
ABSTRACT
Activation of Estrogen receptor (ER) α (α) promotes cell growth and influences the response of cancer cell to chemotherapeutic agents. However, the mechanism by which ERα activation antagonizes cells to chemotherapy-induced cytotoxicity remains unclear. Here, we investigated the effect of cisplatin on ERα activation. In addition, we examined whether down-regulation of ERα modulate cisplatin-mediated cytotoxicity using 2 human ovarian cancer cells (Caov-3 and Ovcar-3) transduced with ERα short hairpin RNA (shRNA). The proliferation assay showed that 17β-estradiol (E2) induced cell proliferation via activation of Akt and extracellular signal-regulated kinase (ERK) cascades, while shRNA mediated downregulation of ERα inhibited the cell proliferation. Immunoblot analysis revealed that cisplatin induced the phosphorylation of ERα at serine 118 via ERK cascade. Luciferase assay showed that cisplatin increases transcriptional activity of estrogen-responsive element (ERE). The E2-stimulated ERα activation attenuated cisplatin-induced cytotoxicity. Meanwhile, down-regulation of ERα inhibited E2-induced protective effect on cisplatin toxicity as determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays. Moreover, Pretreatment with E2 followed by cisplatin decreased the expression of cleaved PARP, and increased the expression of anti-apoptotic protein Bcl-2. Collectively, our findings suggest that activation of ERα by E2 and cisplatin can induce platinum-resistance by increasing the expression of anti-apoptotic protein in ovarian cancer cells. Therefore, our findings provide valuable information that ERα might be a promising therapeutic target for platinum-resistant ovarian cancer.
Abbreviations
| ER | = | estrogen receptor |
| ERK | = | extracellular signal regulated protein kinase |
| PI3K | = | phosphatidylinositol 3-kinase. |
Introduction
Ovarian cancer is the most lethal gynecological cancer, and approximately 70% of patients are diagnosed with advanced stage, International Federation of Gynecology and Obstetrics (FIGO) stage 3-4.Citation1 Currently, standard therapy of ovarian cancer is based on primary debulking surgery followed by platinum-based chemotherapy.Citation2 Although the majority of patients with ovarian cancer respond to initial chemotherapy, most of them relapse during the treatment.Citation3 Recurrence of ovarian cancer has a poor prognosis, with a 5 y survival rate of 23% and 14% for FIGO stage 3 and 4, respectively.Citation4 A strong predictive factor for recurrent ovarian cancer patients is the sensitivity of their tumor to platinum.Citation5 Therefore, it is very important to understand how cancer becomes platinum-resistant and to develop molecular targeting therapies for platinum-resistant ovarian cancer.
Estrogen receptor (ER) α (α) is a member of the nuclear hormone receptor superfamily and classified as a ligand activated transcriptional factor.Citation6 ERα is expressed in more than 50% of ovarian cancersCitation7 and ERα expression is associated with poor prognosis in ovarian cancer patients.Citation7,8 We previously reported that common ovarian tumors frequently produce 17β-estradiol (E2).Citation9 Plasma levels of E2 have positive correlation with tumor volume and stage in malignant ovarian tumors.Citation10 It has been shown that E2 production takes place in tumor stroma, whereas ERα is localized to tumor cells in ovarian tumors.Citation11 These findings suggest that E2-mediated ERα activation may have a role in proliferating the tumor cells by a paracrine fashion in ovarian tumors. Experimentally, E2 stimulates the growth of ovarian cancer cell lines expressing ERα.Citation12 E2 bound ERα activates the expression of genes involved in cell proliferation and survival, thus promoting tumor growth and progression. E2-activated ER transcriptional activity was thought to be the major mechanism by which ER regulates cell behavior, and has been termed genomic action of the ER. Recently, cross talk between estrogen bound ER and signal transduction are suggested to affect ER-mediated functions more than genomic action of the ER. This alternative mechanism is termed non-genomic actions of the ER. Several studies show that E2-stimulated ERα promotes cell proliferation via activation of the extracellular signal regulated protein kinases (ERK) in breast cancer cells.Citation13,14 We previously reported that the phosphatidylinositol 3-kinase (PI3K)-Akt cascade is activated by E2 bound ERα in ovarian cancer cells.Citation15 The Ras/Raf/ERK and PI3K/Akt pathways are involved in proliferation, invasion, metastasis and survival in cancer cells.Citation16
The balance between cellular survival and apoptosis can determine the sensitivity of tumor cells to chemotherapeutic agents. Cisplatin activates the PI3k/Akt or ERK cascades, which promote cell survival, via inactivation of the proapoptotic proteins Bcl-2-associated death protein (BAD) and caspase-9.Citation17 We found that both the PI3K-Akt and ERK pathways are activated by cisplatin in vitro and in vivo, and are involved in resistance to cisplatin and paclitaxel.Citation18-21 Interestingly, E2-mediated ERα activation antagonizes the cytotoxicity of paclitaxel, doxorubicin and cisplatin in breast cancer cells.Citation22-24 Under in vitro condition, the ER antagonist ICI 182,780 (ICI) can improve the efficacy of cisplatin in ovarian cancer cells.Citation25 However, it has been unknown if ERα activation induces platinum resistance in ovarian cancer. In this study, we examined whether cisplatin induces the phosphorylation of ERα via activation of the ERK or Akt cascade. We also investigated the effects of E2-induced ERα activation on sensitivity to cisplatin.
