
Abstract
Following a genotoxic stress, the tumor suppressor p53 translocates to mitochondria to take part in direct induction of apoptosis, via interaction with BCL-2 family members such as BAK and BAX. We determined the kinetics of the mitochondrial translocation of p53 in HCT-116 and PA-1 cells exposed to different genotoxic stresses (doxorubicin, camptothecin, UVB). This analysis revealed an early escalation in the amount of mitochondrial p53, followed by a peak amount and a decrease of mitochondrial p53 at later time points. We show that the serine 20 phosphorylated form of p53 is present at the mitochondria and that the decrease of p53 mitochondrial level during late apoptosis correlates with a decrease of Ser-20 phosphorylation. Moreover, the S20A p53 mutant translocates well to mitochondria after a genotoxic stress but its mitochondrial localization is very low during late apoptosis when compared to wt p53. The S20A mutant also appears to be compromised for interaction with BAK. We propose here that the level of serine 20 phosphorylation is influential on p53 mitochondrial localization during late apoptosis. Additionally, we report the presence of a new ≃45 kDa caspase-cleaved fragment of p53 in the cytosolic and mitochondrial fractions of apoptotic cells.
Abbreviations
| BAK | = | BCL-2 antagonist/killer |
| BAX | = | BCL-2-associated X protein |
| BCL-2 | = | B-cell lymphoma protein 2 |
| BID | = | BH3 interacting-domain death agonist |
| BMH | = | Bismaleimidohexane |
| CARP | = | Caspase 8/10-associated RING domain proteins |
| CHX | = | Cycloheximide |
| CPT | = | Camptothecin |
| DBD | = | DNA-binding domain |
| Dox | = | Doxorubicin |
| DUSP26 | = | dual-specificity phosphatase 26 |
| GRP75 | = | Glucose-regulated protein 75 |
| HIPK2 | = | Homeodomain interacting protein kinase 2 |
| HSP10 | = | Heat shock 10 kDa protein |
| HSP60 | = | Heat shock 60 kDa protein |
| MCL1 | = | Myeloid cell leukemia-1 |
| MDM2 | = | Mouse double minute 2 |
| NES | = | Nuclear export signal |
| OMM | = | Outer mitochondrial membrane |
| PARP | = | Poly [ADP-ribose] polymerase |
| PCNA | = | Proliferating cell nuclear antigen |
| PI | = | Propidium iodide |
| Pin1 | = | peptidylprolyl cis/trans isomerase NIMA-interacting 1 |
| tBID | = | Truncated BH3 interacting-domain death agonist |
| UVB | = | ultraviolet B |
| Z-VAD(OMe)-FMK | = | Z-Val-Ala-DL-Asp(OMe)-fluoromethylketone. |
Introduction
TP53, the gene encoding the tumor suppressor p53, is the most frequently mutated gene in human cancer, with an overall mutation rate of about 50%.Citation1-5 Prevention of malignant transformation by p53 relies on its ability to induce cell death by apoptosis in response to a wide variety of stimuli, including oncogene activation and DNA damage. In presence of an apoptotic stimulus, p53 acts as a transcription factor and induces the expression of pro-apoptotic proteins (KILLER/DR5, BAX, PUMA, NOXA, FAS/CD95, p53AIP1 and others) while it repress that of proteins displaying survival functions such as BCL-2 and MDR1.Citation6,7 In addition, p53 has a transcription-independent role in death, as it can induce apoptosis in the presence of transcription and translation inhibitors.Citation8 Moreover, some mutants of p53 deficient for transcription remain able to trigger apoptosis.Citation9 This transcription-independent pathway of p53-mediated apoptosis relies on its ability to translocate to mitochondria and trigger outer mitochondrial membrane (OMM) permeabilization.Citation10-15 OMM permeabilization is often considered to be a point of “no return” in the course of apoptosis since it allows the release into the cytosol of different apoptosis effectors located in the transmembrane space, such as cytochrome c.Citation16-18 At the mitochondria, p53 interacts directly with different BCL-2 family members and modulate their activity. Its binding with the anti-apoptotic proteins BCL-2 and BCL-XL inhibits their activity and favors OMM permeabilization.Citation19 Importantly, when localized at the OMM, p53 can behave like activating BCL-2 family members, such as the truncated form of BID, tBID.Citation20 p53 directly binds BAK, displacing its inhibitor MCL1.Citation13,15 Displacement of MCL1 is favored when p53 is acetylated on lysine 120 and this form of p53 is enriched at the mitochondria.Citation21 Ultimately, p53 interaction triggers a conformation change of the BAK protein that allows its oligomerization, formation of BAK pores and OMM permeabilization.Citation13,15,20
Little is known about the mechanism(s) that regulate p53 mitochondrial localization during a stress. We previously showed that BAK is a mitochondrial anchor for p53. For example, in the presence of an apoptotic stimulus, BAK −/− mouse embryo fibroblasts display a markedly reduced mitochondrial p53 level, as compared to their wt counterpart.Citation13,22 Thus, the level of BAK is itself influential on the mitochondrial localization of p53. The mitochondrial localization of p53 is also impacted by a common polymorphism. The Arg-72 variant has a greater apoptotic potential and shows a more abundant mitochondrial translocation during a stress than the Pro-72 variant.Citation11 This is linked to the greater affinity of p53 Arg-72 for the ubiquitin E3 ligase MDM2, resulting in enhanced ubiquitination, CRM-1 interaction and nuclear export of this variant.Citation11,23 Moreover, mono-ubiquitination of cytosolic p53 by MDM2 triggers its mitochondrial localization.Citation24 These combined data therefore indicate that ubiquitination is a signal that modulates the mitochondrial translocation of the tumor suppressor.
In this study, we analyzed the kinetics of the mitochondrial localization of p53 in different cellular models. We show that mitochondrial p53 level is not constant during apoptosis. We observed an early peak of mitochondrial p53, followed by a plateau phase and then a marked decrease of p53 during late apoptosis. BAK homo-oligomerization and OMM permeabilization are not responsible for mitochondrial p53 decrease during late apoptosis since these events occur earlier than the loss of mitochondrial p53. Rather, this decrease is concomitant to a loss of phosphorylation of p53 on serine 20. Moreover, while the S20A p53 mutant translocates well to the mitochondria after a stress, its mitochondrial localization is reduced during late apoptosis by comparison to wt p53. Our data suggest that Ser-20 phosphorylation is important for a long term presence of p53 at the mitochondria. We also report the presence of caspase-cleaved fragments of p53 at the mitochondria as well as in the cytosol. However, the decrease of mitochondrial p53 at late apoptosis does not appear to result from the proteolytic activity of caspases.
