackett N, McMahill M, Sphicas E, Lampen N, et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005; 65:11061-70; PMID:16322256; http://dx.doi.org/10.1158/0008-5472.CAN-05-1083
- Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N, Eullit P, Hubbard N, Tye G, Burow ME, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Research. 2011; 71: 4955-4967; PMID:21622715; http://dx.doi.org/10.1158/0008-5472.CAN-11-0898
- Codogno P, Meijer MA. Authophagy and Signaling: Their role in cell survival and cell death. Cell Death and Differentiation. 2005; 12: 1509-1518; PMID:16247498; http://dx.doi.org/10.1038/sj.cdd.4401751
- Ishdorj G, Li L, Gibson SB. Regulation of autophagy in hematological malignancies: role of reactive oxygen species. Leuk Lymphoma. 2012;53:26-33; PMID:21749305; http://dx.doi.org/10.3109/10428194.2011.604752
- Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523-40; PMID:22187934; http://dx.doi.org/10.1042/BJ20111451
- Kung CP, Budina A, Balaburski G, Bergenstock MK, Murphy M. Autophagy in tumor suppression and cancer therapy.Crit Rev Eukaryot Gene Expr. 2011;21: 71-100; PMID:21967333; http://dx.doi.org/10.1615/CritRevEukarGeneExpr.v21.i1.50
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nature Reviews Drug Discovery 9, 883-897; PMID:21031003; http://dx.doi.org/10.1038/nrd3248
- Brinkmann, V., Pinschewer D, Chiba K, Feng, L. FTY720: a novel transplantation drug that modulates lymphocyte traffic rather than activation. Trends Pharmacol. Sci. 2000; 21, 49-52
- Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003; 554:189-93; http://dx.doi.org/10.1016/S0014-5793(03)01168-2
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002; 277, 21453-21457; PMID:11967257; http://dx.doi.org/10.1074/jbc.C200176200
- Zemann B, Kinzel B, Müller M, Reuschel R, Mechtcheriakova D, Urtz N, Bornancin F, Baumruker T, Billich A. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood 2006; 107, 1454-1458; PMID:16223773; http://dx.doi.org/10.1182/blood-2005-07-2628
- Graler MH, Goetzl EJ. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004; 18, 551-553; PMID:14715694
- Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013; 23:107-20; PMID:23273921; http://dx.doi.org/10.1016/j.ccr.2012.11.013
- Lahiri S, Park H, Laviad EL, Lu X, Bittman R, Futerman AH. Ceramide synthesis is modulated by the sphingosine analog FTY720 via a mixture of uncompetitive and noncompetitive inhibition in an Acyl-CoA chain length-dependent manner. J Biol Chem. 2009; 284:16090-8; PMID:19357080; http://dx.doi.org/10.1074/jbc.M807438200
- Liao A, Hu R, Zhao Q, Li J, Li Y, Yao K, Zhang R, Wang H, Yang W, Liu Z. Autophagy induced by FTY720 promotes apoptosis in U266 cells. Eur J Pharm Sci. 2012; 45:600-5; PMID:22281442; http://dx.doi.org/10.1016/j.ejps.2011.12.014
- Wallington-Beddoe CT, Hewson J, Bradstock KF, Bendall LJ. FTY720 produces caspase-independent cell death of acute lymphoblastic leukemia cells. Autophagy. 2011; 7:707-15; PMID:21460633; http://dx.doi.org/10.4161/auto.7.7.15154
- Zhang N, Qi Y, Wadham C, Wang L, Warren A, Di W, Xia P. FTY720 induces necrotic cell death and autophagy in ovarian cancer cells: a protective role of autophagy. Autophagy. 2010; 6:1157-67; PMID:20935520; http://dx.doi.org/10.4161/auto.6.8.13614
- Cristobal I, Manso R, Rincon R, Carames C, Senin C, Borrero A, Martinez-Useros J, Rodriguez M, Zazo S, Martinez-Aguilera O, et al. PP2A inhibition is a common event in colorectal cancer and its restoration using FTY720 shows promising therapeutic potential. Mol Cancer Ther. 2014 Apr;13(4):938–47; PMID:24448818; http://dx.doi.org/10.1158/1535-7163.MCT-13-0150
- Hung JH, Lu YS, Wang YC, Ma YH, Wang DS, Kulp SK, Muthusamy N, Byrd JC, Cheng AL, Chen CS. FTY720 induces apoptosis in hepatocellular carcinoma cells through activation of protein kinase C delta signaling. Cancer Res. 2008;68:1204-12; PMID:18281497; http://dx.doi.org/10.1158/0008-5472.CAN-07-2621
- Ng KT, Man K, Ho JW, Sun CK, Lee TK, Zhao Y, Lo CM, Poon RT, Fan ST. Marked suppression of tumor growth by FTY720 in a rat liver tumor model: the significance of down-regulation of cell survival Akt pathway. Int J Oncol. 2007;30:375-80; PMID:17203219
- Lee TK, Man K, Ho JW, Sun CK, Ng KT, Wang XH, Wong YC, Ng IO, Xu R, Fan ST. FTY720 induces apoptosis of human hepatoma cell lines through PI3-K-mediated Akt dephosphorylation. Carcinogenesis. 2004; 25:2397-405; PMID:15297371; http://dx.doi.org/10.1093/carcin/bgh250
