
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Lung squamous cell carcinoma(LSCC) is the most common and aggressive lung tumor with poor clinical outcome. Previously studies showed that deregulation of long noncoding RNAs (lncRNAs) were involved in LSCC. We intended to figure out the role of lncRNAs in the regulation process of cancer-related genes and pathways they are involved. Data of 552 samples, including 501 cancer samples and 51 normal ones, were extracted from The Cancer Genome Atlas (TCGA). Differentially expressed lncRNAs (DEIs) were screened out (FDR<0.05, |logFC|>1) and then followed by GO ontology and KEGG annotation analysis. Oncogenes from COSMIC data set and Tumor suppressor genes (TSGs) from TSGene data set were collected and analyzed by gene Set Enrichment Analysis (GSEA) . The differentially expressed oncogenes and tumor supressor gene (TSGs) were obtained and co-expression analysis was conducted to generate co-expression lncRNA-gene pairs, which can be helpful in figuring out the role of lncRNA in the regulation of oncogenes and tumor suppressor genes. A total of 31 lncRNAs with low expression levels and 37 lncRNAs with high expression levels were screened out and most of them were enriched in pathways such as meiosis, male gamete generation, defensins. Of note, SFTA1P and CASC2 were found to be related with most of the oncogenes and TSGs by co-expression analysis. We suggested SFTA1P and CASC2 played important role in the regulation of both oncogene and TSGs during the carcinogenesis of LSCC and have the potential to be applied in future diagnosis, prognostic process and target therapy of LSCC.
Introduction
Lung squamous cell carcinoma is one of the most common type of cancer, accounting for 40∼50% of all primary lung cancer.Citation1 Currently, the prognosis of NSCC patients is still very poor. There was not much research about the prognostic and predictive markers for LSCC other than TP53 mutations, which was identified as a causative somatic aberration.Citation2
Long noncoding RNA(lncRNA), with the length of 200 nt and pervasively transcribed in the mammalian genome,Citation3 is involved in a series of processes regulating tumor biology.Citation4, 5 It was reported that lncRNAs are differentially expressed in many kinds of cancer, exerting important regulations on tumor biology via regulating oncogenes or tumor suppressors.Citation6 Given the fact that the number and functions of all kinds of lncRNAs are not well known, differences in IncRNA levels from one specific type of lung cancer might help us better in identifying their potential role as biomarker of LSCC. Therefore, the subtypes of NSCLC and SCLC were not included in our study. We could expound role of the differentially expressed lncRNA in the occurrence and development of cancer more convincingly. Several reviews have emphasized that the role of ncRNAs in different cellular processes has been largely underestimated. It's reported that non-coding RNAs may be involved in the silence of tumor suppressor genes (TSGs) epigenetically, which can inhibit normal cellular growth in cancer.Citation7 LncRNAs mainly played roles in regulation of transcriptional, posttranscriptional and epigenetic mechanisms in the process of tumorigenesis.Citation8 Several studies confirmed the deregulation role of lncRNA in cancers, such as hepatocellular carcinoma, breast,Citation9 osteosarcoma,Citation10 colon and prostate cancers.Citation11, 12
Differentially expressed IncRNAs provide essential evidence to figure out the difference between cancer samples and normal specimens, which can help us in exploring the relevant pathways they were involved. For example, in previous studies, NOTCH1 or NOTCH2 were found to be 2 tumor suppressor specific to epithelial malignancies, thus coming to the conclusion that targeted inhibition of the Notch pathway may affect squamous epithelial malignancies.Citation4
In spite of many studies on the potential important roles of lncRNAs in various kinds of cancer, there had not been much report on lncRNAs associated with LSCC except one study which indicated that lower expression of CASC2 was accompanied by poor prognosis of non small cell lung cancer(NSCLC).Citation13 In this study, we attempted to provide large-scale survey of lncRNAs within LSCC, the subtype of non-small cell lung cancer, and to figure out the role of lncRNA in the regulation process of cancer-related genes of LSCC patients, which may help us in further analysis of identifying pathways they are involved, and thus to identify prognostic marker and novel therapy for LSCC patients.
Materials and methods
Source of data and data pre-processing
A total of 552 expression data (501 LSCC samples and 51 normal ones) were extracted from TCGA (The Cancer Genome Atlas), based on the platform of IlluminaHiSeq. A total of 175 lncRNAs were screened out by annotation profiles from ensemble data set and FRKM values were extracted out and used in the formation of data matrix.Citation14, 15
Identification of differentially expressed lncRNA
Differentially expressed lncRNAs (DEIs) were extracted out from the expression matrix by the algorithm method of limma package.Citation16 The thresholds were FDR<0.05 and |logFC|>1.
GO annotation and KEGG pathway analysis
We applied lncRNA2 function Citation17 software which include functional annotation of 9625 human lncRNA genes to annotate the DEIs. All nodes in Gene Ontology (GO) and a total of 4380 function pathways were included in this functional pathway. GO functional annotation and KEGG pathway analysis of DEIs involved was analyzed by lncRNA2 function.