Results
shRNA mediated downregulation of ERα attenuates E2-induced cell proliferation in ovarian cancer cells
We first examined the expression of ERα in ovarian cancer cell lines. MCF-7 cells which expressing ERα were used as a positive control. Immunoblot analysis showed that ERα is highly expressed in Caov-3 and Ovcar-3 cells (). Next, we investigated the effects of E2 on cell proliferation in Caov-3 and Ovcar-3 cells (). E2 significantly induced cell growth at 10−8 M in both cell lines. Although the pure antiestrogen ICI182780 had no effect on the basal cell growth, it significantly inhibited E2-induced cell growth at 10−8 M in both cell lines. To confirm that E2 induced cell proliferation via ERα, we down-regulated ERα expression in Caov-3 and Ovcar-3 cells using lentiviral shRNA and generated batch clonal lines. The nontarget shRNA served as the control. Immunoblot analysis showed that shRNA targeting ERα markedly decreased the expression of ERα compared to cells transduced with control shRNA in both cell lines (). E2 induced cell proliferation in both cell lines transduced with control shRNA as well as wild type (, left upper and lower panels). In addition, shRNA mediated the down-regulation of ERα in both cell lines and inhibited the E2-induced proliferative effect (, right upper and lower panels). We previously reported that E2 induced cell proliferation via ERα mediated activation of the ERK and PI3K-Akt cascade, both of which are associated with cell proliferation and survival (20). Therefore, we confirmed that E2 induced phosphorylation of ERK and Akt ().
Figure 1. 17β-Estradiol (E2) induced proliferation of Caov-3 and Ovcar-3 cells and down-regulation of estrogen receptor (ER) attenuated E2-induced proliferative effect in these cells. (A) Expression of ERα was examined in Caov-3, Ovcar-3 and A2780 cells. The lysates were analyzed by protein gel blotting using anti-ERα antibody. Cellular lysate from MCF-7 were positive control for ERα. β-actin was used as an internal control. (B) Caov-3 cells (3 × 104 cell per well) and Ovcar-3 cells (6 × 104 cells per well) were in 12-well plates. Cells were allowed to attach overnight. After serum-free starvation for 24 h, cells were cultured with vehicle, E2 (10−8 M), ICI182780 (ICI, 10−6 M), or E2 (10−8 M) + ICI (10−6 M) for 6 d. (C) Specific shRNA for ERα or control shRNA were transfected into Caov-3 and Ovcar-3 cells. Knockdown of ERα expression by specific shRNA was confirmed using western blotting. (D) After control or ERα shRNA transfection, cells were starved with serum-free medium for 24 h and treated with vehicle or E2 (10−8 M) for 6 d. Culture media were exchanged every 48 h. Cells were counted using a cell counter. Values shown are the means ± SEM, n = 3, **P < 0.01 compared with vehicle. E, After serum-free starvation for 24 h, Caov-3 cells were treated with 10−8M E2 for indicated times. Lysates were subjected to protein gel blotting using anti-phospho-Akt antibody, anti-Akt antibody, anti-phospho-ERK antibody or anti-ERK antibody.
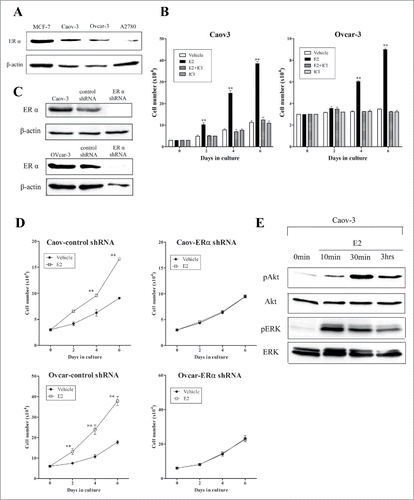
Cisplatin induced the phosphorylation of ERα at serine 118 via ERK cascade
We previously showed that cisplatin activated the ERK and Akt cascade,Citation27 which are known to activate ERα in breast cancer cells.Citation28 Therefore, we determined whether cisplatin induces the activation of ERα in ovarian cancer cells. Immunoblot analysis showed that cisplatin induced phosphorylation of ERα at serine 118 in Caov-3 cells (). We also examined the effects of cisplatin on the transcriptional activation of ERE via ERα. We transfected the ER-responsive receptor plasmid, ptk-ERE-luc, into Caov-3 cells and performed a luciferase assay. Cisplatin caused an increase of approximately 3-fold in luciferase activity compared with vehicle-treated cells. In addition, cotreatment with ICI inhibited the cisplatin-induced increase in luciferase activity in cells (). These results suggest that cisplatin activated ERα and affected its transcriptional activity. In addition, we examined the effect of LY294002 and PD98059 (inhibitors of PI3K/Akt and MEK, respectively) on the cisplatin-induced phosphorylation of ERα. Pretreatment with LY294002 had no effect on the cisplatin-induced phosphorylation of ERα. However, pretreatment with PD98059 attenuated the cisplatin-induced phosphorylation of ERα at serine 118 (). These results suggest that ERK may be a dominant cascade for cisplatin-induced ERα activation. We also examined whether E2 activates the ERα in ovarian cancer cells. Immunoblot analysis showed that E2 induced the phosphorylation of ERα at serine 118 and 167 in Caov-3 cells (supplementary data).
