
ABSTRACT
Nearly 100% of melanomas have a defect in the p16INK4A:cyclin D-CDK4/6:RB pathway, leading to abnormal cell cycle control and unregulated cellular proliferation. Here, we report that P1446A-05, a novel multi-CDK inhibitor has significant inhibitory activity against cutaneous and uveal melanoma. Mechanistic studies revealed that P1446A-05 inhibits phosphorylation targets of CDK members, and induces cell cycle arrest and apoptosis irrespective of melanoma genotype or phenotype. Additionally, we show preclinical evidence that P1446A-05 can synergize with other small molecule inhibitors previously studied in melanoma. Collectively, these data demonstrate that targeting cell cycle and transcriptional CDKs with a small molecule multi-CDK inhibitor is a viable approach for developing novel anti-melanoma therapeutics.
Introduction
The astounding progress made in melanoma therapeutics over the past several years, with successful targeted therapies and immune checkpoint antagonists, has not been without setbacks. The most prevailing limitations, the innate resistance of some melanomas to available treatments and the emergence of acquired resistance to treatment have propelled the search for novel agents. One area of intense interest for targeted therapies is the p16INK4A:cyclin D-CDK4/6:RB pathway (CDK4 pathway), a major regulatory pathway that helps maintain normal cellular proliferation by moderating cell cycle progression. An overwhelming majority of melanomas harbor genomic alterations in the CDK4 pathway, and it has been demonstrated that such alterations may be drivers of melanomagenesis or tumor progression, which would suggest a dependency on the key oncogenes in this pathway.Citation1-5 The success of vemurafenib and dabrafenib, selective BRAFV600E inhibitors, in melanoma gave credence to the strategy of targeting oncogene addiction, and in this light, many inhibitors of the CDK4 pathway are being investigated for potential therapeutic benefit.
Dysregulated cell cycle control unleashes cells from normal proliferation and can result in unrestricted cell division and tumorigenesis.Citation6 Proper cell cycle progression is controlled by a network of interactions between cyclin-dependent kinases (CDK), cyclins, and CDK inhibitors, which are synthesized and complex with each other during specific phases of the cell cycle. The CDK4 pathway is responsible for regulating the critical transition from the G1 to the S phase of the cell cycle. As cells receive mitogenic stimuli to proliferate, the expression of D-type cyclins increases, and they then complex with and activate CDK4, or the homologous CDK6, during the G1 phase. CDK4/6-cyclin D complexes phosphorylate retinoblastoma protein (RB), which effectively lifts the brake on gene transcription and allows for cell cycle progression.Citation3,7,8 p16INK4A, a CDK inhibitor, is part of the negative regulatory loop that keeps the activity of this pathway in check.Citation2 Alterations in this pathway that lead to amplified CDK4 pathway signaling and aberrant cell proliferation have been clinically correlated to increased melanoma risk. Patients harboring germline homozygous deletions and loss-of-function (LOF) mutations in CDKN2A (the gene encoding the p16INK4A protein) and activating mutations in CDK4 are 50 times more likely to develop melanoma.Citation5
The established role that these mutations have in melanoma biology has led to the synthesis of many CDK inhibitors in the preclinical and clinical testing pipelines for potential melanoma therapy. The focus of this study is P1446A-05, a unique multi-CDK inhibitor that has specific affinity for CDK4-cyclin D1, CDK1-cyclin B, and CDK9-cyclin T complexes with half-maximal inhibitory concentrations (IC50) of 90 nM, 25 nM, and 22 nM, respectively.Citation9,10 CDK1 plays a role in the later stages of the cell cycle, where it is believed to regulate the initiation of mitosis, when bound to cyclin A, and direct cells through mitosis, when complexed with cyclin B.Citation8,11 CDK9 is not a canonical cell cycle CDK; rather, CDK9-cyclin T participates in transcription by phosphorylating the C-terminal domain (CTD) of RNA polymerase II's Rpb1 subunit and promoting elongation.Citation8,12,13 P1446A-05 was previously shown to have potent antitumor activity across 30 human cancer cell lines, including non-small-cell lung (NSCL) cancer, colorectal carcinoma, and prostate cancer.Citation9,10 More recently, in 2 phase I clinical studies in patients with advanced refractory tumors, P1446A-05 was deemed to have an acceptable safety profile (NCT00840190, NCT00772876). In this study, we investigate the anti-melanoma activity of P1446A-05 and report that it has significant inhibitory activity against genotypically and phenotypically diverse human melanoma cell lines by promoting cell cycle arrest and inducing apoptosis, and additionally demonstrate preclinical evidence of synergistic cytotoxicity when P1446A-05 is combined with other targeted therapies.