Results
Decrease of p53 mitochondrial level during the late apoptosis of colon and ovarian cancer cells
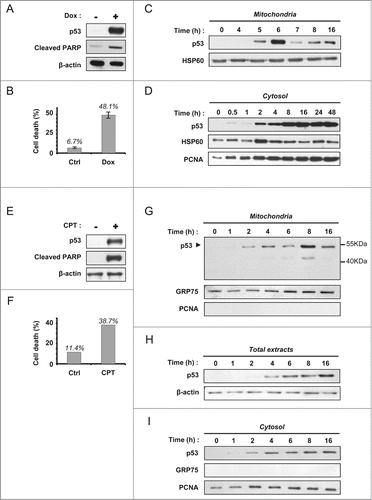
We treated wt p53 HCT-116 colon carcinoma cells with 1 µM doxorubicin (Dox). This genotoxic agent markedly induced p53 as well as PARP cleavage (). Flow cytometry analysis (LDS751/PI co-labeling) indicated a death rate close to 50% at 24 hours (). The level of p53 in the mitochondrial fraction of treated cells peaked at 6 hours and decreased at later time points (7, 8 and 16 hours) (). We next tested whether the observed kinetics were specific for this subcellular compartment. Analysis of cytosolic fractions showed an early increase of p53 level and a sustained level of p53 activation. At 48 hours, no decrease of p53 was recorded (). To confirm these data, we used a second apoptotic stimulus. Exposure of HCT-116 cells to 1 µM camptothecin, a topoisomerase I inhibitor, resulted in marked induction of p53 and cell death by apoptosis as observed by the cleavage of PARP (). Co-labeling of the cells with LDS751 and propidium iodide (PI) followed by flow cytometry analysis allowed determining a death rate close to 40% after 24 hours treatment (). In order to study the kinetics of the mitochondrial localization of p53, mitochondrial fractions were prepared at 0, 1, 2, 4, 6, 8 and 16 hours (). p53 was readily detectable at 2 hours in the mitochondrial fraction of the stressed cells. Mitochondrial p53 level increased up to 8 hours. Interestingly, a decrease in p53 was recorded at 16 hours. We also noted the formation of a lower molecular weight band (between 40–55 kDa) detected by the anti-p53 DO-1 antibody that recognizes the N-terminus (). We next examined p53 level in total extracts and cytosolic fractions. In total extracts, induction of p53 was detectable at 4 hours following camptothecin treatment and p53 level showed a steady increase until 16 hours (). A similar evolution of p53 level was observed in the cytosolic fraction (). These data suggest that the decrease in mitochondrial p53 at 16 hours was specific to mitochondria and does not reflect a globally decreased p53 protein level.
Figure 1. Decrease of mitochondrial p53 during the late apoptosis of HCT-116 cells treated with DOX or CPT. (A) Western blot analysis of p53 induction and PARP cleavage in HCT-116 cells, treated or not for 24 hours with 1 µM Doxorubicin. (B) Cell viability of HCT-116 cells treated (Dox) or not (Ctrl) as in (A) was assessed by flow cytometry (LDS751/PI co-labeling). (C) Kinetic of mitochondrial p53 level in HCT-116 cells. Mitochondrial fractions were isolated after treatment with 1 µM doxorubicin during 0, 4, 5, 6, 7, 8 and 16 hours. HSP60 was used as a loading control. (D) Kinetic of cytosolic accumulation of p53 in HCT-116 cells. Cytosolic fractions were isolated after incubation for different periods of time with 1 µM doxorubicin as indicated in the figure. HSP60 and PCNA were used as loading controls. (E) Western blot analysis of p53 induction and PARP cleavage in HCT-116 cells, treated or not for 24 hours with 1 µM camptothecin. (F) Viability of untreated HCT-116 cells (Ctrl) or cells treated for 24 hours with 1 µM camptothecin (CPT) was assessed by flow cytometry with LDS751/PI co-labeling. (G–I), HCT-116 cells were incubated with 1 µM camptothecin. Mitochondrial fractions (G), total extracts (H) and cytosolic fractions (I) were prepared at the indicated times. Western blot analysis was performed using DO-1 anti-p53 antibody. Purity of the fractions and loading controls were established by detection of GRP75, PCNA and β-actin.
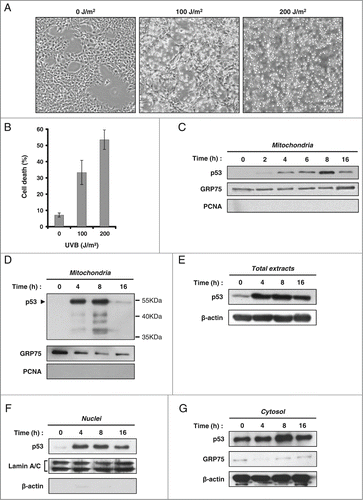
In order to extend these observations to a different cellular model, we treated PA-1 ovarian cancer cells (wt p53) with UVB. Cell death was observed in cultures exposed to 100 or 200 J/m2 UVB (). Flow cytometry analysis (LDS751/PI co-labeling) indicates a death rate of 34%, 24 hours after treatment with 100 J/m2 (). Annexin-V/7-AAD co-labeling of the cells confirmed that cell death occurred by apoptosis (data not shown). From cells treated with 100 J/m2 UVB, we prepared mitochondrial fractions 0, 2, 4, 6, 8 and 16 hours after exposure. As previously observed, we noted an early increase of mitochondrial p53 with a peak at 8 hours. However, a sharp decrease of p53 level in the mitochondrial fraction was noted at 16 hours (). Based on this profile, we selected 4 different time points (0, 4, 8 and 16 hours) to analyze p53 level in different subcellular compartments. In mitochondrial fractions, we repeatedly observed the presence of bands of lower molecular weights recognized by the anti-p53 DO-1 antibody, suggesting that mitochondrial p53 could be the target of a degradation process (). A similar feature was observed in HCT-116 cells treated with camptothecin (). In total extracts as well as nuclear and cytosolic fractions, an early increase of p53 level was observed after the UVB stress and p53 level remained more or less stable until 16 hours ().