Figure 2. 17β-Estradiol (E2) induced phosphorylation of Akt and extracellular signal-regulated protein kinases (ERK), while cisplatin induced phosphorylation of estrogen receptor (ER) at serine 118 via ERK cascade and enhanced estrogen responsive element (ERE) transcriptional activity in Caov-3 cells. (A) After serum-free starvation for 24 h, Caov-3 cells were treated with 100μM cisplatin for indicated times. Lysates were analyzed by western blotting using anti-phospho-ERα (serine 118) antibody. (B) Caov-3 cells (10 × 104 cells per well) were plated in 6-well plates 24 h before transfection. Caov-3 cells were transfected with ptk-ERE-luc and pRLCMV as internal control. After 24 h of serum-free starvation, cells were treated with vehicle, E2 (10−8M), E2 (10−8 M) + ICI182780 (ICI, 10−6 M), cisplatin (100 μM) and cisplatin (100 μM) + ICI (10−6 M) for 24 h. Cell lysates were assayed for luciferase activity. Luciferase activity was normalized against Renilla lucifearse using the pRLCMV control vector. Values shown are the means ± SEM, n = 3, *P < 0.05, **P < 0.01 compared with vehicle. (C) After serum-free starvation for 24 h, Caov-3 cells were treated with or without 10 μM LY294002, or 10 μM PD98059 prior to 100 μM cisplatin for 3 h. Lysates were analyzed by protein gel blotting using anti-phospho-ERα (serine 118) antibody, anti-phospho-ERα (serine 167) antibody, anti-phospho-Akt antibody, anti-Akt antibody, anti-phospho-ERK antibody or anti-ERK antibody.
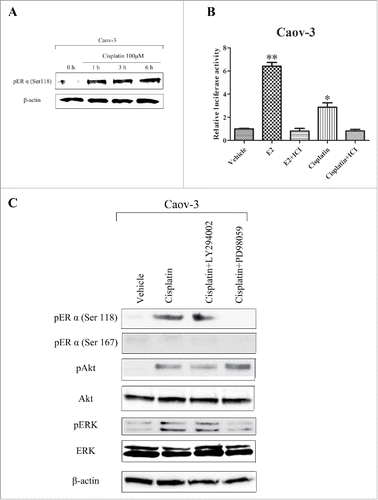
E2 antagonized cisplatin-induced cytotoxicity via ERα in ovarian cancer cells
A previous study reported that E2 antagonized cisplatin-induced cytotoxicity in breast cancer cells.Citation29 We tested the sensitivity of Caov-3 and Ovcar3 cells to cisplatin and the effect of E2 on cisplatin-induced cytotoxicity using the MTS assay. Cells were treated with vehicle, E2, ICI or E2 + ICI for 24 h, followed by the indicated concentrations of cisplatin for 24 h. Cisplatin inhibited cell viability dose-dependently in both cell lines (). Pretreatment with E2 significantly antagonized cisplatin-mediated cytotoxicity, while ICI inhibited the E2-induced protective effect on cisplatin cytotoxicity in both cell lines (). Further titrations revealed half-maximal inhibitory concentration (IC50) values of 109.9 ± 5.41 and 185.0 ± 9.08 μM in Caov-3 cells and 49.2 ± 0.36 and 92.7 ± 0.14 μM in Ovcar-3 cells for treatment with cisplatin and E2 + cisplatin, respectively (). Next, we examined whether downregulation of ERα inhibits the E2-induced protective effect on cisplatin toxicity. The ERα downregulated and nontarget control cells were treated with E2, followed by the indicated concentrations cisplatin for 24 h. E2 significantly antagonized cisplatin in Caov-3 cells transduced with the control shRNA (). However, shRNA mediated down-regulation of ERα inhibited the E2-induced protective effect on cisplatin toxicity (). Similar results were observed in Ovcar-3 cells transduced with control or ERα shRNA (). These results suggest that E2 promoted resistance to cisplatin via ERα in ovarian cancer cells.
Figure 3. 17β-Estradiol (E2) antagonized cisplatin-induced cytotoxicity in Caov-3 and Ovcar-3 cells. (A) Caov-3 and Ovcar-3 cells were treated with vehicle, E2 (10−8 M) or E2 (10−8 M) + ICI182780 (ICI, 10−6 M) for 24 h, followed by incubation with increasing concentrations of cisplatin (0.1–1000 μM) for 24 h. Values of cell viability were determined as described in Materials and Methods. (B) IC50 values (right panel) were determined by direct titration of viability with cisplatin. Values shown are the means ± SEM, n = 9, **P < 0.01 compared with cisplatin.