Materials and methods
Reagents and antibodies
P1446A-05 was provided by Piramal Healthcare Limited (Mumbai, India). Dabrafenib and trametinib were purchased from Selleck Chemicals (Houston, TX). Primary antibodies used for western blots were purchased from Cell Signaling Technology (CST; Danvers, MA), Santa Cruz Biotechnology (SCB; Dallas, TX), or Abcam (Cambridge, MA), as follows: GAPDH (Abcam cat# ab8245), CDK4 (CST cat# 2906), CDK9 (SCB cat# sc-484), total RB (CST cat# 9309), phospho-RB Ser780 (CST cat# 9307), total Rpb1 CTD (CST cat# 2629), phospho-Rpb1 CTD Ser2 (CST cat# 8798), cleaved PARP (CST cat# 9541). HRP-conjugated secondary antibodies were purchased from CST (cat #'s 7074 and 7076).
Human melanoma cells and cell culture
Human melanoma cell lines used in this study including BRAFV600E/NRASWT genotypes (A373-C6, A375, K1, K4, SK-MEL-37, WM1158, and WM793), NRASQ61K/L/BRAFWT genotypes (Mel Juso, MGH-SW-1, and SK-MEL-63), a BRAFWT/NRASWT genotype (CHL-1), and several uveal phenotypes (C918, Mel202, Mel205, MEL270, OCM-1, and OMM 2.3). A375 and CHL-1 were purchased from American Type Culture Collection (Rockville, MD); A375-C6 was purchased from Sigma-Aldrich (Natick, MA); WM793 and WM1158 were gifted from Meenhard Herlyn (Wistar Institute, Philadelphia, PA); C918 and OCM-1 were gifted from Elisabeth Seftor (Children's Memorial Hospital, Chicago, IL); OMM2.3, Mel202, Mel205, and Mel270 were gifted from Bruce Ksander (Schepens Eye Research Institute, Boston, MA); and the following cell lines were previously published, with respective citations: SK-MEL-63,Citation14 K1,Citation15 SK-MEL-37,Citation16 Mel Juso,Citation17 and MGH-SW-1.Citation18 Cutaneous melanoma cells were cultured in vitro in Dulbecco's Modified Eagle Medium (Corning Life Sciences, Tewksbury, MA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA), 100 units/mL penicillin (Life Technologies), and 100 µg/mL streptomycin (Life Technologies). Uveal melanoma cell lines were cultured in vitro in RPMI-1640 with L-glutamine (Lonza, Walkersville, MD) supplemented with 10% fetal bovine serum, 1% HEPES (Lonza), 100 units/mL penicillin, 100 µg/mL streptomycin, and 0.1% β-mercaptoethanol (Sigma-Aldrich). The A375 shTP53 and shGFP lines, as well as vemurafenib-resistant lines, were previously generated and described by our laboratory.Citation19-21 All cells were maintained in incubators at 37°C with an atmosphere of 95% room air and 5% CO2.
2D cell viability assays
Melanoma cells were seeded in 96-well, white-walled, tissue culture plates at a density of 2 × 103 cells/well; all treatments were performed in triplicate. Drug compounds were added 24 hours after initial cell seeding and then cells were incubated for another 72 hours. Cell viability was measured with the CellTiter-Glo luminescence assay (Promega, Madison, WI). In brief, 30 µL of reconstituted reagent was added to each well, plates were incubated, protected from light, for 10 minutes at room temperature on a shaking platform (low speed), and luminescence (total light emission) was measured on either a Molecular Devices Spectramax M5 or Spectramax Plus 384 plate reader (Sunnyvale, CA) with an integration time of 500 ms. Raw data were normalized to DMSO-treated controls and GI50 values were calculated via nonlinear regression curve fit, with error bars representing 95% confidence intervals (GraphPad Prism 6, La Jolla, CA). CompuSyn software (ComboSyn, Inc.,) was used to calculate synergy based on the methodology and algorithms of Chou.Citation22
3D cell culture
Pre-chilled 96 well plates were coated with matrigel (BD Biosciences, San Jose, CA) (100 µl/well) and incubated for 30 minutes at 37°C. Melanoma cells were seeded on the gel (100 cells in 100 µl media/well) and incubated for 30 min at 37°C. Then 100 µl media containing 10% matrigel was added to each well and incubated at 37°C. One week after culture at 37°C with an atmosphere of 95% room air and 5% CO2, the P1446A-05 was added. After 72 hours of treatment, pictures of colonies were taken and the sizes of the colonies were measured and analyzed.
