
Abstract
DNA is the prime target of anticancer treatments. DNA damage triggers a series of signaling cascades promoting cellular survival, including DNA repair, cell cycle arrest, and autophagy. The elevated basal and/or stressful levels of both DNA repair and autophagy observed in tumor cells, in contrast to normal cells, have been identified as the most important drug-responsive programs that impact the outcome of anticancer therapy. The exact relationship between DNA repair and autophagy in cancer cells remains unclear. On one hand, autophagy has been shown to regulate some of the DNA repair proteins after DNA damage by maintaining the balance between their synthesis, stabilization, and degradation. One the other hand, some evidence has demonstrated that some DNA repair molecular have a crucial role in the initiation of autophagy. In this review, we mainly discuss the interplay between DNA repair and autophagy in anticancer therapy and expect to enlighten some effective strategies for cancer treatment.
Abbreviations
| DDR | = | DNA damage response |
| ATM | = | ataxia-telangiectasia mutated |
| ATR | = | ATM and Rad3-related |
| IR | = | ionizing radiation |
| PI3K | = | phosphoinositide 3-kinase |
| Chk1 | = | check-point kinase 1 |
| Chk2 | = | check-point kinase 2 |
| SSBs | = | single-strand break |
| DSBs | = | double-strand breaks |
| MRN | = | Mre11-Rad50-Nbs1 |
| γ-H2AX | = | phosphorylated histone |
| MGMT | = | O6 methylguanine –DNA methyltransferase |
| MMR | = | mismatch repair |
| NER | = | nucleotide excision recombination |
| BER | = | base excision repair |
| HR | = | homologous recombination |
| NHEJ | = | non-homologous end joining |
| DNA-PKcs | = | DNA-dependent protein kinase catalytic subunit |
| PARP-1 | = | poly (ADP-ribose) polymerase 1 |
| AMPK | = | adenosine monophosphate-activated protein kinase |
| TSC2 | = | tuberous sclerosis complex 2 |
| mTOR | = | mammalian target of rapamycin |
| ATG5 | = | autophagy-related gene 5 |
| TMZ | = | temozolomide |
| PML | = | promyelocytic leukemia |
| HDAC | = | histone deacetylases |
| OGG1 | = | 8-oxoguannine DNA glycosidase |
Introduction
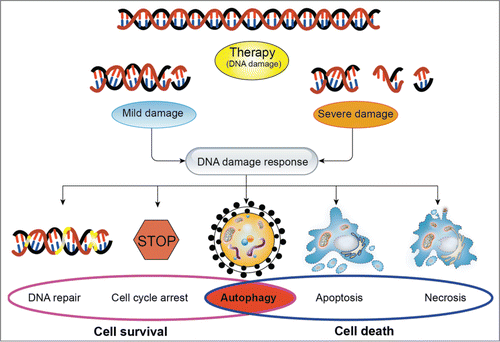
The increasing incidence rate and mortality rate of cancers is becoming one of the biggest problems worldwide.Citation1 There are 3 typical anticancer treatment modalities, including surgical resection, chemotherapy, and radiotherapy.Citation2 Due to the advanced discovery in the majority of patients with tumor, chemotherapy and/or radiotherapy commonly used alternative or supplement scheme are playing an important role in anticancer therapy.Citation3-5 Both of them can interfere with a plethora of cellular functions and may damage a variety of cellular structures. Most importantly, genomic DNA is the pivotal cellular target of chemotherapy and/or radiotherapy. DNA damage can be caused by exogenous and endogenous factors such as chemotherapeutic agents, ionizing radiation (IR), and reactive oxygen species (ROS). In response to DNA damage, cells have evolved a sophisticated signaling transduction network for cellular destiny. Depending on the extent of DNA damage, cellular fate appears entirely differently. When the DNA damage is mild and repairable, cells will trigger a series of signaling cascades to restore the damage, thereby promoting cellular survival. However, when the severe damage is beyond repair, cells will execute the cell death programs such as apoptosis and necrosis to restrain the damaged cell from further expansion.Citation6,7 Although chemotherapy and radiotherapy have been widely used in the treatment of patients with tumor, the resistance of tumor cells to them is becoming a common clinical phenomenon. In general, the failure of DNA targeted anticancer therapy mainly attributes to the survival mechanisms of tumor cells, including the activation of DNA repair pathways, downregulation of pro-apoptotic genes, and overexpression of pro-survival genes.Citation8 Some evidence have suggested that, in contrast to normal cells, the elevated level of the capacity of DNA repair is observed in tumor cells. Furthermore, a number of studies have shown that, in response to DNA damage caused by chemotherapeutic agents and/or ionizing radiation, the activation of DNA-repair signaling pathway is involved in the resistance of tumor cells to these therapies. Furthermore, the efficacy of anticancer treatment can be improved by inhibiting the DNA repair pathway. Thus, targeting the repair mechanisms of DNA damage has been becoming an exciting concept.Citation9-11
Autophagy, a lysosome-dependent degradation process, has been demonstrated to be involved in DNA damage.Citation12-14 In response to DNA damage, autophagy can be induced to degrade aggregated proteins, damaged mitochondria and even ribosome. Increasing evidence has shown that the elevated basal and/or stressful levels of autophagy have been observed in tumor cells.Citation11 Some studies have suggested that autophagy plays a crucial role in the chemoresistance and/or radioresistance of tumor cells. And autophagy inhibition can sensitize tumor cells to apoptosis induced by chemotherapeutic drugs and/or ionizing radiation, thereby enhancing the efficacy.Citation15-17 Because of the characteristics of DNA repair and autophagy in tumor cells, they have been identified as the most important drug-responsive programs that impact the outcome of anticancer therapy. However, the relationship between DNA repair and autophagy remains unclear. Therefore, it is of great significance to exploit the mutual relationship between autophagy and DNA repair. This will be relevant to current clinical efforts to exploit novel therapeutic strategy for enhancing the response of malignancies to chemotherapy and/or radiotherapy. In this review, we mainly focus on the possible relationship between DNA repair and autophagy and expect to improve the efficacy of anticancer treatments.