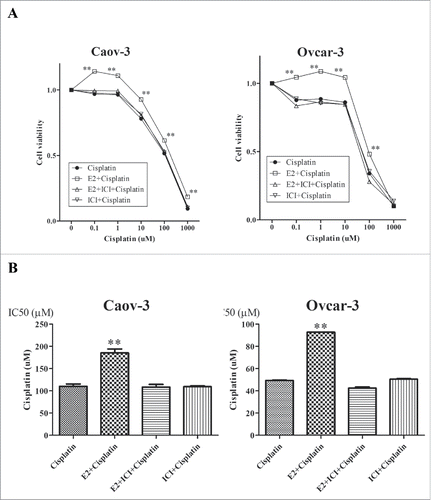
Figure 4. Knockdown of estrogen receptor (ER) α (α) expression attenuated the effect of 17β-Estradiol (E2) on cisplatin sensitivity in Caov-3 and Ovcar-3 cells. (A) After control or ERα shRNA transfection in Caov-3 cells, they were treated with vehicle or E2 (10−8 M) for 24 h, followed by incubation with increasing concentrations of cisplatin (0.1–1000 μM) for 24 h. Values of cell viability were determined as described in Materials and Methods. Values shown are the means ± SEM, n = 9, **P < 0.01 compared with cisplatin. (B) Same experiment was done using Ovcar-3 cells.
E2 induced cisplatin-resistance by increasing Bcl-2 expression
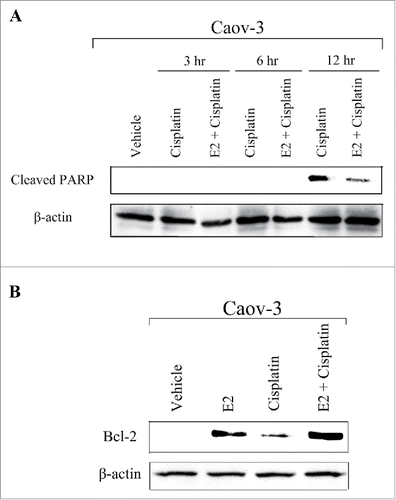
Next, we explored the potential mechanisms by which E2 promoted cisplatin-resistance. We examined the possibility that E2 decreases cisplatin-induced apoptosis using anti-cleaved PARP antibody. Immunoblot analysis showed that pretreatment with E2 decreased the expression of cleaved PARP induced by cisplatin in Caov-3 cells at 12 h (). Several studies have reported that E2 induced cisplatin-resistance by altering the expression of anti-apoptotic proteins in breast cancer cells.Citation29-31 Therefore, we examined the effects of E2 on the expression of anti-apoptotic proteins in Caov-3 cells. Pretreatment with E2 followed by cisplatin markedly increased the expression of Bcl-2 (). These results suggest that E2 promoted cisplatin-resistance by increasing the expression of anti-apoptotic protein Bcl-2.
Figure 5. 17β-Estradiol (E2) inhibited cisplatin-induced apoptosis in Caov-3 cells. (A) Caov-3 cells were treated with vehicle or E2 (10−8M) for 24 h, followed by 100 μM cisplatin for indicated times. Lysates were analyzed by western blotting using anti-Cleaved PARP antibody. (B) Caov-3 cells were treated with vehicle or E2 (10−8 M) for 24 h, followed by 100 μM cisplatin for 24 h. Lysates were analyzed by protein gel blotting using anti-Bcl-2 antibody.
Discussion
We discovered that cisplatin activates ERα via the ERK cascade. Furthermore, E2-induced ERα activation attenuates cisplatin-mediated cytotoxicity by increasing the expression of anti-apoptotic protein Bcl-2 in ovarian cancer cells. We also showed that shRNA mediated downregulation of ERα inhibited E2-induced protective effect on cisplatin toxicity. These findings suggest that activation of ERα by E2 and cisplatin can induce platinum-resistance in ovarian cancer cells. To the best of our knowledge, this is the first report to elucidate the association between ERα activation and cisplatin sensitivity in ovarian cancer cells.
We showed that the E2 induced cell proliferation via ERα was mediated by activation of the Akt and ERK cascades. In addition, the shRNA mediated down-regulation of ERα attenuated the E2-induced cell proliferation (). These results are consistent with other reports, which suggest that E2 bound ERα induced cell proliferation via activation of ERK and Akt.Citation12-14,32 Cisplatin induces activation of the ERK and Akt cascades, which promote cell survival and correlate with platinum resistance.Citation33 However, it was not previously known that cisplatin activates ERα in ovarian cancer cells. In this study, cisplatin induced the phosphorylation of ERα at serine 118 (). ERK and Akt signaling cascades phosphorylate ERα at serine 118 and 167, respectively.Citation28,34 PD98059 (ERK inhibitor) but not LY294002 (PI3K/Akt inhibitor) decreased cisplatin-induced ERα activation. These findings suggest that ERK may be the dominant cascade for the cisplatin-induced ERα activation in ovarian cancer cells. We also showed that E2 phosphorylated ERα at serine 118 and 167 (supplementary data). E2 and cisplatin activate the ERα via the same ERK pathways, therefore they may have addictive effects on ERα activation.