Cell cycle profiling and apoptosis assay
For both cell cycle and apoptosis experiments, cells were seeded in 6-well tissue culture plates at a density of 3 × 105 cells per well; all treatments were performed in triplicate. P1446A-05 and DMSO were added 24 hours after initial cell seeding and incubated for another 24 or 48 hours. To prepare cells for cell cycle profiling, all cells were collected, washed twice with PBS, fixed in 70% ethanol (v/v) and stored at −20°C overnight. Cells were then centrifuged, washed with PBS, and suspended in a 100 μg/ml propidium iodide (Life Technologies) solution with 100 μg/ml of RNAse (BD Biosciences) and 0.1% (v/v) IGEPAL CA-630 (Sigma-Aldrich) in PBS and incubated, protected from light, for 30 minutes at room temperature. Flow cytometry was then performed on a BD FACSCalibur (BD Biosciences), and data were analyzed using FlowJo 7.6.5 software (Ashland, OR).
For further apoptosis analysis, the manufacturer's protocol for an Alexa Fluor 488 annexin V/PI staining kit was followed (Life Technologies). In brief, cells were collected after P1446A-05 treatment, washed twice with PBS, and stained with Alexa Fluor annexin V and propidium iodide (PI) for 15 minutes, protected from light, at room temperature. Flow cytometry was then performed on a BD FACSAria (BD Biosciences), and data were analyzed using FlowJo 7.6.5 software. Early apoptosis (annexin V+/PI-) and late apoptosis (annexin V+PI+) populations were considered in aggregate in .
Western blot analysis
Western blot was performed using standard protocol previously described by our laboratory.Citation19-21 In brief, equal amounts of protein lysate (10–20 µg) were run on pre-cast 4–20% SDS-polyacrylamide gels and transferred to PVDF membranes (Bio-Rad, Hercules, CA). The membranes were blocked in 5% milk in TBS-Tween for 1 hour, and incubated with primary and secondary antibodies for 2 hours and 1 hour, respectively; blocking and antibody incubation were performed at room temperature. Proteins were detected using chemiluminescence substrate (Pierce, Rockford, IL) and X-ray film (Denville Scientific, South Plainfield, NJ).
Statistics
The data points represent the mean and error bars correspond to standard error of the mean (SEM) with n = 3, except in the GI50 bar graph, where they represent 95% confidence intervals. 2-way ANOVA was used to compare cell cycle profiles and induction of apoptosis. Statistical analyses were performed using GraphPad Prism 6 (La Jolla, CA).