Figure 2. Marked decrease of mitochondrial p53 during late apoptosis in PA-1 cells treated with UVB. (A) Phase contrast microscopy images displaying populations of PA-1 cells untreated or exposed to 100 and 200 J/m2 UVB. Images were taken 24 hours after UVB treatment. (B) Determination of cell death by flow cytometry. PA-1 cells were labeled with LDS751/PI, 24 hours after exposure or not to 100 and 200 J/m2 UVB. (C–G) Assessment of p53 level by western blot (anti-p53 DO-1 antibody) in different cell compartments in PA-1 cells irradiated or not with 100 J/m2 UVB. Detection of GRP75, PCNA, Lamin A/C and β-actin was performed to control for the purity of the fractions and to monitor loading of the wells. Evolution of p53 level was determined at the indicated time points in mitochondrial fractions (C and D) total extracts (E), nuclear extracts (F) and cytosolic fractions (G).
Decrease of mitochondrial p53 during late apoptosis results from its dephosphorylation on serine 20
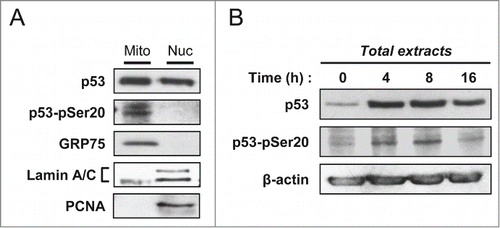
Previously, we and others published that interaction of p53 with the E3 ligase MDM2 and its subsequent ubiquitination was important for its mitochondrial translocation.Citation11,24 Interaction between MDM2 and p53 is influenced by post-translational modifications. Of note, phosphorylation of p53 on Ser-20 and Thr-18 are known to modulate MDM2 binding and p53 ubiquitination. Both modifications occur in the minimal region of p53 required for MDM2 binding.Citation25-27 We treated PA-1 ovarian cancer cells with 100 J/m2 UVB. At 16 hours post-irradiation, nuclear and mitochondrial fractions were isolated. For each fraction, we analyzed by western blot an equal amount of “total” p53 and found that the Ser-20 phosphorylated form of p53 (P-Ser20) was present at the mitochondria (). Thr-18 phosphorylation was not detected (data not shown). We performed a similar analysis on total cell extracts at 0, 4, 8 and 16 hours following the irradiation (). We observed that Ser-20 phosphorylation follows the same kinetics than mitochondrial p53 level, with a bell-shaped curve and a decrease during late apoptosis (Compare and ). Together, these data suggested a potential role for Ser-20 phosphorylation of p53 in its mitochondrial localization.
Figure 3. Presence of the serine 20 phosphorylated form of p53 in the mitochondrial fraction. (A) Mitochondrial and nuclear fractions of PA-1 cells were isolated at 16 hours after irradiation with 100 J/m2 UVB. Equal amounts of “total” p53 were loaded for each fraction on SDS-PAGE. Western blot analysis was performed with an antibody specific to P-Ser20 p53 and the anti-p53 DO-1 monoclonal antibody. GRP75, Lamin A/C and PCNA were used to control for the purity of the fractions. (B) Kinetics of p53 level and its serine 20 phosphorylated form in total extracts of PA-1 cells treated with 100 J/m2 UVB. At 0, 4, 8 and 16 hours post-irradiation, total extracts were isolated. Evolution of total p53 level as well as of its serine 20 phosphorylated form were assessed by western blot analysis.
To unveil the possible impact of Ser-20 phosphorylation on the mitochondrial localization of p53 during a stress, we transiently transfected H1299 p53 −/− cells with wt p53, the S20A p53 mutant or the empty pRc/CMV vector. The next day, protein expression was blocked with 10 μg/ml cycloheximide (CHX) and cells were treated with 1 μM camptothecin. Total protein extracts, cytosolic and mitochondrial fractions were analyzed by western blot after 4 and 16 hours of treatments (). After 4 hours, both wt and S20A p53 were present at the mitochondria. Moreover, although the overall level of transfected S20A p53 is lower than that of wt p53, both forms were similarly distributed in the different compartments analyzed (). This suggests that, in presence of a genotoxic stress, Ser-20 phosphorylation of p53 does not impact on its mitochondrial translocation. After 16 hours of treatment, relative to the amount of “total p53,” we observed a depletion of S20A p53 in the mitochondrial fraction, when compared to wt p53 (). This suggested that Ser-20 phosphorylation favors a long-term presence of p53 at the mitochondria.
Figure 4. Serine 20 phosphorylation is required for a prolonged mitochondrial localization of p53. (A) Assessment of p53 level by western blot (anti-p53 DO-1 antibody) in the mitochondrial and cytosolic fractions as well as total extracts of H1299 cells transfected with wt p53, the S20A mutant or the empty vector (E). Twenty-four hours after transfection, protein synthesis was blocked with 10 µg/mL cycloheximide (CHX) and cells were treated simultaneously with 1 µM camptothecin (CPT). We analyzed p53 levels after 4 and 16 hours of treatment. Detection of GRP75, PCNA and β-actin was performed to control for the purity of the fractions and to monitor loading of the wells. Densitometry analysis was based on the ratio of p53 signal to that of the respective loading control, and the signal recorded for wt p53 was set to 1. (B) Determination of p53 level by western blot (anti-p53 DO-1 antibody) in the mitochondrial fractions as well as total extracts of H1299 cells transfected with wt p53 or the S20A mutant. Twenty-four hours after transfection, protein synthesis was blocked with 10 µg/mL cycloheximide (CHX) and cells were treated simultaneously with 1 µM camptothecin (CPT). We analyzed p53 levels after 0, 4, 8 and 16 hours of treatment. Detection of GRP75, PCNA and β-actin was performed to control for the purity of the fractions and to monitor loading of the wells. Densitometry analysis was based on the ratio of p53 signal to that of the respective loading control, and the signal recorded at 0 hour was set to 1.
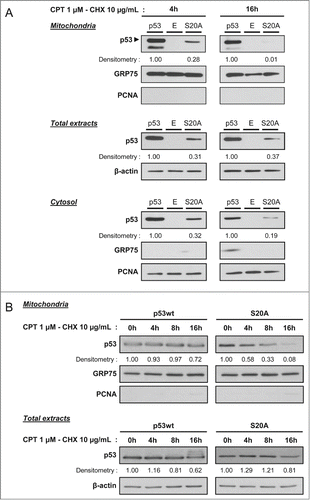
In order to strengthen this data, we performed a time course experiment to compare the level of wt p53 and the S20A mutant in total protein extracts and mitochondrial fractions at different time points after the stress (). H1299 cells were transfected with wt and S20A p53. The next day, protein expression was blocked with 10 μg/ml cycloheximide (CHX) and cells were treated with 1 μM camptothecin. In mitochondrial fractions, wt p53 level was stable between 0 and 8 hours, and a ≃30% reduction of its level was recorded after 16 hours of treatment (). In contrast, already after 4 hours of treatment, we observed a marked decrease of the level of the S20A mutant in the mitochondrial fraction, and it was virtually absent from this subcellular compartment after 16 hours. Of note, evolution during time of wt and S20A p53 levels was similar in total protein extracts ().