DNA, DNA damage and DNA damage response
DNA is the target, directly or indirectly, for a large number of our cancer chemotherapeutics and irradiation.Citation18-20 DNA damage is of obvious importance in determining the efficacy of these therapies. In response to DNA damage, cells trigger a sophisticated signal transduction network, known as DNA damage response (DDR), including the detection of the DNA damage and activation of transcription factors involved in DNA damage repair, cell cycle arrest, and cell death.Citation7,21,22 It is well known that cellular fate in response to DNA damage has been shown to be determined by the ability of DNA repair to restore DNA integrity which if unrepaired would lead to the activation of cell death programs.Citation11,22 Therefore, avid DNA repair could be a detriment to successful anticancer therapy.Citation10 There was once a time when the complexity and redundancy of DNA repair made its components less attractive targets for cancer drug development. Fortunately, our growing understanding of mutational complexity of most solid tumors make it clear that DNA will remain important among the therapeutic targets for the foreseeable future.
In response to DNA damage, the first signal transduction wave is governed by rapid activation of checkpoint kinases which are best studied ataxia-telangiectasia mutated (ATM), and ATM and Rad3-related (ATR).Citation23-25 Although structurally related to the phosphoinositide 3-kinase (PI3K) family members, ATM and ATR, which are central components of the DNA damage response, are protein kinases.Citation26,27 ATM plays an important role in the response to DNA double strand breakage caused by irradiation, controlling the initial phosphorylation of several key proteins such as check-point kinase 2 (Chk2), p53, and BRCA1 in response to DNA damage.Citation28,29 ATR is likely to be regulated in some fashion because it controls the late phosphorylation of p53 in response to irradiation and the phosphorylation of check-point kinase 1 (Chk1).Citation30,31 More importantly, ATM and ATR control different downstream DNA damage responses by controlling Chk1 and Chk2 kinases: 2 serine/threonine kinases that are structurally unrelated but that share some overlapping substrate specificity.Citation32 As we known, DNA double-strand breaks (DSBs) potently caused by ionizing radiation, are the highest lethal DNA lesions.Citation33,34 Following DNA double-strand breaks, the activation of ATM is induced and subsequently becomes recruited as an inactive dimmer to the DNA lesion by the Mre11-Rad50-Nbs1 (MRN) complex formation.Citation35,36 The MRN complex and ATM bind with each other through multiple protein-protein interactions, and locate at the damaged DNA foci marked by phosphorylated histone (γ-H2AX) to regulate numerous downstream mediators for coordinating the DNA damage response.Citation37-39 By contrast, the ATR protein kinase is recruited to the DNA lesion by the ATR-interacting protein in response to DNA single-strand break (SSBs).Citation40,41 After the phosphorylation of ATM and/or ATR, checkpoint kinases including Chk1 and Chk2 are activated to transmit the damage signaling to the effectors molecules such as the tumor suppressor protein p53. Depending on the cellular context and the extent of DNA damage, p53 induces transcription of different sets of target genes, leading to apoptosis, cell cycle arrest or cellular senescence ().Citation42-45
Figure 1. The effects of the DNA damage response on cellular fate in response to DNA damage. In response to DNA damage, cells trigger a series of signaling cascades. Depending on the extent of DNA damage, cellular fate will be determined by DNA damage response. When the DNA damage is mild and repairable, cells will activate the signaling pathways of promoting cell survival, including DNA repair, cell cycle arrest, and autophagy. However, when the severe damage is beyond repair, cells will execute the cell death programs such as autophagy, apoptosis and necrosis to restrain the damaged cell from further expansion. Notably, autophagy plays a dual role in determining cell fate. In general, depending on the extent of DNA damage and the tumor type, the type of stimuli, autophagy could be a survival mechanism or induce programmed cell death.
Targeting of DNA repair as anticancer therapy
In response to DNA damage, cellular fate has been shown to be determined by the capacity of DNA-repair pathways to restore DNA integrity, which if unrepaired would lead to the activation of cell death programs. The impairment of DNA repair systems may predispose cells to genomic instability, which is considered a hallmark of cancer.Citation46,47 Depending on the specific type of DNA damage and the phase of the cell cycle, different repair mechanisms are potentiated to restore the damaged DNACitation11,48 In general, the unmodified complementary strand of the DNA or the sister chromatid is selected as a template to restore the original strand. Without accessing to a template, cells use an error-prone repair mechanism known as translesion synthesis as the last resort.Citation49
In general, DNA repair systems are divided into direct and indirect ones. The direct protein-mediated incorporate repair during replication, catalyzed by the main DNA polymerase, the O6 methylguanine–DNA methyltransferase (MGMT) enzyme repair for damages in O6 position of guanine caused by endogenous and alkylating agents and finally the repair of pyrimidine dimmers.Citation50,51 There are several types of DNA indirect repair systems,Citation52 including (1) the mismatch repair (MMR) pathway, (2) the nucleotide excision recombination (NER) pathway, (3) the base excision repair (BER) pathway, (4) the homologous recombination (HR) pathway, and (5) the non-homologous end joining (NHEJ) pathway. In brief, MMR recognizes and repairs small loops in DNA either by base-base mismatches or by insertion/deletion loops.Citation53,54 MMR system is considered to be critical for maintaining the overall integrity of the genome, whose defect is associated with an increased risk of cancer. In NER, XPC enzyme spots the damage and then DNA is unfolding from both sides in the site of damage because of the presence of helicases XPA, XPG, and TFIIH. Finally, the removal of the damaged DNA follows the synthesis of a new strand.Citation55,56 BER is of great importance for repairing the small base lesions induced by oxidation and alkylation damage. Apurinic/apyrimidinic endonuclease 1 (APE1) recognizes the abasic site and hydrolyzes the phosphodiester bond. At a last step, the repair is catalyzed by DNA polymerase β and DNA is joined by DNA ligase.Citation57,58 The majority of NER lesions involve exogenous sources, whereas BER is mostly, but not exclusively concerned with damage of endogenous origin. Both systems repair damages affecting just one DNA strand (SSBs). Most problematic are DSBs, as both DNA strands are affected. DSBs are primarily repaired by 2 different pathways: HR that is a template-guided, error-free pathway predominantly operating in the S and G2 phases of the cell cycle and NHEJ that tends to be error prone and is operational in all phases of the cell cycle.Citation39,59,60