Results
P1446A-05 significantly reduces proliferation of human melanoma cell lines in 2D and 3D culture
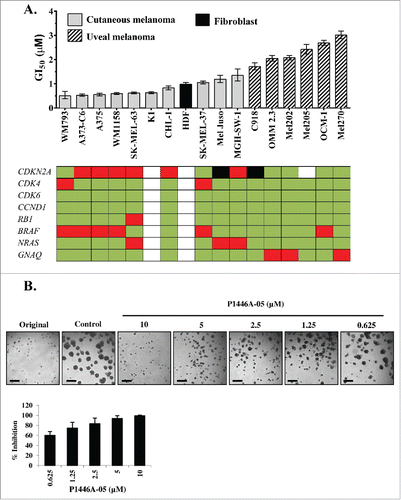
P1446A-05 was designed and synthesized as a novel multi-CDK inhibitor. To determine the activity of P1446A-05 against melanoma, we treated a panel of human cutaneous and uveal melanoma cell lines with P1446A-05 at a series of concentrations (0.1 to 30 µM) for 72 hours. The results demonstrated that P1446A-05 had growth inhibiting effects across the full panel of cell lines tested, with a 6-fold range in the concentration that inhibited cellular proliferation by 50% (GI50); the most sensitive (WM793) and least sensitive (MEL270) cell lines had GI50's of 0.51 µM and 3.02 µM, respectively (). In fact, the 6 uveal melanoma lines were the least sensitive lines based on GI50 values. The mean GI50 of these uveal lines was 2.33 µM, which is significant higher compared to a mean GI50 of 0.78 µM among the cutaneous melanoma lines (P < 0.0001). The four most sensitive lines were BRAFV600E-mutant cell lines, although there does not appear to be a strict genotype-phenotype correlation. The compiled dose-response curves for all lines tested are presented in the supplementary data (Fig. S1). The mutation information of the cell lines used is shown in Table S1. P1446A-05 also had growth inhibitory and cytotoxic effects on A375 and Mel-Juso spheroid formation in a 3D model. Following 72 hours of P1446A-05 treatment, the colonies were significantly reduced in size in a dose-dependent manner (, Fig. S2).
Figure 1. P1446A-05 strongly reduces proliferation of human melanoma cell lines in 2D and 3D culture. (A) P1446A-05 exhibits antiproliferative effects across different melanoma genotypes and phenotypes. Sensitivity ranking placed cutaneous melanoma lines ahead of uveal melanoma lines. Cells were incubated with a range of P1446A-05 concentrations for 72 hours, and cell viability was analyzed using the CellTiter-Glo luminescence assay. GI50 values were calculated using nonlinear regression curve fit in GraphPad Prism 6 (error bars show 95% CI). (B) Escalating doses of P1446A-05 demonstrate strong inhibition of 3D melanoma spheroids. Mel Juso cells were cultured as spheroids in matrigel and micrographs were taken after 72 hours of drug treatment. Representative images are shown; scale bars represent 50 µm in spheroid images. Analysis of spheroid size was used to calculate % inhibition relative to control.
P1446A-05 arrests cell cycle progression and induces apoptosis irrespective of melanoma genotype or phenotype
Having established the efficacy of P1446A-05 against the panel of melanoma cell lines, we sought to characterize the mechanism underlying the observed growth inhibiting response. Treatment of A375 (BRAFV600E/NRASWT), SK-MEL-63 (NRASQ61K/BRAFWT), and OCM-1 (uveal) revealed increases in both sub-G1 fractionation and G2/M arrest at 5 µM P1446A-05; both changes were statistically significant compared to DMSO-treated controls (P<0.001) (). At lower P1446A-05 concentrations (e.g. 0.5 µM), there appeared to be an increase in the G1 population (Fig. S3). The substantial rise in the sub-G1 population for the A375, SK-MEL-63 and OCM-1 lines at 24 hrs was largely preserved at 48 hrs, except for the OCM-1 line (). There also appeared to be partial dose dependence in terms of the apoptotic response.
Figure 2. P1446A-05 induces cell cycle arrest and apoptosis. (A, B) P1446A-05 causes G2-phase cell cycle arrest and an increase in sub-G1-phase cells at 24 and 48 hours. Cells were treated with 1 µM and 5 µM P1446A-05 for 24 hours and 1 µM P1446A-05 for 48 hours. Cell cycle progression was assessed using PI staining and flow cytometry. (C) P1446A-05 induces apoptosis, which correlates to the increase in sub-G1-phase cells. A375 and SK-MEL-63 cells were treated with 1 µM P1446A-05, and OCM-1 cells were treated with 1.5 µM P1446A-05. Apoptosis was assessed using annexin VPI staining and flow cytometry. (D) Compared to DMSO-treated controls, A375 cells treated with 1 µM P1446A-05 revealed a robust increase in cleaved PARP. GAPDH served as a loading control. Error bars represent SEM from triplicates. *P < 0.0001, #P < 0.001.