We showed that the E2-stimulated ERα activation attenuated cisplatin-induced toxicity in ovarian cancer cells (). These results are consistent with a previous report that E2 antagonizes the cytotoxicity of cisplatin in breast cancer cells expressing ERα.Citation29 We also showed that knockdown of ERα inhibited the E2-induced protective effect on cisplatin (). This inhibition suggests that ERα may be a potential target in developing alternative therapies for platinum-resistant ovarian cancer. Several studies have reported that E2 induced platinum-resistance by altering the expression of anti-apoptotic proteins in breast cancer cells.Citation29-31 We discovered that pretreatment with E2 followed by cisplatin treatment decreased apoptosis by increasing Bcl-2 expression (). In addition, we showed that cisplatin increased the transcriptional activity of ERα in ovarian cancer cells (). Several studies have shown that ERα activates the expression of genes associated with cell proliferation and survival, including Bcl-2.Citation35,36 Collectively, these findings suggest that ERα-targeted therapy may improve platinum-resistance in ovarian cancer.
A previous study demonstrates that ICI improve the efficacy of cisplatin in ovarian cancer cells,Citation25 which is inconsistent with our results. In our study, ICI did not increase cisplatin-induced cytotoxicity but attenuated the E2-induced protective effect of cisplatin in ovarian cancer cells (). These results suggest that E2-induced ERα activation might be important in the mechanisms mediating platinum resistance in ERα-positive ovarian cancer cells. Previously, we reported that common ovarian tumors frequently produced estrogen from the tumor stroma.Citation9 E2 released from the stroma of ovarian cancer cells may have a role in antagonizing platinum agents via ERα activation in a paracrine fashion.
Hormonal ERα-targeted therapy prevents disease recurrence and reduces mortality from ERα-positive breast cancer. However, the positive response to ERα-targeted therapy in ovarian cancer is limited. The clinical response rate of tamoxifen (a selective estrogen receptor modulator) for recurrent ovarian cancer was 48%.Citation37 Furthermore, combination therapy with first line chemotherapeutic agents and tamoxifen did not improve the efficacy of the cytotoxic agents in ovarian cancer patients.Citation38 However, the limitation of previous clinical studies is that ERα expression was not investigated. Further studies are required to evaluate the association between ERα status and response to tamoxifen. Fulvestrant is a novel estrogen receptor antagonist that competitively binds to ER with approximately 100 times greater than that of tamoxifen.Citation39 This binding induces a rapid degradation of ER, resulting in inhibition of transcriptional activity induced by E2. In phase II study of fulvestrant in ERα-positive recurrent ovarian cancer, almost half of the patients experienced clinical benefits and none developed grade 2, 3 or 4 toxicities.Citation40 Fulvestrant is well-tolerated and efficacious in patients with recurrent ovarian cancer. In this study, fulvestrant (ICI) could not increase the efficacy of cisplatin. It might have a weakness for combination therapy with cisplatin in clinical study. However, we showed that fulvestrant inhibited E2-induced protective effect on cisplatin cytotoxicity. Therefore, it may be useful for combination therapy with cisplatin in ovarian cancer cells releasing high levels of E2. Well-designed prospective trials are needed to identify suitable patients for ERα-targeted therapy in ovarian cancers.
In summary, we concluded that cisplatin activated ERα mediated by ERK cascade and E2-induced ERα activation attenuated cisplatin-induced toxicity. This attenuation was mediated by the inhibition of apoptosis in ovarian cancer cells. We also discovered that downregulation of ERα expression inhibited the cisplatin-resistance induced by E2. ERα may, therefore, be a promising therapeutic target for platinum-resistant ovarian cancer. Furthermore, previous clinical studies showed that the effect of ERα-targeted therapy is limited in ovarian cancer. It is therefore, important to develop new strategies using fulvestrant which is a new ER blocker, and select the appropriate patients in ERα-targeted therapy, which would be beneficial to ovarian cancer patients.
Materials and methods
Reagents
Cisplatin, E2 and ICI were obtained from Sigma-Aldrich (St. Louis, MO, USA). Anti-ERα antibody (SC-542) for western blotting was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho ERα (Ser118) (#2511), anti-phospho ERα (Ser167) (#5587), anti-phospho Akt (Ser473) (#4058), anti-Akt (#9272), anti-phospho-p44/42 mitogen-activated protein kinases (MAPK, ERK 1/2, Thr202/Tyr204) (#9106), anti-p44/42 MAPK (ERK 1/2) (#4695), anti-cleaved poly-adenosine diphosphate (ADP) ribose polymerase (PARP) (#9541), and anti-Bcl-2 (#2876) antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA).
Cell culture
Human ovarian (Caov-3) and human breast (MCF-7) cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). The human ovarian cancer cell line Ovcar-3 was kindly provided by Dr. Shridhar (Mayo Clinic, Rochester, MN, USA). The human ovarian cancer cell line A2780, derived from a patient prior to treatment, was kindly provided by Drs. Tsuruo (Institute of Molecular and Cellular Bioscience, Tokyo, Japan), R.F. Ozols and T.C. Hamilton (NCI, National Institutes of Health, Bethesda, MD, USA). The Caov-3, A2780 and MCF-7 cells were cultured in Dulbecco's modified Eagle medium (Sigma Aldrich, St. Louis, MO, USA), while Ovcar-3 cell line was cultured in M199:105 medium at 37°C. The culture media were both supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin and cells were kept in a humidified atmosphere of 95% air and 5% CO2.
shRNAs
ERα targeting shRNA and nontargeting shRNA (control) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). According to shRNA transfection protocol, the control shRNA or ERα shRNA were transfected into Caov-3 and Ovcar-3 cells, and stably expressing cells were selected with puromycin at a concentration of 1 μg/mL.