More and more evidence show that the DNA-repair signaling pathway is involved in cellular resistance to anticancer therapy. The MRN is essential in recognizing DNA double strand breaks caused by ionizing radiation and acts as an important part of the DNA repair machinery, which also recognizes viral genetic materials and initiates a cellular response that results in the concatemerization and inactivation of nonself DNA.Citation36,61 The inhibition of MRN complex prevents the repair of radiation induced double strand breaks and their constant accumulation leads to the increased cell death.Citation62 ATM, a sensor of DNA damage, is required for the phosphorylation of p53 and DNA-dependent protein kinase catalytic subunit (DNA-PKcs), both of which are key regulators in DNA DSBs repair.Citation63,64 It is reported that ATM inhibition sensitizes cells to apoptosis caused by chemotherapeutic drugs or ionizing radiation.Citation65,66 Many studies have reported that HR plays an important role in cell replication.Citation67,68 Because of the generation of DSBs during replication that is not properly repaired, cells with impaired HR exhibit cell proliferation defects. In addition, in support of this, the essential role of HR in replication is illustrated by embryonic lethality of mice with knockout of genes required for HR, including the Rad51 recombinase or the breast cancer susceptibility genes BRCA1 or BRCA2.Citation69 The inhibition of DNA-PKcs, a DNA-repair protein involved in NHEJ pathways of DNA DSBs, is shown to sensitize human glioma cells to radiation-induced apoptosis.Citation70,71 It has been reported that poly (ADP-ribose) polymerase 1 (PARP-1) participates in many molecular and cellular processes including DNA damage detection, DNA repair, and carcinogenesis.Citation72 PARP-1 activation is shown to contribute to cell survival during DNA damage.Citation73 Additionally, the involvement of PARP-1 activation in cell survival has also been confirmed in human breast cancer tissue.Citation74
Presently, it seems like that, in contrast to conventional chemotherapy or radiotherapy regimens, the concurrent strategy using DNA repair inhibitors with radical chemoradiotherapy is more effective and conducive to the success of anticancer therapy. However, caution must be given when entertaining the addition of a DNA repair inhibitor with DNA-damaging agents. In general, there may be a difficulty in using the combined regimen if the chemotherapeutic agents also systemic DNA damage in normal tissues. Furthermore, the benefit of using DNA repair inhibitors could be problematic since it may suppress the repair of late-acting normal tissues or lead to the accumulation of DNA breaks in normal tissues, finally resulting in the increased possibility of genetic instability and second malignancies. Thus, further studies are needed to provide novel cancer therapies without undue harm.
Functions of autophagy
Autophagy, a ubiquitous highly conserved pathway in eukaryotic cells, has been shown to be induced in adverse conditions including nutrient starvation,Citation75 hypoxia,Citation76 oxidative stress,Citation77 DNA damageCitation7 and other stress conditions. It is widely accepted that autophagy is normally considered a cellular homeostatic mechanism for quality control by promoting the degradation and eliminated of damaged organelles, long-lived proteins, and other damaged cellular constituents. In addition to recycling cellular components, autophagy plays crucial roles in a variety of physiologic and pathologic processes, including adaptive response to starvation, anti-aging, antigen presentation, development of placenta, elimination of intracellular microbes, genomic stabilityCitation78 and tumorigenesis, whereas dysregulation of autophagy is involved in human diseases including neurodegenerative disorders, cardiovascular, and cancer.Citation79,80
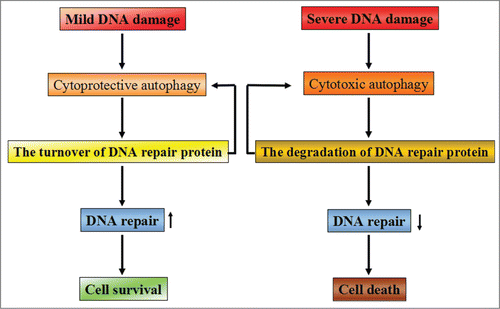
Recent studies suggest that autophagy plays a dual role in determining cell fate, which means it could be a survival mechanism or induce programmed cell death under different cellular stresses. In some conditions, the activation of autophagy provides cellular protection presumably by eliminating dysfunctional organelles and proteins and maintaining energy balance. This pro-survival function of autophagy could contribute to tumor cell survival and growth in the face of adverse conditions, such as hypoxia and nutrient starvation. Apparently, a form of protective autophagy is also induced in response to irradiation as well as cancer chemotherapeutic drugs, where pharmacological or genetic inhibition of autophagy has been shown to result in enhanced sensitivity of tumor cells to various therapeutic modalities. To a large extent, the cytoprotective function of autophagy appears to be a consequence of the fact that autophagy induction inhibits apoptosis through cross-talk between autophagy and apoptosis regulatory pathways. On the other hand, autophagy is also considered as a type of cell death program.Citation81 It is well known that there are 3 types of cell death. Necrosis, a type of unprogrammed cell death, evokes cellular inflammation reaction by immunological activation and subsequently leads to cell death in neighboring cells. Apoptosis, which is programmed cell death type I, is characterized by the formation of chromatin condensation, DNA fragmentation, and apoptotic bodies. Cellular death by over-activated autophagy has been shown as programmed cell death type II. Upregulation of autophagy has been shown to lead to cell death in order to eliminate damaged or abnormal cells. It would appear reasonable to predict that cytotoxic autophagy should be a consequence of higher levels or a more prolonged process of cellular self-digestion than for cytoprotective autophagy. However, there is no explicit experimental evidence suggests that autophagy which is cytotoxic is uniquely different from autophagy which is cytoprotective. In general, depending on the type of the tumor and stimuli and the extent of DNA damage, autophagy can have both cytoprotective and cytotoxic functions ().