Collection of oncogenes and Tumor suppressor genes (TSGs)
Data of all the 634 oncogenes were from COSMIC data set,Citation17, 18 which included information of somatic mutation associated with all kinds of cancers. The whole 1452 tumor suppressor gene data were extracted from TSGene data set and TAG data set.Citation19 There were 1217 TSGs collected from more than 9000 studies in TSGene 2.0 (1018 protein coding genes and 199 non-coding genes) while 265 TSGs were included in TAG data set.
Identification of differentially expressed oncogenes and TSGs
Cluster heatmap of the top 100 most significantly differentially expressed oncogenes and TSGswere painted by heatmap 2.0 of Limma package in R language with the thresholds of FC = 2 and FDR<0.05.
Enrichment analysis of gene set
Differentially expressed oncogenes and TSGs were annotated by GSEA (Gene Set Enrichment Analysis).Citation20, 21 GO, KEGG and cancer gene neighborhood were all included in GSEA, which can help us in obtaining the pathways and functions of differentially expressed cancer genes.
Co-expression analysis
Pearson correlation coefficient of each lncRNA and cancer gene, as well as confidence interval and p-value, was calculated out using the function of cor ()andcor.test() of R fuction. The formula were as follow,
Co-expression pair of lncRNA-gene was screened out with the threshold of |correlation coefficient|>0.5 and FDR<0.05 to analyze the regulation role of lncRNApalyed on oncogenes and TSGs.Citation22
Results
Identification of DEIs
DEIs were identified by Limma package with the thresholds of FDR<0.05 and |logFC|>0. As a result, a toal of 32 downregulated lncRNA and 37 upregulated lncRNA were screened out (). Multiple of the top 10 listed lncRNAs of all the up-and-downregulated genes was extremely significant, indicating expression of these lncRNAs were quite different between cancer samples and normal ones. Full information of DEIs were in Table S1 and results of TSG difference in detail were listed in Table S2 while the difference of oncogene in detail was listed in Table S3.
Table 1. The top 10 most enriched up and downregulated lncRNAs.
GO and KEGG annotation of lncRNAs
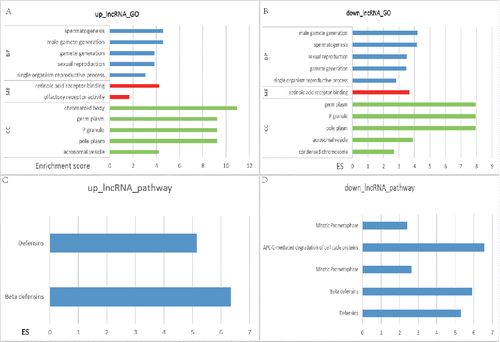
There were 45 biologic processes in mostly enriched GO annotation of upregulated lncRNA, along with 11 cellular_component, 2molecular_function and 2 KEGG annotations (). The most enriched GO terms were spermatogenesis while the most enriched KEGG wasdefensins. There were 58 biologic processes in the mostly enriched GO terms of lncRNA, as well as 20 cellular_component and one molecular_function. Of all the 8 KEGG annotations, the most significant GO term was spermatogenesis while the most significant KEGG term was APC-C-mediated degradation. The 2 terms were both GO annotation of spermatogenesis, indicating that these significant lncRNAs were quite different from those normal samples, thus affecting the regulation of reproduction and inheritance process. lncRNAs in these pathways were involved in regulation of cellular cycles and pathways of defensins.
Figure 1. The top 5 pathways of GO and KEGG enrichment analysis in differentially expressed IncRNA. The horizontal axis included the score of enrichment while the vertical axis represented the enriched pathways. GO annotation of upregulated IncRNA was displayed in (A) while the downregulated ones were in (B). (C) was the KEGG annotation of upregulated lncRNA while (D) was that of downregulated lncRNA. The higher the enrichment score was, the more the pathways were involved in cancer regulation.
Identification of differentially expressed oncogene and TSGs
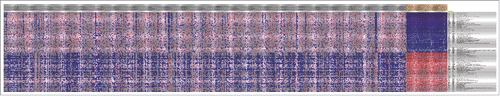
A total of 518 downregulated TSGs and 503 upregulated TSGs were screened out by Limma package with the thresholds of FDR<0.05 and |logFC|>1. The top 10 most genes enriched in TSGs were list in Similarly, there were 177 upregulated oncogenes and 234 downregulated oncogenes (). Heatmap of the top 100 differentially expressed TSGs and oncogeneswas shown in and , respectively, indicating that there was significant difference of cancer related genes between normal samples and tumor specimens.
Table 2. The top 10 most enriched genes of TSGs.
Table 3. The top 10 up and downregulated oncogenes.
Figure 2. Heatmap of the top 100 differentially expressed TSGs between normal samples and tumor specimens. The horizontal axis were different samples, the gray ones were tumor samples while the yellow ones were normal specimens. The vertical axis was genes, which indicated that there was obvious difference among normal samples and tumor ones.

Figure 3. The top 100 differentially exprssed oncogenes between normal samples and tumor specimens. The horizontal axis was samples whie the gray ones were tumor samples and the yellow ones were normal specimens. The vertical axis stands for different genes.