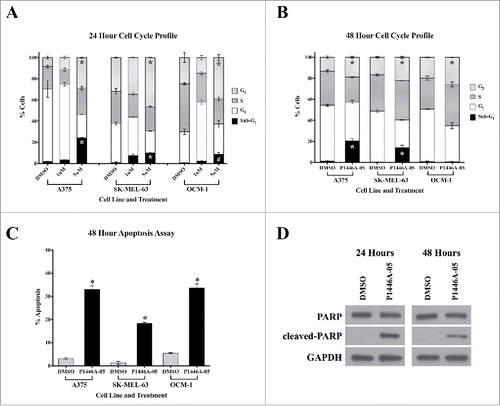
In order to further confirm P1446A-05-induced apoptosis, we next analyzed the cellular response after P1446A-05 treatment using FITC annexin V () and PARP cleavage () assays. The results showed that P1446A-05 significantly increased the apoptotic cell percentage (), and increased the PARP cleavage (). These results both corroborated the presence of a robust apoptotic response. Taken together, these findings indicate that P1446A-05 has dramatic anti-melanoma activity by arresting cell cycle at G2/M and inducing cell apoptosis.
P1446A-05 inhibits CDK family members
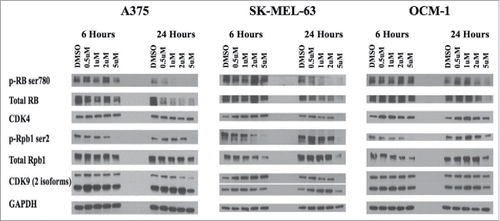
We next validate the intracellular targets of P1446A-05. Two cutaneous melanoma lines (A375 and SK-MEL-63) and one uveal melanoma line (OCM-1) were treated with either DMSO or P1446A-05 (0.5, 1, 2, and 5 µM) for 6 and 24 hours (). In terms of transcription, P1446A-05 is known to inhibit CDK9, which is known to phosphorylate Rpb1 and regulate transcription.Citation13 As shown in , exposure to P1446A-05 dramatically reduced pRpb1Ser2 by 6 hours, suggesting that P1446A-05 could rapidly block CDK9. In terms of cell cycle effects, P1446A-05 reduced pRBSer780, a target of CDK4 and CDK6, by 24 hours; total RB was also decreased by the drug, perhaps through its potential anti-transcriptional effects. Interestingly, the effect on pRBSer780 was more pronounced in the A375 line compared to the OCM1 line, which is consistent with the viability results (). In addition, pCDK7Thr170 was diminished with drug treatment (Fig. S4), suggesting that CDK1 may also be targeted although pCDK7Thr170 is not solely phosphorylated by CDK1. Thus, P1446A-05 does appear to be a multi-CDK inhibitor.
Figure 3. Intracelluar target validation. P1446A-05-mediated inhibition of CDK4 and CDK9 was confirmed via protein gel blotting for relevant phosphotargets in both CDK pathways. CDK4s phosphotarget on RB protein (ser780) becomes inhibited by 24 hours across all 3 cell lines in a dose dependent manner. CDK9s phosphotarget on RNAPII's Rpb1 (ser2) revealed better inhibition at 6 hours, and was only inhibited with the highest dose of P1446A-05 at 24 hours. The housekeeping protein GAPDH served as a loading control.
P1446A-05 has synergistic potential with other anti-melanoma small molecule inhibitors
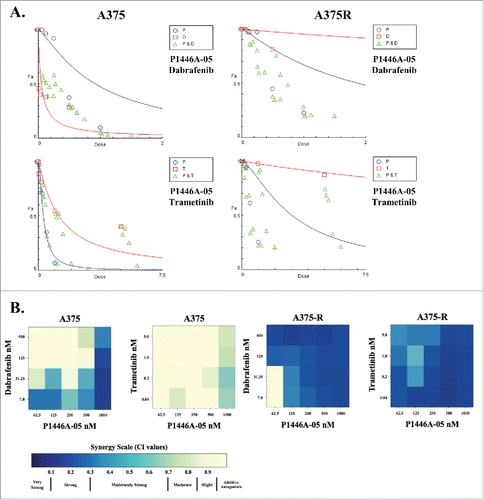
The growing interest in combination therapies aimed toward therapeutic resistance led us to investigate the potential of P1446A-05 to synergize with other MAPK pathway-targeted therapies to inhibit the proliferation of paired BRAFV600E vemurafenib sensitive (A375 and K4) and resistant (A375-R and K4-R) melanoma lines; resistant cell lines were generated via chronic exposure to increasing concentrations of vemurafenib. These cell lines were treated with either dabrafenib (BRAF inhibitor) or trametinib (MEK inhibitor) and P1446A-05 for 72 hours. P1446A-05 and dabrafenib demonstrated pockets of moderately strong to strong synergism in naive A375 cells. However, the combinations of P1446A-05 and dabrafenib or trametinib were highly synergistic at almost all dose combinations in the A375-R cells (). While a consistent pattern of synergy was not identified with the K4 lines (both BRAFV600E), our results suggest that P1446A-05 and dabrafenib were more effective against vemurafenib-resistant K4 cells, while P1446A-05 and trametinib produced better synergy in vemurafenib-sensitive K4 cells ().