DNA damage and autophagy
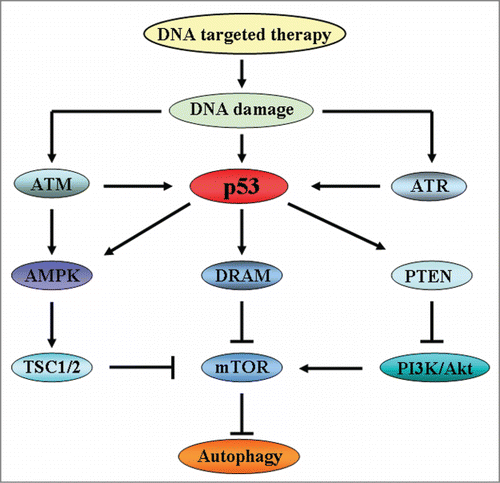
As we known, many different signaling pathways participate in the regulation of autophagy modulated. The mammalian target of rapamycin (mTOR), as the most extensively studied regulator of autophagy, is regulated by the class I phosphoinositide 3-kinase/Akt (PI3K/Akt) that down-regulates autophagy.Citation82,83 In addition, adenosine monophosphate-activated protein kinase (AMPK), which can be activated by its upstream effectors LKB1, TAK1, and CaMKKβ,Citation84 negatively regulates mTOR by either directly inhibiting mTOR or activating tuberous sclerosis 2 (TSC2) proteins, which is an upstream factor of mTOR, thereby activates autophagy.Citation85
Recently, some researchers suggested that autophagy could be induced by DNA damage.Citation12,13 ATM, as one of the sensors at the forefront of the DNA damage response, in response to DNA damage, is activated and recruited as an inactive dimmer to the DNA lesion by a sensor complex comprising the proteins Mre11, Rad50, and NBS1, thereby contributing to ATM autophosphorylation for its fully activation to promote the repair of damaged DNA in the nucleus. Recent studies have reported that exposing cells to genotoxic stress induces ATM activation causing repression of mTOR signaling through AMPK pathway and finally activates autophagy for cell survival or cell death. Depending on the cellular context and the extent of DNA damage-which determines whether or not damage is reparable-autophagy can trigger different cellular fate. The protein p53, one of the central regulators of apoptosis induced by DNA damage, plays a bidirectional role in the regulation of autophagy depending on its subcellular localization. On one hand, p53 can either activate AMPK, which inactivates mTOR thereby inducing autophagy, or promote the transcriptional expression of damage-regulated autophagy modulator (DRAM), which is a lysosomal protein that facilitates the autophagic process.Citation86,87 Additionally, p53 activation also results in the expression of the phosphatidylinositol phosphate PTEN, which is an inhibitor of PI3K/Akt signaling pathway, to contribute to the suppression of mTOR thereby activating autophagy. On the other hand, in contrast to nuclear p53, cytoplasmic p53 functions as a repressor of autophagy.Citation88 Therefore, depending on the cellular context and the extent of DNA damage, autophagy may play a cytoprotective or cytotoxic role in the determination cellular fate ().
Figure 2. The signaling pathways of autophagy induced by DNA damage. In response to DNA damage, ATM regulates autophagy through signaling of AMPK and TSC1/2. Meanwhile, ATM and ATR also activate p53 to induce the activation of autophagy. p53 plays an important role in autophagy regulation. On one hand, p53 positively activates AMPK and DRAM to inhibit mTOR which is an inhibitor of autophagy, thereby inducing autophagy activation. One the other hand, p53 activates PTEN, a repressor of PI3K/Akt, to inhibit mTOR and induces autophagy initiation.
Targeting of autophagy as anticancer therapy
Although the prosurvival or anticancer role of autophagy in anticancer therapy is still controversial, the studies in vitro and in vivo are more likely to support the idea that autophagy contributes to the cancer cells' resistance to anticancer drugs. Recently, several studies have shown that resistance to anticancer therapies can be augmented through the elevated expression of autophagy in different tumor cell lines.Citation89,90 Furthermore, increasing evidence shows that inhibition of autophagy in cells treated with DNA–damaging agents leads to the increased cell death. Thus, autophagy is becoming a potential target for cancer therapy. More importantly, we should be careful when autophagic inhibitors augments the cytotoxicity in combination with anticancer drugs, since it may vary according to the cellular context such as cell type, the extent of DNA damage, and other circumstances.
The Interaction Between Autophagy and DNA Repair
Presently, both autophagy and DNA repair are thought to be the most closely associated with the resistance of anticancer therapies including chemotherapy and radiation therapy. Increasing evidence has shown that, in contrast to normal cells, the elevated basal and/or stressful levels of autophagy and DNA repair has been observed in tumor cells. Recent studies have demonstrated that autophagy and DNA repair have multiple and often overlapping and complementary functions in cancer, both in terms of affecting tumor development and in modulating the response to chemotherapy and/or radiation. However, how autophagy and the DNA-repair pathways interact with each other remains unclear. Thus, exploring the relationship between autophagy and DNA repair may allow us to develop a promising therapeutic strategy to enhance the present efficacy of anticancer therapy and improve clinical outcomes ().
Figure 3. The possible relationship between DNA repair and autophagy. Depending on the extent of DNA damage, cellular fate appears entirely differently by activating different signaling pathways. When DNA damage is mild and repairable, cytoprotective autophagy will be activated to promote cellular survival via enhancing the turnover of DNA repair proteins. Meanwhile, some of the DNA repair proteins are able to promote autophagy activation, thereby contributing to cell survival. However, when severe DNA damage is beyond repair, cells will activate cytotoxic autophagy to execute the cell death program by accelerating the degradation of DNA repair proteins. The decreased capacity of DNA repair will aggravate the induction of cytotoxic autophagy and finally result in cell death.
Autophagy as the regulator of DNA repair
The activation of autophagy was shown to precede phosphorylation of ATM and p53 and activation of DNA repair proteins in response to DNA-damaging agents, revealing an intricate pathway in which autophagy acts upstream, not just as a consequence, of DDR activation. A recent study observed increased radiosensitivity of autophagy-related gene 5 (ATG5) or BCN1-depleted cells and reduced ATG5−/− tumor growth after irradiation, suggesting that autophagy could affect trafficking of damaged DNA.Citation91 Furthermore, another study also uncovered that the damaged nuclear DNA caused by DNA-damaging agents is exported to the outside of nuclear and ultimately cleared by autophagy.Citation92 And massive damaged DNA was found to accumulate in autophagy-deficient cells. Thus, these findings support that autophagy activation can attenuate the extent of the DNA damage induced by endogenous and/or exogenous factors, thereby reducing the cytotoxicity of the damaged DNA and finally contributing to cell survival.