Plasmids
The estrogen responsive element (ERE)-containing reporter gene, ptk-ERE-luc, was kindly provided by Dr. Hayashi (Tohoku University, Sendai, Japan).Citation26
Cell proliferation assay
Cells were plated at a density of 3 × 104 cells per well in 12-well plates and allowed to attach overnight. Following serum starvation, cells were treated with vehicle, E2 (10−8 M), ICI (10−6 M), or E2 + ICI with 2% charcoal-stripped serum by exchanging the medium for fresh medium containing the indicated agent every 48 h for 6 d. A TC20 ™ automated cell counter (Bio Rad Laboratories, Berkeley, CA, USA) was used to count the cells.
Western blotting
The cells were washed twice in phosphate-buffered saline (PBS) and scraped into lysis buffer containing of 50 mM HEPES (pH 7.5), 150 mM sodium chloride (NaCl), 10% glycerol, 1% Triton X-100, 1.5 mM magnesium chloride (MgCl2), 1 mM enthylenediaminetetraacetic acid (EDTA), 10 mM sodium pyrophosphate, 100 µM sodium orthovanadate, 100 mM sodium fluoride (NaF), 10 µg/ml aprotinin, 10 µg/ml leupeptin, and 1 mM phenylmenthylsulfonyl fluoride (PMSF). The cells were sonicated in the lysis buffer. The lysates were centrifuged at 10000 rpm at 4°C for 10 min, and the protein concentrations of the supernatant were using a protein assay reagent (Bio Rad Laboratories, Berkeley, CA, USA). Equal amounts of proteins were separated using sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Blocking was performed with 5% skimmed milk powder or 3% bovine serum albumin in 1x Tris-buffered saline (TBS). Western blot analyses were performed with various specific primary antibodies. Immunoreacted bands in the immunoblot were visualized with the corresponding secondary horseradish peroxidase-conjugated IgG by using the enhanced chemiluminescence reagents.
Luciferase assay
Cells were plated at a density of 10 × 104 per well in 6-well plates 24 h before transfection. Cells were transfected with 1 μg ptk-ERE-luc and 0.1 μg pRLCMV (ToYo Ink, Tokyo, Japan) as internal control. After 24 h of serum starvation, the cells were treated with vehicle, E2 (10−8 M), cisplatin (100 μM), E2 (10−8 M) + ICI (10−6 M), or cisplatin (100 μM) + ICI (10−6 M) for 24 h. Then the cells were harvested, and a luciferase assay was performed using the Picagene dual-sea pansy luminescence kit (ToYo Ink). Firefly-luciferase and sea pansy-luciferase activites were measured using a luminometer (Lumat LB9507; EG&G, Berthold, Bad Wildbad, Germany). The firefly-luciferase activity was normalized to the sea pansy-luciferase activity to determine the transfection efficiency.
Cell viability assessment
Cells were plated at a density of 6,000 cells per well in 96 well plates in plating medium. The following day, cells were incubated with E2 for 24 h in the treatment medium. After 24h, cisplatin was added for 24h. The number of viable cells was determined by measuring the absorbance of a dissolved formazan product at A490 after the addition of 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) for 2 h as described by the manufacturer (Promega, Madison, WI, USA). All experiments were performed in quadruplicate, and the viability was expressed as the ratio of the viable cells after cisplatin treatment to untreated cells.
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM). Two way comparisons were made with the student's t test. Multiple group comparisons were made using a one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test using GraphPad PRISM software (GraphPad software Inc., La Jolla, CA, USA). Significant differences were defined as P < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_data.zip
Download Zip (588.1 KB)Funding
This work was supported in part by Grants-in-Aid Scientific Research No. 26462512 (T.O.) and No.24592502 (T.S.) from Japan Society for the promotion of science
References
- Hensley ML. Epithelial ovarian cancer. Curr Treat Options Oncol 2002; 3:131-41; PMID:12057076; https://doi.org/10.1007/s11864-002-0059-3
- Han ES, Lin P, Wakabayashi M. Current status on biologic therapies in the treatment of epithelial ovarian cancer. Curr Treat Options Oncol 2009; 10:54-66; PMID:19381822; https://doi.org/10.1007/s11864-009-0100-x
- Tummala MK, McGuire WP. Recurrent ovarian cancer. Clin Adv Hematol Oncol 2005; 3:723-36; PMID:16224447
- Makar AP. Hormone therapy in epithelial ovarian cancer. Endocrine-related cancer 2000; 7:85-93; PMID:10903526; https://doi.org/10.1677/erc.0.0070085