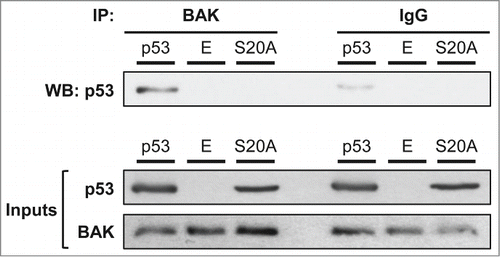
In order to further analyze the link between Ser-20 phosphorylation and p53 mitochondrial localization, we investigated the capacity of both wt and S20A forms of p53 to interact with BAK (). H1299 cells were transfected with wt p53, the S20A mutant or the empty vector and the next day exposed to 1 μM camptothecin. After 4 hours of treatment, BAK was immunoprecipitated and the presence of p53 in the precipitate was analyzed by western blot. Surprisingly, the S20A mutant failed to interact with BAK, while a robust interaction was present between BAK and wt p53 ().
Figure 5. A serine 20 mutated form of p53 is impaired for BAK binding. We transfected p53 −/− H1299 cells with wt p53, the S20A mutant or the empty pRc/CMV vector (E). To obtain an equivalent amount of protein expression, we transfected 2 times more micrograms DNA for the S20A mutant. Twenty-four hours after transfection, cells were stressed with 1 µM camptothecin for 4 hours. After treatment, cells were lysed and total protein extracts were immunoprecipitated with an anti-BAK antibody or irrelevant IgG, as indicated in the materials and methods. We then analyzed by western blot the interaction of BAK with the tumor suppressor using an anti-p53 antibody. Inputs for p53 and BAK (precleared lysates) are also presented.
Presence of caspase-cleaved fragments of p53 in the mitochondrial and cytosolic fractions of cells undergoing apoptosis
The above data indicate that the mitochondrial localization of p53 during apoptosis is characterized by a bell-shaped kinetic curve, showing an early peak followed by a marked decrease at later time points. Moreover, the phosphorylation status of Ser-20 is influential on p53 mitochondrial localization during late apoptosis as well as BAK binding.
Because we consistently observed lower molecular weight p53 fragments in our western blot analysis (), we next examined whether the decreased level of mitochondrial p53 during late apoptosis could be the consequence of a degradation of p53 and more specifically that the tumor suppressor could be the target of mitochondrial caspases. Indeed, although caspases are normally cytosolic they have been observed in other cell compartments. Procaspases-2, -3 and -9 are known to be present at the mitochondria.Citation28 Procaspase-3 is found localized at the inner and outer mitochondrial membranes, in a complex with the chaperones HSP60 and HSP10.Citation29,30 Additionally, we previously identified an interaction between mitochondrial caspase-3 and wt as well as mutant forms of p53.Citation31
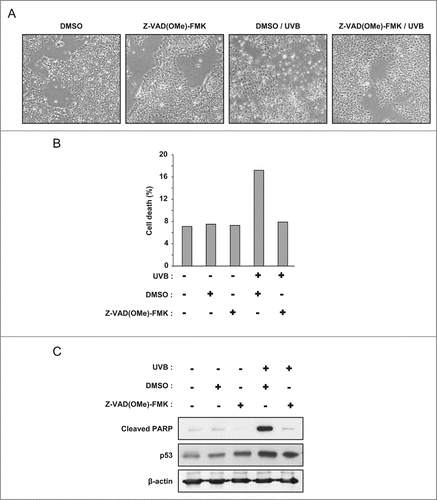
To test the hypothesis that p53 could be degraded by mitochondrial caspases during late apoptosis, we treated PA-1 ovarian cancer cells with 100 J/m2 UVB in the presence or not of the pan-caspase inhibitor Z-VAD(OMe)-FMK. The inhibitor was able to prevent cell death in the UVB treated population (). By flow cytometry analysis, we observed 18% of cell death in the UVB treated cells, and this was inhibited by Z-VAD(OMe)-FMK (). Similarly, UVB caused PARP cleavage and this was abolished by the pan-caspase inhibitor ().
Figure 6. The pan-caspases inhibitor Z-VAD(OMe)-FMK inhibits cell death and PARP cleavage in PA-1 cells exposed to UVB. (A) Phase contrast microscopy images of PA-1 cells treated with 100 J/m2 UVB, in the presence of 50 µM Z-VAD(OMe)-FMK or its vehicle (DMSO). Images were taken 16 hours after UVB treatment. (B) Analysis of the viability of PA-1 cells treated or not with 100 J/m2 UVB by flow cytometry (LDS751/PI co-labeling). Cells were pre-incubated with Z-VAD(OMe)-FMK or its vehicle (DMSO). Analysis was performed 16 hours after treatment. (C) Western blot analysis of cleaved PARP and p53 level in PA-1 cells treated as in (A) and (B).
We prepared mitochondrial and cytosolic fractions of cells treated with 100 J/m2 UVB in the presence or not of Z-VAD(OMe)-FMK. Localization of mitochondrial p53 showed the typical bell-shaped kinetic after UVB treatment, with a marked decrease of mitochondrial p53 at 16 hours (). Notably, this marked decrease at 16 hours was still observed in the presence of the pan-caspase inhibitor. Densitometry analysis confirmed that Z-VAD(OMe)-FMK only had a marginal effect on mitochondrial p53 level at 16 hours (). However, the lower molecular weight (≃45 kDa) p53 band vanished in the presence of Z-VAD(OMe)-FMK, supporting the premise that this band corresponded to a caspase-cleaved p53 fragment ().
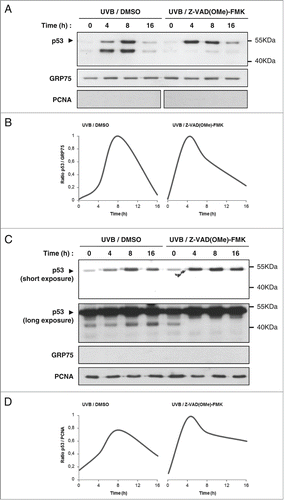
We next sought to examine whether this caspase-cleaved ≃45 kDa p53 fragment was the result of caspase-cleavage of mitochondrial or of cytosolic p53. To test these possibilities, we analyzed the cytosolic fractions at the same time points (). Notably, the same lower molecular weight band was observed in the cytosol, and incubation with Z-VAD(OMe)-FMK resulted in its disappearance. Furthermore, an increase of cytosolic p53 level was observed in the presence of the caspase inhibitor. Thus, unlike in mitochondrial fractions, cytosolic p53 level tends to increase when Z-VAD(OMe)-FMK is added (). These data suggest that this ≃45 kDa caspase-cleaved fragment of p53 may be generated in the cytosol and then translocates to the mitochondria.