So how does autophagy influence DNA damage repair? To date, the possible mechanisms are involved in the following aspects. Firstly, autophagy may provide metabolic precursors for the generation of ATP as well as regulate the supply of dNTPs for DNA synthesis during DNA repair.Citation93,94 As we known, many steps in the process of DNA repair, including DNA unwinding by helicases during NER,Citation95 ATP-dependent chromatin remodeling complexes in DSB repairCitation96 and PARP activity,Citation97 are ATP dependent, and the depletion of ATP results in the failure of DNA repair. Katayama M et al has shown that, in temozolomide (TMZ)-treated glioma cells, the inhibition of autophagy suppresses the generation of ATP and finally increased mitotic catastrophe. The mitotic catastrophe is prevented by adding pyruvate to rescue ATP levels, indicating that autophagy-sustained ATP generation may be employed in some mechanisms that contribute to genomic integrity including DNA repair processes.Citation93 A recent study shows that autophagy, through the degradation of ribonu-cleotide reductase (Rnr) subunits, may fight mutagenesis by keeping a balanced dNTP pool, which is essential for DNA replication and repair.Citation78,98 Secondly, besides ATP generation and dNTP recycling, autophagy has been shown to be involved in the turnover of some of the DNA repair proteins participated in the regulation/processing of genomic stability. Rad51, a key protein of homologous recombination, has been shown to play a vital role in the repair of DNA double-strand breaks (DSBs) induced by radiation or some DNA-damaging agents. Overexpression of Rad51 can influence the chemoradiotherapy treatment outcome as well as potentiate radioresistance in tumor cells. Ning mo et al. have demonstrated that inhibition of autophagy with Atg5-targeted shRNA or an autophagy inhibitor both resulted in decreased expression of Rad51, thereby enhancing the susceptibility of nasopharyngeal carcinoma (NPC) cells to radiation.Citation99 Heekyong Bae suggested that suppression of autophagy by inactivation of FIP200 (200 kDa FAK-family interacting protein) impaired the capacity of DNA damage repair and increased cell death induced by ionizing radiation and anticancer agents such as etoposide and camptothecin in mouse embryonic fibroblasts (MEF).Citation100 A recent report showed that p62 is required for polyubiquitinated protein interaction with promyelocytic leukemia (PML) nuclear bodies involved in multiple DNA damage repair proteins such as BLM/WRN DNA helicases, the Mre11 complex, or TopBP1.Citation101 In this scenario, increased p62 expression caused by deficient autophagy due to FIP200 deletion may be responsible for the defective DNA damage repair and consequent increase in cell death following DNA damage-inducing agents.
Conversely, some studies have shown that autophagy can also promote degradation of DNA damage repair proteins, thus facilitating cell death. A recent report showed that treatment with valproic acid (VPA), an histone deacetylases (HDAC) inhibitor, impaired the activation of Rad53 in response to DSBs in budding yeast.Citation102 In the VPA-treated cells, the first factor recruited to DSB sites, remained bound to the DSB site, accompanied by reduced levels of Sae2, which is responsible for removing Mre11 from the DSB region, a step required for the process of lesion repair. In this scenario, pharmacological or genetic disruption of autophagy increased the acetylated levels of Sae2, whereas rapamycin, which activates autophagy through mTOR inhibition, decreased it, confirming that autophagy induced by VPA could impair DSB processing through degradation of acetylated Sae2. One study conducted in breast cancer cells showed that rapamycin (an mTOR inhibitor) promoted the inhibition of repair of ionizing radiation-induced DSBs by the significant suppression of HR and NHEJ.Citation103 In the rapamycin-treated cells, the decreased DSB repair by HR is correlated with the impaired recruitment of BRCA1 and Rad51to DNA repair foci. Since autophagy has been shown to promote degradation of acetylated Sae2 in VPA-treated yeast cells, thereby influencing the dynamics of DNA DSB repair in an intricate manner, it is possible that the activation of autophagy could contribute to the perseverance of DNA damage and further enhancement of apoptotic signaling in mammalian cells by controlling turnover of certain DNA repair-related enzymes. In this context, in response to nutrient starvation, autophagy activation was shown to degrade the base-excision repair enzyme 8-oxoguannine DNA glycosidase (OGG1) in cardiomyocytes.Citation104
Taken together, these studies indicate that autophagy can influence the resolution of DNA lesions and plays complex roles in the context of DNA damage and repair. Interestingly, it remains unclear when autophagy contributes to DNA repair and when autophagy inhibits DNA repair. In general, in response to reparable DNA damage, autophagy may promote the DNA damage repair by above mentioned approaches. Alternatively, when the DNA damage is beyond repair, autophagy may lead to the initiation of cell death programs including apoptosis and autophagic cell death. Further studies are needed to reveal this exciting yet obscure role of autophagy in DNA repair process.
DNA repair enzymes as the regulator of autophagy
Several studies have also suggested that the important role of some of the DNA-repair proteins in the activation of autophagy after genotoxic stresses. In response to a genotoxic methylating agent, 6-thioguanine, DNA mismatch repair induces autophagy and thus enhances the survival of human colorectal and endometrial cancer cells.Citation105 Interestingly, the inhibition of DNA-PKcs, a key DNA-repair protein, was shown to sensitize human malignant glioma cells to radiation by inducing autophagy, which was considered a form of programmed cell death.Citation106 The degradation of the MRN complex by oncolytic adenoviruses (Ads) proteins results in the inhibition of DSBs repair, which subsequently activates autophagy and leads to the increased cell killing in prostate cancer cell lines.Citation107 MGMT, an important enzyme responsible for repairing monoadducts caused by alkylating agents in DNA, is the major cause of resistance in the treatments of glioma. Inhibiting DNA repair with O6-benzylguanine, an inhibitor of MGMT, leads to the over-activation of autophagy and the increased autophagic cell death in resistant glioma cells treated with temozolomide.Citation108 In general, only in these conditions where DNA repair and apoptosis are inhibited or defective, autophagy seems to play an alternative mechanism of cell death for clearing these damaged organelles and proteins. However, the molecular mechanism of DNA repair influence autophagy is still not well understood. Apparently, further studies are needed to exploit the role of DNA repair proteins in autophagy activation in response to anticancer therapy.