Figure 4. Synergism between P1446A-05 and other small molecule inhibitors. (A) Isobolograms demonstrating synergy between P1446A-05 and dabrafenib or trametinib. The isobolograms were generated using Compusyn software. P, P1446A-05; T, trametinib; D, dabrafenib. (B) Heat maps depicting combination indices (CI's) for dose combinations of P1446A-05 with dabrafenib and trametinib. A375 and K4 cell lines, both sensitive and resistant (A375-R, K4-R) to vemurafenib, were treated. Cells were incubated with all drug combinations at indicated doses for 72 hours, cell viability was analyzed with CellTiter-Glo, and CI's were calculated using CompuSyn software.
Discussion
In this study, we show that a novel multi-CDK inhibitor, P1446A-05, has in vitro activity against a genotypically and phenotypically diverse panel of human melanoma cell lines, produces anti-proliferative effects by modulating G2/M arrest and inducing apoptosis, and synergizes with other small molecule inhibitors. Together, these findings support the use of broad-spectrum CDK inhibitors as a potential therapeutic option for melanoma patients, and are in agreement with preclinical studies that have investigated other multi-CDK inhibitors as anti-melanoma agents.Citation5,12,23,24
We determined that the cellular phenotype underlying the P1446A-05-mediated growth inhibition was a combination of G2/M phase cell cycle arrest and apoptosis. Based on other studies of CDK4/6 inhibitors in melanoma that revealed a predominant G1 phase arrest with little or no apoptosis, we expected that P1446A-05 would cause some G1 phase arrest.Citation5,24 Our results, however, are more in line with those achieved by dinaciclib, a specific inhibitor of CDK1/2/5/9, which also causes G2/M phase cell cycle arrest and apoptosis.Citation12,23
We validated that P1446A-05 inhibits the kinase function of CDK4 and CDK9, so while we are confident that the anti-proliferative effects produced by this drug resulted in some part from appropriate CDK inhibition, it is impossible to rule out contributory off-target effects. The decreases in CDK4 and CDK9 levels after treatment with high-dose P1446A-05 for 24 hours were not expected, but a possible explanation for this effect, as well as the observed decreases in total RB and total Rpb1, is reduced gene expression secondary to P1446A-05-mediated inhibition of CDK9, which is involved in transcriptional regulation. Supporting this explanation is the fact that expression levels of these proteins were least affected in the OCM-1 cell line, which was less sensitive to P1446A-05, in terms of GI50 value, than A375 and SK-MEL-63.
The celebrated success stories of targeted therapies, starting with vemurafenib and being echoed by dabrafenib and trametinib, are incomplete without recognizing that the majority of patients with an initial disease response will eventually relapse as their melanomas develop resistance to inhibition of the MAPK pathway. The paradigm of anti-melanoma targeted therapies is shifting from monotherapy to combination therapy, with the goal of targeting multiple pathways to prevent the emergence of acquired resistance. Increased p16INK4A:cyclin D-CDK4/6:RB pathway signaling is one of many proposed mechanisms behind acquired resistance to BRAF inhibition, and copy number variants in important genes in this pathway have been correlated to worse BRAF inhibitor treatment response.Citation2,3,25 The results of our synergy studies using matched vemurafenib-sensitive and vemurafenib-resistant A375 and K4 cell lines are particularly intriguing and support the therapeutic strategy of combining CDK inhibitors with targeted MAPK therapies. To explain the mechanism of this synergy would be highly speculative, however, further exploration of the physiologic mechanisms responsible for the synergy of these drug combinations is needed to elucidate and fine-tune their potential therapeutic benefits. For instance, in the A375 model, combinations of P1446A-05 with dabrafenib or trametinib worked better against vemurafenib-resistant cells, suggesting that a promising application of either combination would be in patients who have progressed on BRAF inhibitor therapy.