BAK oligomerization and OMM permeabilization occur prior the decrease of mitochondrial p53 during late apoptosis
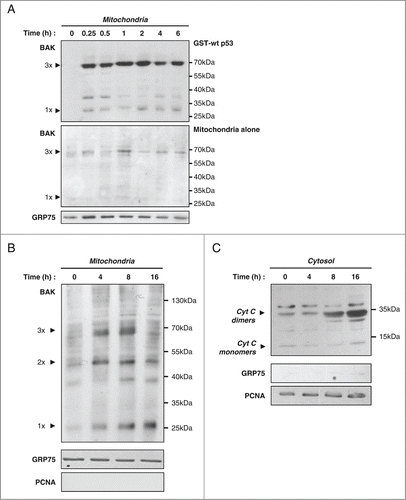
It has been shown that during apoptosis, BAK is an anchor for mitochondrial p53.Citation13,22 The interaction of p53 and BAK triggers homo-oligomerization of the latter.Citation13,15,20 We reasoned that this significant conformational change could disturb the interaction between the 2 partners and thus also lead to the observed decrease of mitochondrial p53 during late apoptosis. To explore this possibility, we analyzed BAK oligomerization when purified mitochondria were incubated with wt p53. Mitochondria isolated from SaOS-2 p53 −/− cells were incubated with recombinant GST-p53 during 0 to 6 hours. After crosslinking with BMH (bismaleimidohexane), the presence of BAK multimers was analyzed by western blot (). We found that formation of BAK multimers occurred very rapidly (15 min) and remained stable over time. We next tested, in cells undergoing apoptosis, whether a correlation existed between the loss of mitochondrial p53 during late apoptosis and BAK oligomerization as well as OMM permeabilization. We prepared the mitochondrial fractions from PA-1 cells treated with 100 J/m2 UVB at 0, 4, 8 and 16 hours. BAK oligomerization was visualized after BMH crosslinking and western blot analysis. BAK dimers and trimers were detected as soon as 4 hours. Longer incubations did not increase their abundance. Moreover, a significant population of BAK monomers remained present (). Determination of cytochrome c release into the cytosol confirmed that OMM permeabilization had occurred (). Although cytochrome c has a molecular weight of 14 kDa, in our assay, the main band was at 35 kDa, corresponding to a cytochrome c dimer not reduced (disulfide bond), as previously described.Citation32 This indicated that cytochrome c release started between 4 and 8 hours, after BAK oligomerization (). In conclusion, these data do not support the hypothesis that the altered conformation of BAK associated with oligomerization is responsible for the loss of p53 mitochondrial localization. The data also indicate that the decrease of mitochondrial p53 level is a late event during the apoptotic process, occurring after OMM permeabilization.
Figure 8. Formation of BAK oligomers and OMM permeabilization precede loss of mitochondrial p53. (A) GST-p53 wt induces in vitro BAK homo-oligomerization. Isolated mitochondria from SaOS-2 p53 null cells were incubated or not (Mitochondria alone) with GST-p53 wt for different incubation times. BMH crosslinker was added to a final concentration of 2.77 mM to visualize BAK multimers on western blot analysis. A detection of GRP75 was also performed in mitochondria incubated in the absence of p53 for the same time points. (B) Mitochondrial fractions of PA-1 cells treated with 100 J/m2 UVB were isolated at 0, 4, 8 and 16 hours after stress. Western blot analysis with an antibody against the BAK protein was performed after crosslinking with 2.77 mM BMH. Purity and loading of the different fractions were verified by detection of GRP75 and PCNA. (C) Western blot analysis of cytochrome c release in cytosolic fractions of PA-1 cells treated as above. Purity and loading of the fractions were controlled by detection of cytosolic/nuclear PCNA and mitochondrial GRP75.
Discussion
We analyzed p53 mitochondrial translocation in 2 cell lines (HCT-116, PA-1) in presence of different genotoxic agents. We observed an early translocation of p53 with consistent detection at mitochondria within 2 hours after the genotoxic stress. After a peak, we observed a marked decrease of p53 mitochondrial levels during late apoptosis. Our data corroborate previous observations. In ML-1 cells treated with camptothecin, a peak of mitochondrial p53 was observed after 6–8 hours of treatment.Citation33 In another study, maximum p53 accumulation at the mitochondria occurred 5 hours after gamma radiation of LnCaP cells.Citation34
Several groups previously showed that p53 mitochondrial localization is regulated by post-translational modifications. MDM2 can mono-ubiquitinate C-terminal lysines of p53, favoring its mitochondrial translocation.Citation11,24,35 In addition, Ser-46 phosphorylation by HIPK2 allows binding to Pin1 (peptidylprolyl cis/trans isomerase, NIMA-interacting 1) whose activity favors p53 mono-ubiquitination and subsequent mitochondrial translocation.Citation36 In our study, we detected P-Ser20 p53 in the mitochondrial fraction of PA-1 cells undergoing apoptosis. We also found that phosphorylation of p53 on serine 20 decreases during late apoptosis. Our observations indicate that the S20A p53 mutant translocates well to mitochondria in cells exposed to a genotoxic stress. However, it is very different from wt p53 with regards to the duration of its anchorage at the mitochondria. Indeed, after a genotoxic stress, it is rapidly depleted from the mitochondrial fraction, suggesting that Ser-20 phosphorylation is important for a durable presence of p53 at the OMM.
Our observations raise different questions. First, what is the mechanism leading to the decrease of Ser-20 phosphorylation? Phosphatases from Dual Specificity Protein Phosphatase (DUSP) family are able to dephosphorylate proteins on tyrosine, threonine and serine residues and are described to be deregulated in cancer and other diseases.Citation37,38 Among them, DUSP26 (dual-specificity phosphatase 26; also called MKP-8, LDP-4 and SKRP3) is localized in the nucleus and cytoplasm and can dephosphorylate serines 20 and 37 of p53.Citation39,40 This suggests the possibility that phosphatases could control the turn-over of mitochondrial p53.