- Azuma H, Horie S, Muto S, Otsuki Y, Matsumoto K, Morimoto J, Gotoh R, Okuyama A, Suzuki S, Katsuoka Y, et al. Selective cancer cell apoptosis induced by FTY720; evidence for a Bcl−dependent pathway and impairment in ERK activity. Anticancer Res. 2003;23:3183-93; PMID:12926052
- Bareford MD, Hamed HA, Allegood J, Cruickshanks N, Poklepovic A, Park MA, Ogretmen B, Spiegel S, Grant S, Dent P. Sorafenib and pemetrexed toxicity in cancer cells is mediated via SRC-ERK signaling. Cancer Biol Ther. 2012;13:793-803; PMID:22673740; http://dx.doi.org/10.4161/cbt.20562
- Yacoub A, Hamed HA, Allegood J, Mitchell C, Spiegel S, Lesniak MS, Ogretmen B, Dash R, Sarkar D, Broaddus WC, et al. PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res. 2010;70:1120-9; PMID:20103619; http://dx.doi.org/10.1158/0008-5472.CAN-09-4043
- Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, Salisbury JD, Cheng AS, Li L, Abbosh PH, Huang TH, et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006; 66:11954-66; PMID:17178894; http://dx.doi.org/10.1158/0008-5472.CAN-06-1666
- Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, James CD. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7:164-76; PMID:15831234; http://dx.doi.org/10.1215/S1152851704000821
- Carón RW, Yacoub A, Li M, Zhu X, Mitchell C, Hong Y, Hawkins W, Sasazuki T, Shirasawa S, Kozikowski AP, et al. Activated forms of H-RAS and K-RAS differentially regulate membrane association of PI3K, PDK-1, and AKT and the effect of therapeutic kinase inhibitors on cell survival. Mol Cancer Ther. 2005;4:257-70; PMID:15713897
- Carón RW, Yacoub A, Zhu X, Mitchell C, Han SI, Sasazuki T, Shirasawa S, Hagan MP, Grant S, Dent P. H-RAS V12-induced radioresistance in HCT116 colon carcinoma cells is heregulin dependent. Mol Cancer Ther. 2005;4:243-55; PMID:15713896
- Ihle NT, Lemos R Jr, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143-50; PMID:19117997; http://dx.doi.org/10.1158/0008-5472.CAN-07-6656
- Price MM, Oskeritzian CA, Falanga YT, Harikumar KB, Allegood JC, Alvarez SE, Conrad D, Ryan JJ, Milstien S, Spiegel S. A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J Allergy Clin Immunol. 2013; 131:501-11; PMID:22939756; http://dx.doi.org/10.1016/j.jaci.2012.07.014
- Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Yamada A, Zhao R, Milstien S, Zhou H, Spiegel S, et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012;72:726-35; PMID:22298596; http://dx.doi.org/10.1158/0008-5472.CAN-11-2167
- Jiang Q, Rao X, Kim CY, Freiser H, Zhang Q, Jiang Z, Li G. Gamma-tocotrienol induces apoptosis and autophagy in prostate cancer cells by increasing intracellular dihydrosphingosine and dihydroceramide. Int J Cancer. 2012;130:685-93; PMID:21400505; http://dx.doi.org/10.1002/ijc.26054
- Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy. 2007;3:635-7; PMID:17786026; http://dx.doi.org/10.4161/auto.4916
- Li Y, Li S, Qin X, Hou W, Dong H, Yao L, Xiong L. The pleiotropic roles of sphingolipid signaling in autophagy. Cell Death Dis. 2014;5:e1415; http://dx.doi.org/10.1038/cddis.2014.383
- Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23:310-22; PMID:23726895; http://dx.doi.org/10.1016/j.semcancer.2013.05.008
- Hamed HA, Das SK, Sokhi UK, Park MA, Cruickshanks N, Archer K, Ogretmen B, Grant S, Sarkar D, Fisher PB, Dent P. Combining histone deacetylase inhibitors with MDA-7/IL-24 enhances killing of renal carcinoma cells. Cancer Biol Ther. 2013;14:1039-49; PMID:24025359; http://dx.doi.org/10.4161/cbt.26110
- Park MA, Mitchell C, Zhang G, Yacoub A, Allegood J, Häussinger D, Reinehr R, Larner A, Spiegel S, Fisher PB, et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer Res. 2010;70:6313-24; PMID:20631069; http://dx.doi.org/10.1158/0008-5472.CAN-10-0999
- Walker T, Mitchell C, Park MA, Yacoub A, Rahmani M, Häussinger D, Reinehr R, Voelkel-Johnson C, Fisher PB, Grant S, et al. 17-allylamino-17-demethoxygeldanamycin and MEK1/2 inhibitors kill GI tumor cells via Ca2+-dependent suppression of GRP78/BiP and induction of ceramide and reactive oxygen species. Mol Cancer Ther. 2010;9:1378-95; PMID:20442308; http://dx.doi.org/10.1158/1535-7163.MCT-09-1131
- Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ, Selvam SP, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med. 2013;5:105-21; PMID:23180565; http://dx.doi.org/10.1002/emmm.201201283
- Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831-8; PMID:22922758; http://dx.doi.org/10.1038/nchembio.1059