In summary, this study presents P1446A-05 as an effective in vitro anti-melanoma agent that produces cytotoxic effects against a range of melanoma genotypes and phenotypes by causing cell cycle arrest and apoptosis. P1446A-05 has previously demonstrated in vitro activity in colorectal, NSCL, and prostate cancer, and has completed 2 phase I clinical trials in patients with advanced refractory malignancies, with an acceptable safety profile (NCT00840190, NCT00772876).Citation9,10 Our work provides further preclinical, mechanistic support for P1446A-05 as a potential novel cancer therapy, especially for advanced melanoma patients. We also demonstrate the potential for P1446A-05 to synergize with other small molecule inhibitors to create enhanced combination therapies for melanoma. Further in vitro and in vivo investigations of specific multi-CDK inhibitors, which target both canonical cell cycle and transcriptional CDKs, alone and in combination, are warranted to expand upon these preliminary findings and enhance synergistic and therapeutic potential.
Disclosure of potential conflicts of interest
SB and SPS are both employees of Piramal Enterprises Limited; neither had control over the design of experimentation, analysis of results, or writing of the manuscript.
Supplementary_Datas.zip
Download Zip (723.3 KB)Acknowledgments
We would like to acknowledge the excellent staff of the Photopathology Core Facility within the Wellman Center for Photomedicine: Dr. Jie Zhao, Danny Cao, Megan Scanlan, Lingxian Wu, and Alyssa Lovas. Their expertise and availability in helping with experimental planning and data acquisition were enormous assets to this project.
References
- Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005; 353:2135-47; PMID:16291983; http://dx.doi.org/10.1056/NEJMoa050092
- Lee B, Sandhu S, McArthur G. Cell cycle control as a promising target in melanoma. Curr Opin Oncol 2015; 27:141-50; PMID:25588041; http://dx.doi.org/10.1097/CCO.0000000000000159
- Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res: Off Am Assoc Cancer Res 2013; 19:5320-8; PMID:24089445; http://dx.doi.org/10.1158/1078-0432.CCR-13-0259
- Walker GJ, Flores JF, Glendening JM, Lin AH, Markl ID, Fountain JW. Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B, or one of their downstream targets. Genes Chromosomes Cancer 1998; 22:157-63; PMID:9598804; http://dx.doi.org/10.1002/(SICI)1098-2264(199806)22:2<157::AID-GCC11>3.0.CO;2-N
- Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, Do H, Mitchell C, Cullinane C, Liu W, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res 2014; 27:590-600; PMID:24495407; http://dx.doi.org/10.1111/pcmr.12228
- Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 2001; 1:222-31; PMID:11902577; http://dx.doi.org/10.1038/35106065
- Miller DM, Flaherty KT. Cyclin-dependent kinases as therapeutic targets in melanoma. Pigment Cell Melanoma Res 2014; 27:351-65; PMID:24405945; http://dx.doi.org/10.1111/pcmr.12211
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9:153-66; PMID:19238148; http://dx.doi.org/10.1038/nrc2602
- Gupta S, Jain MM, Maru A, Nag SM, Somani N, Mehta AO, et al. Abstract: A phase I study of selective cyclin dependent kinase inhibitor P1446A-05 administered on an intermittent schedule in patients with advanced refractory tumors. J Clin Oncol: Off J Am Soc Clin Oncol. 2012; 30(supplement):3011
- Joshi KS, Padgaonkar A, Rathos M, Wagh V, Manohar S, Bhatia D, et al. Abstract 3054: P1446A-05: a new oral cyclin-dependent kinase inhibitor with potent preclinical antitumor activity. Cancer Res 2012; 72(8 Supplement):3054; PMID:22282653; http://dx.doi.org/10.1158/1538-7445.AM2012-3054
- Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 2005; 30:630-41; PMID:16236519; http://dx.doi.org/10.1016/j.tibs.2005.09.005
- Desai BM, Villanueva J, Nguyen TT, Lioni M, Xiao M, Kong J, Krepler C, Vultur A, Flaherty KT, Nathanson KL, et al. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PloS One 2013; 8:e59588; PMID:23527225; http://dx.doi.org/10.1371/journal.pone.0059588
- Fisher RP. The CDK network: linking ycles of cell division and gene expression. Genes Cancer 2012; 3:731-8; PMID:23634260; http://dx.doi.org/10.1177/1947601912473308
- Rudy W, Guckel B, Siebels M, Lindauer M, Meuer SC, Moebius U. Differential function of CD80- and CD86-transfected human melanoma cells in the presence of IL-12 and IFN-gamma. Int Immunol 1997; 9:853-60; PMID:9199968; http://dx.doi.org/10.1093/intimm/9.6.853
- Katagata Y, Aoki T, Hozumi Y, Yoshida T, Kondo S. Identification of K1/K10 and K5/K14 keratin pairs in human melanoma cell lines. J Dermatol Sci 1996; 13:219-27; PMID:9023704; http://dx.doi.org/10.1016/S0923-1811(96)00538-5
- Saito Y, Wang Z, Hagino-Yamagishi K, Civelli O, Kawashima S, Maruyama K. Endogenous melanin-concentrating hormone receptor SLC-1 in human melanoma SK-MEL-37 cells. Biochem Biophys Res Commun 2001; 289:44-50; PMID:11708774; http://dx.doi.org/10.1006/bbrc.2001.5926
- van Ham SM, Tjin EP, Lillemeier BF, Gruneberg U, van Meijgaarden KE, Pastoors L, Verwoerd D, Tulp A, Canas B, Rahman D, et al. HLA-DO is a negative modulator of HLA-DM-mediated MHC class II peptide loading. Curr Biol 1997; 7:950-7; PMID:9382849; http://dx.doi.org/10.1016/S0960-9822(06)00414-3
- Padua RA, Barrass NC, Currie GA. Activation of N-ras in a human melanoma cell line. Mol Cell Biol 1985; 5:582-5; PMID:3887133; http://dx.doi.org/10.1128/MCB.5.3.582
- Ji Z, Kumar R, Taylor M, Rajadurai A, Marzuka-Alcala A, Chen YE, Njauw CN, Flaherty K, Jönsson G, Tsao H. Vemurafenib synergizes with nutlin-3 to deplete survivin and suppresses melanoma viability and tumor growth. Clin Cancer Res 2013; 19:4383-91; PMID:23812671; http://dx.doi.org/10.1158/1078-0432.CCR-13-0074
- Ji Z, Njauw CN, Taylor M, Neel V, Flaherty KT, Tsao H. p53 rescue through HDM2 antagonism suppresses melanoma growth and potentiates MEK inhibition. J Invest Dermatol 2012; 132:356-64; PMID:21993556; http://dx.doi.org/10.1038/jid.2011.313
- Kumar R, Taylor M, Miao B, Ji Z, Njauw JC, Jonsson G, Frederick DT, Tsao H. BAP1 has a survival role in cutaneous melanoma. J Invest Dermatol 2015; 135:1089-97; PMID:25521456; http://dx.doi.org/10.1038/jid.2014.528
- Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58:621-81; PMID:16968952; http://dx.doi.org/10.1124/pr.58.3.10
- Abdullah C, Wang X, Becker D. Expression analysis and molecular targeting of cyclin-dependent kinases in advanced melanoma. Cell Cycle 2011; 10:977-88; PMID:21358262; http://dx.doi.org/10.4161/cc.10.6.15079
- Yadav V, Burke TF, Huber L, Van Horn RD, Zhang Y, Buchanan SG, Chan EM, Starling JJ, Beckmann RP, Peng SB. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer ther 2014; 13:2253-63; PMID:25122067; http://dx.doi.org/10.1158/1535-7163.MCT-14-0257
- Nathanson KL, Martin AM, Wubbenhorst B, Greshock J, Letrero R, D'Andrea K, O'Day S, Infante JR, Falchook GS, Arkenau HT, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clinical Cancer Res 2013; 19:4868-78; PMID:23833299; http://dx.doi.org/10.1158/1078-0432.CCR-13-0827