Second, why a p53 form that is not phosphorylated on Ser-20 has a shortened mitochondrial localization? We investigated the ability of wt and S20A p53 to interact with BAK and surprisingly observed that the S20A mutant was impaired for BAK binding. Previous reports indicate that interaction with BAK relies on p53 DBD and that BAK does not interact with the p53 N-terminal region.Citation13 However, our data raise the possibility that phosphorylation of Ser-20 regulates, likely indirectly, the interaction with BAK. Absence of Ser-20 phosphorylation could promote interactions with other partners in other subcellular compartments and therefore lead to a decreased mitochondrial localization, as seen at later time points of the apoptotic process. Alternatively, mitochondrial p53 could be degraded during late apoptosis. Moreover, dephosphorylation of Ser-20 could be the trigger for its degradation. We detected lower molecular weight forms of p53 in the mitochondrial fractions of cells undergoing apoptosis, including a major fragment with an apparent molecular weight of ≃45 kDa. In presence of Z-VAD(OMe)-FMK, this p53 fragment is not produced, indicating that it is derived from caspase cleavage. However, the caspase inhibitor failed to prevent the decrease of mitochondrial p53 level, suggesting that caspase cleavage is not responsible for the loss of mitochondrial p53 during late apoptosis.
Caspases-2, -3 and -9 are known to be located at the mitochondria and caspase-3 can interact with p53 at the OMM.Citation28–31 Our In silico analysis (CASVM software) revealed 4 putative conserved caspase cleavage sites within p53 (Fig. S1–A). These cleavage sites can theoretically generate 14 different fragments from full length p53. As the ≃45 kDa p53 fragment was recognized by the monoclonal DO-1 anti-p53 antibody, it cannot result from N-terminal cleavage (positions 21/22 and 42/43).Citation41 The ≃45 kDa fragment thus likely corresponds to p53 1–352 aa (Fig. S1–B). This p53 fragment was present in both the mitochondrial and cytosolic fractions. A previous study by Sayan et al. showed that cytosolic p53 can be cleaved by active caspases and that some fragments are able to translocate to mitochondria.Citation41 Additional experiments are thus required to determine in which subcellular compartment the ≃45 kDa p53 fragment is produced.
In summary, we show that the presence of p53 at the mitochondria is not constant during apoptosis. After an early translocation and a peak, a marked decrease of p53 mitochondrial levels is observed during late apoptosis. The apoptotic process is often described as composed of 3 consecutive phases: the initiation phase, the decision phase and the execution (or degradation) phase.Citation16,17 The decrease of mitochondrial p53 levels during late apoptosis clearly occurs during the execution phase. Indeed, loss of mitochondrial p53 is observed after BAK oligomerization, OMM permeabilization, release of cytochrome c and caspases activation. Activation of caspases cannot explain this late reduction since this is still observed when they are inhibited. Interestingly, we observed a decreased Ser-20 phosphorylation during late apoptosis. Moreover, the p53 S20A mutant is impaired for BAK binding and is rapidly depleted from the mitochondria after a genotoxic stress. Our data involve the phosphorylation status of Ser-20 in the decrease of mitochondrial p53 levels during late apoptosis and suggest that this post-translational modification is influential on the p53-BAK interaction.
Materials and Methods
Cell culture and treatments
We used the wt p53 HCT-116 human colorectal carcinoma cell line (ATCC CCL-247), the PA-1 human ovarian teratocarcinoma cell line (wt p53) (ATCC CRL-1572), the p53 null SAOS-2 human osteosarcoma cell line (ATCC HTB-85) and the p53 null H1299 human non-small cell lung carcinoma cell line (ATCC CRL-5803). All cells, routinely tested for mycoplasma contamination, were grown in DMEM (Gibco 42430–082) supplemented with 10% foetal bovine serum and 1% penicillin-streptomycin (Gibco 15140–163). Doxorubicin (Sigma-Aldrich D1515), camptothecin (Sigma-Aldrich C9911) and cycloheximide (Sigma-Aldrich C4859) were used as indicated in the figures. To inhibit caspases, we used Z-VAD(OMe)-FMK (Bachem N-1560) at a concentration of 50 µM. This pan-caspase inhibitor was applied to the cells 1 hour before treatment. For UVB irradiation, cells were grown in 100 mm petri-dishes. Before irradiation, the medium was removed and 2 mL of PBS was added. For treatment, cells were exposed to 100 J/m2 UVB (Vilber Lourmat RMX-3W) without dish lid at room temperature. Whereupon, cells were re-incubated at 37°C in fresh culture medium.
Plasmids
The human p53 cDNA (amino acids 1–393, polymorphic variant R72) is cloned into the pRc/CMV vector (Invitrogen) between the HindIII and XbaI restriction sites. The p53 S20A mutant was generated by site-directed mutagenesis (Stratagene).
Total protein extracts
Proteins were extracted with ice-cold radio-immunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 0.1% SDS, 0.1% Triton X-100) supplemented with protease (Complete Protease Inhibitor Cocktail, Roche Diagnostics 11836170001) and phosphatase inhibitors (Phosphatase Inhibitor Cocktail 2 and 3, Sigma Aldrich P5726 & P0044).
Mitochondria isolation
The method is based on differential centrifugations. Briefly, cells were scraped and centrifuged at 600 g (4°C, 5 min). The pellet was washed with 10 mL PBS. After a second centrifugation, the supernatant was discarded and the pellet was resuspended in 2 mL fractionation buffer (10 mM Hepes pH 7.4, 1 mM EDTA, 0.2 mM EGTA, 300 mM Mannitol) supplemented with proteases (Complete Protease Inhibitor Cocktail, Roche Diagnostics 11836170001) and phosphatases inhibitors (Phosphatase Inhibitor Cocktail 2 and 3, Sigma Aldrich P5726 & P0044)). After manual lysis with a type B Dounce homogenizer, nuclei and lysed membranes were removed by 3 successive centrifugations at 500 g (4°C, 10 min). The supernatant was collected and centrifuged at 9,000 g (4°C, 10 min). The supernatant from this last centrifugation represents the cytosolic fraction. The pellet (mitochondrial fraction) was resuspended in 600 µL fractionation buffer. To maximize the purity, the mitochondrial and cytosolic fractions were once again centrifuged at 9,000 g. The supernatant of the cytosolic fraction was isolated. The pellet of the mitochondrial fraction was resuspended in 100 µL of fractionation buffer and placed at −20°C overnight for complete lysis of mitochondria.