Conclusions and Perspectives
Some evidences have demonstrated that both autophagy and DNA repair are associated with the efficacy of chemotherapy and/or radiotherapy and have a crucial function in resistance to anticancer treatments. However, autophagy inhibition or the blockage of DNA repair fails to obtain the success of anticancer therapy. Excitingly, the use of the therapeutic regimen which means autophagy inhibition combining DNA repair inhibition and chemotherapeutic agents is considered a novel therapeutic strategy. Recently, a study conducted by Liu et al. provides important insights into the consequence of autophagy inhibition on DNA repair.Citation109 They reported that in the absence of autophagy, inhibition of DNA repair resulted in persistence of genomic lesions and rapid cell death. Above all, it is significant to explore the interplay between autophagy and DNA repair to achieve greater efficacy in cancer treatment. And further studies are needed before we can identify potential therapeutic targets that integrate the process of autophagy and DNA repair in anticancer therapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855
- Kahlem P, Dorken B, Schmitt CA. Cellular senescence in cancer treatment: friend or foe? J Clin Invest 2004; 113:169-74; PMID:14722606
- Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol 2005; 205:275-92; PMID:15641020
- Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract 2005; 14 1:35-48; PMID:16103712
- Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006; 5:219-34; PMID:16518375
- Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001; 4:303-13; PMID:11991684
- Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI, Franco R. DNA damage and autophagy. Mutat Res 2011; 711:158-66; PMID:21419786
- Yoon JH, Ahn SG, Lee BH, Jung SH, Oh SH. Role of autophagy in chemoresistance: regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem Pharmacol 2012; 83:747-57; PMID:22226932
- Bates SE. DNA repair: a reinvigorated therapeutic target. Clin Cancer Res 2010; 16:4510; PMID:20823139
- Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer 2012; 12:587-98; PMID:22918414
- Friedberg EC. DNA damage and repair. Nature 2003; 421:436-40; PMID:12540918
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-21; PMID:11099404
- Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; 27:4860-4; PMID:18408756
- Botrugno OA, Robert T, Vanoli F, Foiani M, Minucci S. Molecular pathways: old drugs define new pathways: non-histone acetylation at the crossroads of the DNA damage response and autophagy. Clin Cancer Res 2012; 18:2436-42; PMID:22512979
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14:500-10; PMID:16990848
- Huang S, Sinicrope FA. Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy 2010; 6:256-69; PMID:20104024; http://dx.doi.org/10.4161/auto.6.2.11124
- Chen YS, Song HX, Lu Y, Li X, Chen T, Zhang Y, Xue JX, Liu H, Kan B, Yang G, et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis Esophagus 2011; 24:437-43; PMID:21166739; http://dx.doi.org/10.1111/j.1442-2050.2010.01156.x
- Ren J, Singh BN, Huang Q, Li Z, Gao Y, Mishra P, Hwa YL, Li J, Dowdy SC, Jiang SW. DNA hypermethylation as a chemotherapy target. Cell Signal 2011; 23:1082-93; PMID:21345368; http://dx.doi.org/10.1016/j.cellsig.2011.02.003
- Palchaudhuri R, Hergenrother PJ. DNA as a target for anticancer compounds: methods to determine the mode of binding and the mechanism of action. Curr Opin Biotechnol 2007; 18:497-503; PMID:17988854; http://dx.doi.org/10.1016/j.copbio.2007.09.006
- Chen Z, Peng Y, Chen M, Chen X, Zhang G. DNA as a target for anticancer compounds screening directly by resonance light scattering technique. Analyst 2010; 135:2653-60; PMID:20714524; http://dx.doi.org/10.1039/c0an00386g
- Woods D, Turchi JJ. Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol Ther 2013; 14:379-89; PMID:23380594; http://dx.doi.org/10.4161/cbt.23761
- Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002; 297:547-51; PMID:12142523; http://dx.doi.org/10.1126/science.1074740
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3:155-68; PMID:12612651; http://dx.doi.org/10.1038/nrc1011
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol 2000; 1:179-86; PMID:11252893; http://dx.doi.org/10.1038/35043058
- Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013; 5; a012716; PMID:24003211; http://dx.doi.org/10.1101/cshperspect.a012716
- Jiang M, Zhao L, Gamez M, Imperiale MJ. Roles of ATM and ATR-mediated DNA damage responses during lytic BK polyomavirus infection. PLoS Pathog 2012; 8:e1002898; PMID:22952448; http://dx.doi.org/10.1371/journal.ppat.1002898
- Yan S, Sorrell M, Berman Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell Mol Life Sci 2014; 71:3951-67; PMID:24947324; http://dx.doi.org/10.1007/s00018-014-1666-4
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433-9; PMID:11100718; http://dx.doi.org/10.1038/35044005
- Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol 2004; 24:708-18; PMID:14701743; http://dx.doi.org/10.1128/MCB.24.2.708-718.2004
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448-59; PMID:10859164; http://dx.doi.org/10.1101/gad.840500
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21:4129-39; PMID:11390642; http://dx.doi.org/10.1128/MCB.21.13.4129-4139.2001
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science 1996; 274:1664-72; PMID:8939848; http://dx.doi.org/10.1126/science.274.5293.1664
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell 2007; 28:739-45; PMID:18082599; http://dx.doi.org/10.1016/j.molcel.2007.11.015
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nature08467
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005; 308:551-4; PMID:15790808; http://dx.doi.org/10.1126/science.1108297
- Assenmacher N, Hopfner KP. MRE11/RAD50/NBS1: complex activities. Chromosoma 2004; 113:157-66; PMID:15309560; http://dx.doi.org/10.1007/s00412-004-0306-4
- Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 2008; 36:5678-94; PMID:18772227; http://dx.doi.org/10.1093/nar/gkn550
- Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J 2000; 19:463-71; PMID:10654944; http://dx.doi.org/10.1093/emboj/19.3.463
- Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002; 23:687-96; PMID:12016139; http://dx.doi.org/10.1093/carcin/23.5.687
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542-8; PMID:12791985; http://dx.doi.org/10.1126/science.1083430
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science 2001; 294:1713-6; PMID:11721054; http://dx.doi.org/10.1126/science.1065521
- Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer 2009; 9:714-23; PMID:19730431
- Nelson WG, Kastan MB. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol 1994; 14:1815-23; PMID:8114714
- Meek DW. The p53 response to DNA damage. DNA Repair (Amst) 2004; 3:1049-56; PMID:15279792; http://dx.doi.org/10.1016/j.dnarep.2004.03.027
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2:594-604; PMID:12154352; http://dx.doi.org/10.1038/nrc864
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11:220-8; PMID:20177397; http://dx.doi.org/10.1038/nrm2858
- Kelley MR, Logsdon D, Fishel ML. Targeting DNA repair pathways for cancer treatment: what's new? Future Oncol 2014; 10:1215-37; PMID:24947262; http://dx.doi.org/10.2217/fon.14.60
- Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, Yoshimura M, Orelli BJ, Bishop DK, Hirano S, Ohzeki M, et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res 2005; 65:11704-11; PMID:16357182; http://dx.doi.org/10.1158/0008-5472.CAN-05-1214
- Michailidi C, Papavassiliou AG, Troungos C. DNA repair mechanisms in colorectal carcinogenesis. Curr Mol Med 2012; 12:237-46; PMID:22082485; http://dx.doi.org/10.2174/156652412799218859
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366-74; PMID:11357144; http://dx.doi.org/10.1038/35077232
- Senthong P, Millington CL, Wilkinson OJ, Marriott AS, Watson AJ, Reamtong O, Eyers CE, Williams DM, Margison GP, Povey AC. The nitrosated bile acid DNA lesion O6-carboxymethylguanine is a substrate for the human DNA repair protein O6-methylguanine-DNA methyltransferase. Nucleic Acids Res 2013; 41:3047-55; PMID:23335782; http://dx.doi.org/10.1093/nar/gks1476
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39-85; PMID:15189136; http://dx.doi.org/10.1146/annurev.biochem.73.011303.073723
- Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annu Rev Genet 2000; 34:359-99; PMID:11092832; http://dx.doi.org/10.1146/annurev.genet.34.1.359
- Hsieh P. Molecular mechanisms of DNA mismatch repair. Mutat Res 2001; 486:71-87; PMID:11425513; http://dx.doi.org/10.1016/S0921-8777(01)00088-X
- Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer 2001; 1:22-33; PMID:11900249; http://dx.doi.org/10.1038/35094000
- Truglio JJ, Croteau DL, Van Houten B, Kisker C. Prokaryotic nucleotide excision repair: the UvrABC system. Chem Rev 2006; 106:233-52; PMID:16464004; http://dx.doi.org/10.1021/cr040471u
- Lindahl T. Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat Res 2000; 462:129-35; PMID:10767624; http://dx.doi.org/10.1016/S1383-5742(00)00024-7
- Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett 2000; 476:73-7; PMID:10878254; http://dx.doi.org/10.1016/S0014-5793(00)01674-4
- Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res 2002; 509:49-78; PMID:12427531; http://dx.doi.org/10.1016/S0027-5107(02)00224-5
- Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 2003; 4:712-20; PMID:14506474; http://dx.doi.org/10.1038/nrm1202
- Czornak K, Chughtai S, Chrzanowska KH. Mystery of DNA repair: the role of the MRN complex and ATM kinase in DNA damage repair. J Appl Genet 2008; 49:383-96; PMID:19029686; http://dx.doi.org/10.1007/BF03195638
- Kanaar R, Wyman C. DNA repair by the MRN complex: break it to make it. Cell 2008; 135:14-6; PMID:18854148; http://dx.doi.org/10.1016/j.cell.2008.09.027
- Flassig RJ, Maubach G, Tager C, Sundmacher K, Naumann M. Experimental design, validation and computational modeling uncover DNA damage sensing by DNA-PK and ATM. Mol Biosyst 2014; 10:1978-86; PMID:24833308; http://dx.doi.org/10.1039/c4mb00093e
- Kodama M, Otsubo C, Hirota T, Yokota J, Enari M, Taya Y. Requirement of ATM for rapid p53 phosphorylation at Ser46 without Ser/Thr-Gln sequences. Mol Cell Biol 2010; 30:1620-33; PMID:20123963; http://dx.doi.org/10.1128/MCB.00810-09
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998; 281:1677-9; PMID:9733515; http://dx.doi.org/10.1126/science.281.5383.1677
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 1998; 280:1089-91; PMID:9582124; http://dx.doi.org/10.1126/science.280.5366.1089
- Klein HL, Kreuzer KN. Replication, recombination, and repair: going for the gold. Mol Cell 2002; 9:471-80; PMID:11931756; http://dx.doi.org/10.1016/S1097-2765(02)00493-8
- Costes A, Lambert SA. Homologous recombination as a replication fork escort: fork-protection and recovery. Biomolecules 2012; 3:39-71; PMID:24970156; http://dx.doi.org/10.3390/biom3010039
- Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003; 22:5784-91; PMID:12947386; http://dx.doi.org/10.1038/sj.onc.1206678
- Zhuang W, Li B, Long L, Chen L, Huang Q, Liang ZQ. Knockdown of the DNA-dependent protein kinase catalytic subunit radiosensitizes glioma-initiating cells by inducing autophagy. Brain Res 2011; 1371:7-15; PMID:21108935; http://dx.doi.org/10.1016/j.brainres.2010.11.044
- Callen E, Jankovic M, Wong N, Zha S, Chen HT, Difilippantonio S, Di Virgilio M, Heidkamp G, Alt FW, Nussenzweig A, et al. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol Cell 2009; 34:285-97; PMID:19450527; http://dx.doi.org/10.1016/j.molcel.2009.04.025
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 1999; 342 (Pt 2):249-68; PMID:10455009; http://dx.doi.org/10.1042/0264-6021:3420249
- Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G, Menissier de Murcia J, Almendros A, Ruiz de Almodovar M, Oliver FJ. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009; 5:61-74; PMID:19001878; http://dx.doi.org/10.4161/auto.5.1.7272
- Wang H, Lu C, Tan Y, Xie J, Jiang J. Effect of adriamycin on BRCA1 and PARP-1 expression in MCF-7 breast cancer cells. Int J Clin Exp Pathol 2014; 7:5909-15; PMID:25337234
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A 2011; 108:4788-93; PMID:21383122; http://dx.doi.org/10.1073/pnas.1100844108
- Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol 2010; 22:177-80; PMID:20022734; http://dx.doi.org/10.1016/j.ceb.2009.11.015
- Wu SB, Wu YT, Wu TP, Wei YH. Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochim Biophys Acta 2014; 1840:1331-44; PMID:24513455; http://dx.doi.org/10.1016/j.bbagen.2013.10.034
- Vessoni AT, Filippi-Chiela EC, Menck CF, Lenz G. Autophagy and genomic integrity. Cell Death Differ 2013; 20:1444-54; PMID:23933813; http://dx.doi.org/10.1038/cdd.2013.103
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463-77; PMID:15068787; http://dx.doi.org/10.1016/S1534-5807(04)00099-1
- Ventruti A, Cuervo AM. Autophagy and neurodegeneration. Curr Neurol Neurosci Rep 2007; 7:443-51; PMID:17764636; http://dx.doi.org/10.1007/s11910-007-0068-5
- Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol 2007; 78:217-45; PMID:17338918; http://dx.doi.org/10.1016/S0070-2153(06)78006-1
- Esclatine A, Chaumorcel M, Codogno P. Macroautophagy signaling and regulation. Curr Top Microbiol Immunol 2009; 335:33-70; PMID:19802559
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/10.1016/j.ceb.2009.11.014
- Li Y, Zhang J, Chen X, Liu T, He W, Chen Y, Zeng X. Molecular machinery of autophagy and its implication in cancer. Am J Med Sci 2012; 343:155-61; PMID:21709535; http://dx.doi.org/10.1097/MAJ.0b013e31821f978d
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121-34; PMID:16839881; http://dx.doi.org/10.1016/j.cell.2006.05.034
- Crighton D, Wilkinson S, Ryan KM. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007; 3:72-4; PMID:17102582; http://dx.doi.org/10.4161/auto.3438
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol 2010; 22:181-5; PMID:20044243; http://dx.doi.org/10.1016/j.ceb.2009.12.001
- Hu YL, Jahangiri A, Delay M, Aghi MK. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res 2012; 72:4294-9; PMID:22915758; http://dx.doi.org/10.1158/0008-5472.CAN-12-1076
- Zou Z, Yuan Z, Zhang Q, Long Z, Chen J, Tang Z, Zhu Y, Chen S, Xu J, Yan M, et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012; 8:1798-810; PMID:23026799; http://dx.doi.org/10.4161/auto.22110
- Ko A, Kanehisa A, Martins I, Senovilla L, Chargari C, Dugue D, Marino G, Kepp O, Michaud M, Perfettini JL, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ 2014; 21:92-9; PMID:24037090; http://dx.doi.org/10.1038/cdd.2013.124
- Lan YY, Londono D, Bouley R, Rooney MS, Hacohen N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep 2014; 9:180-92; PMID:25284779; http://dx.doi.org/10.1016/j.celrep.2014.08.074
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007; 14:548-58; PMID:16946731; http://dx.doi.org/10.1038/sj.cdd.4402030
- Dyavaiah M, Rooney JP, Chittur SV, Lin Q, Begley TJ. Autophagy-dependent regulation of the DNA damage response protein ribonucleotide reductase 1. Mol Cancer Res 2011; 9:462-75; PMID:21343333; http://dx.doi.org/10.1158/1541-7786.MCR-10-0473
- de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev 1999; 13:768-85; PMID:10197977; http://dx.doi.org/10.1101/gad.13.7.768
- Bao Y, Shen X. Chromatin remodeling in DNA double-strand break repair. Curr Opin Genet Dev 2007; 17:126-31; PMID:17320375; http://dx.doi.org/10.1016/j.gde.2007.02.010
- Rodriguez-Vargas JM, Ruiz-Magana MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodriguez MI, Munoz-Gamez JA, de Almodovar MR, Siles E, Rivas AL, et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 2012; 22:1181-98; PMID:22525338; http://dx.doi.org/10.1038/cr.2012.70
- Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, Chabes A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res 2011; 39:1360-71; PMID:20961955; http://dx.doi.org/10.1093/nar/gkq829
- Mo N, Lu YK, Xie WM, Liu Y, Zhou WX, Wang HX, Nong L, Jia YX, Tan AH, Chen Y et al. Inhibition of autophagy enhances the radiosensitivity of nasopharyngeal carcinoma by reducing Rad51 expression. Oncol Rep 2014, 32(5):1905-12; PMID:25175062
- Bae H, Guan JL. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res 2011; 9:1232-41; PMID:21807966; http://dx.doi.org/10.1158/1541-7786.MCR-11-0098
- Pankiv S, Lamark T, Bruun JA, Overvatn A, Bjorkoy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem 2010; 285:5941-53; PMID:20018885; http://dx.doi.org/10.1074/jbc.M109.039925
- Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011; 471:74-9; PMID:21368826; http://dx.doi.org/10.1038/nature09803
- Chen H, Ma Z, Vanderwaal RP, Feng Z, Gonzalez-Suarez I, Wang S, Zhang J, Roti Roti JL, Gonzalo S. The mTOR inhibitor rapamycin suppresses DNA double-strand break repair. Radiat Res 2011; 175:214-24; PMID:21268715; http://dx.doi.org/10.1667/RR2323.1
- Siggens L, Figg N, Bennett M, Foo R. Nutrient deprivation regulates DNA damage repair in cardiomyocytes via loss of the base-excision repair enzyme OGG1. FASEB J 2012; 26:2117-24; PMID:22302830; http://dx.doi.org/10.1096/fj.11-197525
- Zeng X, Yan T, Schupp JE, Seo Y, Kinsella TJ. DNA mismatch repair initiates 6-thioguanine–induced autophagy through p53 activation in human tumor cells. Clin Cancer Res 2007; 13:1315-21; PMID:17317843; http://dx.doi.org/10.1158/1078-0432.CCR-06-1517
- You H, Kong MM, Wang LP, Xiao X, Liao HL, Bi ZY, Yan H, Wang H, Wang CH, Ma Q, et al. Inhibition of DNA-dependent protein kinase catalytic subunit by small molecule inhibitor NU7026 sensitizes human leukemic K562 cells to benzene metabolite-induced apoptosis. J Huazhong Univ Sci Technolog Med Sci 2013; 33:43-50; PMID:23392706; http://dx.doi.org/10.1007/s11596-013-1069-z
- Rajecki M, af Hallstrom T, Hakkarainen T, Nokisalmi P, Hautaniemi S, Nieminen AI, Tenhunen M, Rantanen V, Desmond RA, Chen DT, et al. Mre11 inhibition by oncolytic adenovirus associates with autophagy and underlies synergy with ionizing radiation. Int J Cancer 2009; 125:2441-9; PMID:19672857; http://dx.doi.org/10.1002/ijc.24608
- Kanzawa T, Bedwell J, Kondo Y, Kondo S, Germano IM. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J Neurosurg 2003; 99:1047-52; PMID:14705733; http://dx.doi.org/10.3171/jns.2003.99.6.1047
- Liu EY, Xu N, O'Prey J, Lao LY, Joshi S, Long JS, O'Prey M, Croft DR, Beaumatin F, Baudot AD et al: Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci U S A 2015, 112(3):773-8; PMID:25568088; http://dx.doi.org/10.1073/pnas.1409563112