- Eng KH, Hanlon BM, Bradley WH, Szender JB. Prognostic factors modifying the treatment-free interval in recurrent ovarian cancer. Gynecol Oncol 2015; 139:228-35; PMID:26383827; https://doi.org/10.1016/j.ygyno.2015.09.011
- Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 1994; 63:451-86; PMID:7979245; https://doi.org/10.1146/annurev.bi.63.070194.002315
- Matsuo K, Sheridan TB, Mabuchi S, Yoshino K, Hasegawa K, Studeman KD, Im DD, Rosenshein NB, Roman LD, Sood AK. Estrogen receptor expression and increased risk of lymphovascular space invasion in high-grade serous ovarian carcinoma. Gynecol Oncol 2014; 133:473-9; PMID:24674832; https://doi.org/10.1016/j.ygyno.2014.03.563
- Schlumbrecht MP, Xie SS, Shipley GL, Urbauer DL, Broaddus RR. Molecular clustering based on ERalpha and EIG121 predicts survival in high-grade serous carcinoma of the ovary/peritoneum. Mod Pathol 2011; 24:453-62; PMID:21102415; https://doi.org/10.1038/modpathol.2010.211
- Matsumura S, Ohta T, Takahashi T, Yamazaki T, Takahashi K, Kurachi H. Non-sex cord-stromal ovarian tumors frequently produce and secrete estrogen in postmenopausal women: impact on bone metabolism and abnormal endometrial histology. J Clin Endocrinol Metab 2013; 98:2775-82; PMID:23780377; https://doi.org/10.1210/jc.2013-1267
- Mahlck CG, Backstrom T, Kjellgren O. Plasma level of estradiol in patients with ovarian malignant tumors. Gynecol Oncol 1988; 30:313-20; PMID:3391418; https://doi.org/10.1016/0090-8258(88)90245-4
- Yamagata S, Yamamoto K, Yamamoto K, Tsuchida S, Kawamura N, Matsumoto Y, Ueki S, Sugawa T. Estrogen production in epithelial tumors of the ovary–localization of estrogen-synthesizing cells. Nihon Sanka Fujinka Gakkai zasshi 1989; 41:1776-82; PMID:2687406
- Choi JH, Lee KT, Leung PC. Estrogen receptor alpha pathway is involved in leptin-induced ovarian cancer cell growth. Carcinogenesis 2011; 32:589-96; PMID:21173433; https://doi.org/10.1093/carcin/bgq276
- Osborne CK, Shou J, Massarweh S, Schiff R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 2005; 11:865-70; PMID:15701879
- Kirsammer G, Strizzi L, Margaryan NV, Gilgur A, Hyser M, Atkinson J, Kirschmann DA, Seftor EA, Hendrix MJ. Nodal signaling promotes a tumorigenic phenotype in human breast cancer. Semin Cancer Biol 2014; 29:40-50; PMID:25073112; https://doi.org/10.1016/j.semcancer.2014.07.007
- Kimura A, Ohmichi M, Kawagoe J, Kyo S, Mabuchi S, Takahashi T, Ohshima C, Arimoto-Ishida E, Nishio Y, Inoue M, et al. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene 2004; 23:4505-15; PMID:15048073; https://doi.org/10.1038/sj.onc.1207582
- Henson ES, Gibson SB. Surviving cell death through epidermal growth factor (EGF) signal transduction pathways: implications for cancer therapy. Cell Signal 2006; 18:2089-97; PMID:16815674; https://doi.org/10.1016/j.cellsig.2006.05.015
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91:231-41; PMID:9346240; https://doi.org/10.1016/S0092-8674(00)80405-5
- Kimura A, Ohmichi M, Kurachi H, Ikegami H, hayakawa J, Tasaka K, Kanda Y, Nishino Y, Jikihara H, Matsuura N, et al. Role of mitogen-activated protein kinase/extracellular signal-regulated kinase cascade in gonadotropin-releasing hormone-induced growth inhibition of a human ovarian cancer cell line. Cancer Res 1999; 59:5133-42; PMID:10537288
- Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishino Y, Adachi K, Tasaka K, Kanzaki T, Murata Y. Inhibition of extracellular signal-regulated protein kinase or c-Jun N-terminal protein kinase cascade, differentially activated by cisplatin, sensitizes human ovarian cancer cell line. J Biol Chem 1999; 274:31648-54; PMID:10531373; https://doi.org/10.1074/jbc.274.44.31648
- Ohta T, Ohmichi M, Hayasaka T, Mabuchi S, Saitoh M, Kawagoe J, Takahashi K, Igarashi H, Du B, Doshida M, et al. Inhibition of phosphatidylinositol 3-kinase increases efficacy of cisplatin in in vivo ovarian cancer models. Endocrinology 2006; 147:1761-9; PMID:16396982; https://doi.org/10.1210/en.2005-1450
- Mabuchi S, Ohmichi M, Kimura A, Hisamoto K, Hayakawa J, Nishino Y, Adachi K, Takahashi K, Arimoto-Ishida E, Nakatsuji Y, et al. Inhibition of phosphorylation of BAD and Raf-1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem 2002; 277:33490-500; PMID:12087097; https://doi.org/10.1074/jbc.M204042200
- Chang J, Sui M, Fan W. Estrogen receptor α attenuates therapeutic efficacy of paclitaxel on breast xenograft tumors. Breast Cancer Res Treat 2012; 134:969-80; PMID:22374518; https://doi.org/10.1007/s10549-012-1994-8