Preparation of nuclear extracts
Cells were scraped and centrifuged at 600 g (4°C, 5 min). The pellet was washed with 10 mL PBS and resuspended in 2 mL hypotonic buffer (10 mM Hepes-NaOH pH 7.9, 0.1 mM EDTA, 10 mM KCl) supplemented with 1 mM DTT as well as proteases (Complete Protease Inhibitor Cocktail, Roche Diagnostics 11836170001) and phosphatases inhibitors (Phosphatase Inhibitor Cocktail 2 and 3, Sigma Aldrich P5726 & P0044). After 15 min incubation on ice, cells were lyzed with a type B Dounce homogenizer. Next, cells were centrifuged at 4,000 g (4°C, 2 min). The supernatant (cytoplasmic fraction) was collected and again centrifuged at 4,000 g (4°C, 2 min) to maximize purity. The pellet was washed with 750 µL hypotonic buffer and then resuspended in 1 mL hypertonic buffer (20 mM Hepes-KOH pH 7.9, 400 mM NaCl, 1 mM EDTA, 20% Glycerol) supplemented with 1 mM DTT as well as proteases and phosphatases inhibitors. The pellet was incubated on ice for 45 min with brief vortexing every 10 min. Next, samples were centrifuged at 14,000 g (4°C, 5 min) and the supernatant (soluble nuclear fraction) was collected.
Western blot
Protein concentrations were determined using the Dc Protein Assay Kit (Bio-Rad Laboratories). Proteins were resolved on polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked for 30 min with PBS containing 0.2% Tween-20 (PBS-T) and 5% non-fat dry milk or alternatively 5% BSA. After blocking, membranes were incubated with the primary antibodies either 1 hour at room temperature or overnight at 4°C. The following primary antibodies were used: mouse anti-actin antibody (dilution of 1:6,000; Sigma-Aldrich A1978), mouse anti-cleaved PARP (Asp214) (dilution of 1 : 1,000; Cell Signaling 9546), mouse anti-p53 (DO-1) (dilution of 1 : 1,000; Millipore OP43); rabbit anti-pSer20 p53 (dilution of 1 : 1,000; Cell Signaling 9287); rabbit anti-lamin A/C (dilution of 1 : 800; Santa Cruz sc-20681); mouse anti-PCNA (dilution of 1:2,500; Santa Cruz sc-56); goat anti-GRP75 (dilution of 1 : 400; Santa Cruz sc-1058); rabbit anti-HSP60 (dilution of 1:400; Santa Cruz sc-1722); rabbit anti-BAK, NT (dilution of 1 : 500; Millipore 06–536) and mouse anti-cytochrome C (dilution of 1:1,000; BD Pharmingen 556433). After 3 washes with PBS-T, membranes were incubated with the appropriate peroxydase-conjugated secondary antibodies at a 1:2,500 dilution: goat anti-mouse (Dako P0447); goat anti-rabbit (Dako P0448). Immunoreactive bands were detected using the Western Lightning Kit (Perkin Elmer).
Production of recombinant GST-p53
The human p53 cDNA (amino acids 1–393) cloned into the pGEX-2T plasmid (GE Healthcare) was transformed into BL21 E. coli cells (GE Healthcare). Protein production was induced by addition of 0.1 mM IPTG for 5 hours at 30°C. Recombinant GST-tagged p53 protein was purified as describedCitation13 using glutathione-Sepharose 4B beads according to the manufacturer's protocol for batch purification (GE Healthcare). Recombinant proteins were eluted from the beads with 20 mM glutathione overnight at 4°C, followed the next day by 4 short glutathione elutions of 5 min. Purified recombinant GST-p53 was concentrated by ultrafiltration (Merck Millipore Amicon Ultra-4 Centrifugal Filter Unit UFC803008).
BAK oligomerization
The in vitro assays were performed essentially as described.Citation13 Briefly, 20 µg of purified mitochondria were incubated with 1 µg of recombinant GST-p53. After incubation, BAK oligomers were crosslinked with freshly made 2.77 mM BMH (Bismaleimidohexane; Pierce 22330) during 30 min at room temperature.
Flow cytometry
Cell viability was determined by co-labeling with LDS751 and propidium iodide using the Guava Viacount reagent (Millipore, USA) according to the instructions of the manufacturer, on a Guava easyCyte 6HT-2L flow cytometer (Millipore, USA).
Immunoprecipitation
Cells were lysed in NP-40 lysis buffer (50 mM Tris–HCl pH 8, 5 mM EDTA, 150 mM NaCl, 1% NP-40) supplemented with protease inhibitors. After lysis, 500 μg of proteins were precleared with PureProteome™ protein A magnetic beads (Millipore LSKMAGA02) and the irrelevant rabbit IgG. After preclearing, lysates were incubated for 2h at 4°C with 2 μg of rabbit anti-BAK antibody (Millipore, 06–536) or irrelevant IgG. Protein complexes were immunoprecipitated with protein A magnetic beads for 30 min at room temperature. The beads were then washed 2 times in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 0.1% SDS, 0.1% Triton X-100), followed by 6 washes in high-salt wash buffer (50 mM Tris-HCl pH 7.4, 1% NP-40, 1 M NaCl). Bound proteins were eluted with Laemmli sample buffer and analyzed by western blot.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Supplemental Figure
Download MS Power Point (155.9 KB)Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
The laboratory of P Dumont is financed by the FNRS-TELEVIE (Fonds National de la Recherche Scientifique) (Brussels, Belgium) as well as the FSR (Fonds Spéciaux de Recherche UCL, Louvain-la-Neuve). C. Castrogiovanni is the owner of a grant from the FNRS-TELEVIE (grant Télévie N°7458713F).
References
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009; 9:862-73; PMID:19935675; http://dx.doi.org/10.1038/nrc2763.
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 2006; 6:909-23; PMID:17128209; http://dx.doi.org/10.1038/nrc2012.
- Donehower LA, Lozano G. Twenty years studying p53 functions in genetically engineered mice. Nat Rev Cancer 2009; 9:831-41; PMID:19776746; http://dx.doi.org/10.1038/nrc2731.
- Petitjean A, Achatz MIW, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 2007; 26:2157-65; PMID:17401424; http://dx.doi.org/10.1038/sj.onc.1210302.
- Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9:701-13; PMID:19693097.
- Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ 2006; 13:951-61; PMID:16575405; http://dx.doi.org/10.1038/sj.cdd.4401916.
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8:275-83; PMID:17380161; http://dx.doi.org/10.1038/nrm2147.
- Arima Y, Nitta M, Kuninaka S, Zhang D, Fujiwara T, Taya Y, Nakao M, Saya H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J Biol Chem 2005; 280:19166-76; PMID:15753095; http://dx.doi.org/10.1074/jbc.M410691200.
- Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev 1995; 9:2170-83; PMID:7657168; http://dx.doi.org/10.1101/gad.9.17.2170.
- Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 2000; 275:16202-12; PMID:10821866; http://dx.doi.org/10.1074/jbc.275.21.16202.
- Dumont P, Leu JI-J, Della Pietra AC 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 2003; 33:357-65; PMID:12567188; http://dx.doi.org/10.1038/ng1093.
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577-90; PMID:12667443; http://dx.doi.org/10.1016/S1097-2765(03)00050-9.
- Leu JI-J, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004; 6:443-50; PMID:15077116; http://dx.doi.org/10.1038/ncb1123.
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303:1010-4; PMID:14963330; http://dx.doi.org/10.1126/science.1092734.
- Pietsch EC, Perchiniak E, Canutescu AA, Wang G, Dunbrack RL, Murphy ME. Oligomerization of BAK by p53 utilizes conserved residues of the p53 DNA binding domain. J Biol Chem 2008; 283:21294-304; PMID:18524770; http://dx.doi.org/10.1074/jbc.M710539200.
- Kroemer G, Petit P, Zamzami N, Vayssière JL, Mignotte B. The biochemistry of programmed cell death. FASEB J 1995; 9:1277-87; PMID:7557017.
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87:99-163; PMID:17237344; http://dx.doi.org/10.1152/physrev.00013.2006.
- Van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ 2002; 9:1031-42; PMID:12232790; http://dx.doi.org/10.1038/sj.cdd.4401088.
- Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta 2009; 1787:414-20; PMID:19007744; http://dx.doi.org/10.1016/j.bbabio.2008.10.005.
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9:47-59; PMID:18097445; http://dx.doi.org/10.1038/nrm2308.
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006; 24:841-51; PMID:17189187; http://dx.doi.org/10.1016/j.molcel.2006.11.026.
- Pietsch EC, Leu JI-J, Frank A, Dumont P, George DL, Murphy ME. The tetramerization domain of p53 is required for efficient BAK oligomerization. Cancer Biol Ther 2007; 6:1576-83; PMID:17895645; http://dx.doi.org/10.4161/cbt.6.10.4719.
- Lohrum MAE, Woods DB, Ludwig RL, Bálint É, Vousden KH. C-Terminal Ubiquitination of p53 Contributes to Nuclear Export. Mol Cell Biol 2001; 21:8521-32; PMID:11713287; http://dx.doi.org/10.1128/MCB.21.24.8521-8532.2001.
- Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J 2007; 26:923-34; PMID:17268548; http://dx.doi.org/10.1038/sj.emboj.7601560.
- Olsson A, Manzl C, Strasser A, Villunger A. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ 2007; 14:1561-75; PMID:17627286; http://dx.doi.org/10.1038/sj.cdd.4402196.
- Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ 2006; 13:941-50; PMID:16601750; http://dx.doi.org/10.1038/sj.cdd.4401925.
- Maclaine NJ, Hupp TR. The regulation of p53 by phosphorylation: a model for how distinct signals integrate into the p53 pathway. Aging 2009; 1:490-502; PMID:20157532.
- Zhivotovsky B, Samali A, Gahm A, Orrenius S. Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ 1999; 6:644-51; PMID:10453075; http://dx.doi.org/10.1038/sj.cdd.4400536.
- Mancini M, Nicholson DW, Roy S, Thornberry NA, Peterson EP, Casciola-Rosen LA, Rosen A. The Caspase-3 Precursor Has a Cytosolic and Mitochondrial Distribution: Implications for Apoptotic Signaling. J Cell Biol 1998; 140:1485-95; PMID:9508780; http://dx.doi.org/10.1083/jcb.140.6.1485.
- Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J 1999; 18:2040-8; PMID:10205158; http://dx.doi.org/10.1093/emboj/18.8.2040.
- Frank AK, Pietsch EC, Dumont P, Tao J, Murphy ME. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biol Ther 2011; 11:740-5; PMID:21307660; http://dx.doi.org/10.4161/cbt.11.8.14906.
- Mareninova OA, Sung K-F, Hong P, Lugea A, Pandol SJ, Gukovsky I, Gukovskaya AS. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem 2006; 281:3370-81; PMID:16339139; http://dx.doi.org/10.1074/jbc.M511276200.
- Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 2004; 24:6728-41; PMID:15254240; http://dx.doi.org/10.1128/MCB.24.15.6728-6741.2004.
- Chen X, Wong JYC, Wong P, Radany EH. Low-dose valproic acid enhances radiosensitivity of prostate cancer through acetylated p53-dependent modulation of mitochondrial membrane potential and apoptosis. Mol Cancer Res MCR 2011; 9:448-61; PMID:21303901; http://dx.doi.org/10.1158/1541-7786.MCR-10-0471.
- Brooks CL, Gu W. p53 Ubiquitination: Mdm2 and Beyond. Mol Cell 2006; 21:307-15; PMID:16455486; http://dx.doi.org/10.1016/j.molcel.2006.01.020.
- Sorrentino G, Mioni M, Giorgi C, Ruggeri N, Pinton P, Moll U, Mantovani F, Del Sal G. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell Death Differ 2013; 20:198-208; PMID:22935610; http://dx.doi.org/10.1038/cdd.2012.112.
- Ducruet AP, Vogt A, Wipf P, Lazo JS. Dual specificity protein phosphatases: therapeutic targets for cancer and Alzheimer disease. Annu Rev Pharmacol Toxicol 2005; 45:725-50; PMID:15822194; http://dx.doi.org/10.1146/annurev.pharmtox.45.120403.100040.
- Patterson KI, Brummer T, O'Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J 2009; 418:475-89; PMID:19228121.
- Shang X, Vasudevan SA, Yu Y, Ge N, Ludwig AD, Wesson CL, Wang K, Burlingame SM, Zhao Y-J, Rao PH, et al. Dual-specificity phosphatase 26 is a novel p53 phosphatase and inhibits p53 tumor suppressor functions in human neuroblastoma. Oncogene 2010; 29:4938-46; PMID:20562916; http://dx.doi.org/10.1038/onc.2010.244.
- Vasudevan SA, Skoko J, Wang K, Burlingame SM, Patel PN, Lazo JS, Nuchtern JG, Yang J. MKP-8, a novel MAPK phosphatase that inhibits p38 kinase. Biochem Biophys Res Commun 2005; 330:511-8; PMID:15796912; http://dx.doi.org/10.1016/j.bbrc.2005.03.028.
- Sayan BS, Sayan AE, Knight RA, Melino G, Cohen GM. p53 Is Cleaved by Caspases Generating Fragments Localizing to Mitochondria. J Biol Chem 2006; 281:13566-73; PMID:16531411; http://dx.doi.org/10.1074/jbc.M512467200.