- Jiang Z, Guo J, Shen J, Jin M, Xie S, Wang L. The role of estrogen receptor alpha in mediating chemoresistance in breast cancer cells. J Exp Clin Cancer Res 2012; 31:42; PMID:22553917; https://doi.org/10.1186/1756-9966-31-42
- Lee MT, Ho SM, Tarapore P, Chung I, Leung YK. Estrogen receptor β isoform 5 confers sensitivity of breast cancer cell lines to chemotherapeutic agent-induced apoptosis through interaction with Bcl2L12. Neoplasia 2013; 15:1262-71; PMID:24339738; https://doi.org/10.1593/neo.131184
- Ercoli A, Battaglia A, Raspaglio G, Fattorossi A, Alimonti A, Petrucci F, Caroli S, Mancuso S, Scambia G. Activity of cisplatin and ICI 182,780 on estrogen receptor negative ovarian cancer cells: cell cycle and cell replication rate perturbation, chromatin texture alteration and apoptosis induction. Int J Cancer 2000; 85:98-103; PMID:10585591; https://doi.org/10.1002/(SICI)1097-0215(20000101)85:1%3c98::AID-IJC18%3e3.0.CO;2-A
- Niikawa H, Suzuki T, Miki Y, Suzuki S, Nagasaki S, Akahira J, Honma S, Evans DB, Hayashi S, Kondo T, et al. Intratumoral estrogens and estrogen receptors in human non-small cell lung carcinoma. Clin Cancer Res 2008; 14:4417-26; PMID:18579664; https://doi.org/10.1158/1078-0432.CCR-07-1950
- Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishino Y, Adachi K, Tasaka K, Kanzaki T, et al. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res 2000; 60:5988-94; PMID:11085518
- Anbalagan M, Rowan BG. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol Cell Endocrinol. 2015; 418:264-72; PMID:25597633; https://doi.org/10.1016/j.mce.2015.01.016
- LaPensee EW, LaPensee CR, Fox S, Schwemberger S, Afton S, Ben-Jonathan N. Bisphenol A and estradiol are equipotent in antagonizing cisplatin-induced cytotoxicity in breast cancer cells. Cancer Lett 2010; 290:167-73; PMID:19796866; https://doi.org/10.1016/j.canlet.2009.09.005
- Mlynarcikova A, Macho L, Fickova M. Bisphenol A alone or in combination with estradiol modulates cell cycle- and apoptosis-related proteins and genes in MCF7 cells. Endocr Regul 2013; 47:189-99; PMID:24156707; https://doi.org/10.4149/endo_2013_04_189
- Perillo B, Sasso A, Abbondanza C, Palumbo G. 17beta-estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol 2000; 20:2890-901; PMID:10733592; https://doi.org/10.1128/MCB.20.8.2890-2901.2000
- Armaiz-Pena GN, Mangala LS, Spannuth WA, Lin YG, Jennings NB, Langley RR, Schmandt R, Lutgendorf SK, Cole SW, et al. Estrous cycle modulates ovarian carcinoma growth. Clin Cancer Res 2009; 15:2971-8; PMID:19383821; https://doi.org/10.1158/1078-0432.CCR-08-2525
- Ohmichi M, Hayakawa J, Tasaka K, Kurachi H, Murata Y. Mechanisms of platinum drug resistance. Trends Pharmacol Sci 2005; 26:113-6; PMID:15749154; https://doi.org/10.1016/j.tips.2005.01.002
- Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, Taylor J, Epstein RJ, Fuller-Pace FV, Egly JM, et al. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene 2002; 21:4921-31; PMID:12118371; https://doi.org/10.1038/sj.onc.1205420
- Liu Y, Ludes-Meyers J, Zhang Y, Munoz-Medellin D, Kim HT, Lu C, Ge G, Schiff R, Hilsenbeck SG, Osborne CK, et al. Inhibition of AP-1 transcription factor causes blockade of multiple signal transduction pathways and inhibits breast cancer growth. Oncogene 2002; 21:7680-9; PMID:12400010; https://doi.org/10.1038/sj.onc.1205883
- Li ZL, Ueki K, Kumagai K, Araki R, Otsuki Y. Regulation of bcl-2 transcription by estrogen receptor-alpha and c-Jun in human endometrium. Med Mol Morphol 2014; 47:43-53; PMID:23665993; https://doi.org/10.1007/s00795-013-0043-y
- Simpkins F, Garcia-Soto A, Slingerland J. New insights on the role of hormonal therapy in ovarian cancer. Steroids 2013; 78:530-7; PMID:23402742; https://doi.org/10.1016/j.steroids.2013.01.008
- Schwartz PE, Chambers JT, Kohorn EI, Chambers SK, Weitzman H, Voynick IM, MacLusky N, Naftolin F. Tamoxifen in combination with cytotoxic chemotherapy in advanced epithelial ovarian cancer. A prospective randomized trial. Cancer 1989; 63:1074-8; PMID:2917311; https://doi.org/10.1002/1097-0142(19890315)63:6%3c1074::AID-CNCR2820630606%3e3.0.CO;2-0
- Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res 1991; 51:3867-73; PMID:1855205
- Argenta PA, Thomas SG, Judson PL, Downs LS Jr, Geller MA, Carson LF, Jonson AL, Ghebre R. A phase II study of fulvestrant in the treatment of multiply-recurrent epithelial ovarian cancer. Gynecol Oncol 2009; 113:205-9; PMID:19239974; https://doi.org/10.1016/j.ygyno.2009.01.012