GSEA analysis of differentially expressed oncogenes
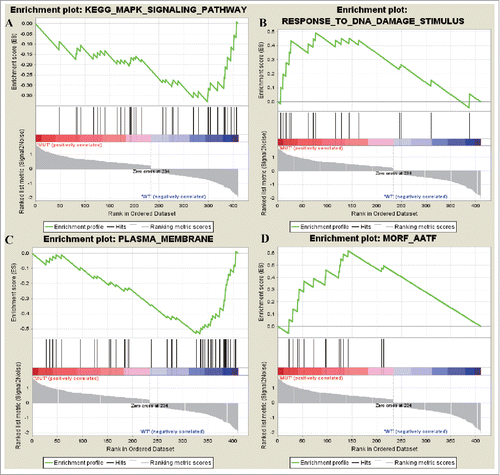
No upregulated KEGG gene set but 3 downregulated ones were screened out with the threshold of NOM p-val <0.25. downregulated oncogene was mainly enriched in pathways as CYTOKINE_CYTOKINE_RECEPTOR_INTERACTION,KEGG_MAPK_SIGNALING_PATHWAY and KEGG_JAK_STAT_SIGNALING_PATHWAY.A total of 3 upregulated GO gene sets and 14 downregulated ones were enriched. The 3 upregulated Go gene sets were RESPONSE_TO_DNA_DAMAGE_STIMULUS, DNA_METABOLIC_PROCESS, RESPONSE_TO_ENDOGENOUS_STIMULUS, while the downregulated ones were PLASMA_MEMBRANE, PLASMA_MEMBRANE_PART, MEMBRANE. There were 14 gene sets in upregulated cancer neighborhood gene while there was no gene set in downregulated genes. Genes sets of upregulated cancer neighborhood gene were MORF_AATF, MORF_DEK, MORF_UBE2I. On the other hand, upregulated gene of differentially expressed oncogene wereRESPONSE_TO_DNA_DAMAGE_STIMULUS and MORF_AATF (). AATF was a kind of transcription factor against apoptosis process, indicating that the main cause in the occurrence of NCSC was DNA damage and cell obtained the characteristic of anti-apoptosis.
Figure 4. The most enriched data set of differentially expressed up and downregulated oncogenes. (A) represents the downregulated genes in KEGG_MAPK_SIGNALING_PATHWAY data set and (B) represented how many genes were upregulated in GO data set RESPONSE_TO_DNA_DAMAGE_STIMULUSj. (C) represents the downregulated genes in PLASMA_MEMBRANE data set and (D) represented how many genes were upregulated in MORF_AATF.
GSEA analysis of differentially expressed TSGs
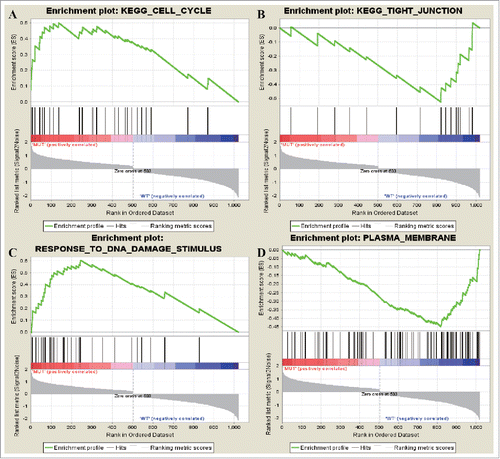
Two upregulated KEGG gene sets and 5 downregulated KEGG gene sets were enriched. The upregulated gene sets were KEGG_CELL_CYCLE and KEGG_P53_SIGNALING_PATHWAY while the downregulated ones were KEGG_TIGHT_JUNCTION, KEGG_LEUKOCYTE_TRANSENDOTHELIAL_MIGRATION, and KEGG_MAPK_SIGNALING_PATHWAY.A total of 17 upregulated GO terms and 25 downregulated GO terms were screened out. The upregulated terms were RESPONSE_TO_DNA_DAMAGE_STIMULUS,RESPONSE_TO_ENDOGENOUS_STIMULUS, DNA_METABOLIC_PROCESS while the downregulated ones were PLASMA_MEMBRANE, INTRINSIC_TO_MEMBRANE, INTEGRAL_TO_MEMBRANE.
Cancer neighborhood gene sets
A total of 24 upregulated cancer neighborhood gene sets and 2 downregulated gene sets were figured out. The upregulated genes sets of cancer neighborhood gene were MORF_BUB3,MORF_EIF3S2 and MORF_XRCC5 while the downregulated ones were GCM_MAP4K4 and MORF_CDC2L5.
The most significant terms in TSG were mainly enriched in KEGG_CELL_CYCLEand RESPONSE_TO_DNA_DAMAGE_STIMULUS (). Genes were downregulated in KEGG_TIGHT_JUNCTION and PLASMA_MEMBRANE.
Figure 5. The most enriched data sets of tumor supressor gene. (A) represented upregulated genes in KEGG_CELL_CYCLE and (B) represented downregulated genes in KEGG_TIGHT_JUNCTION. (C) represented upregulated genes in RESPONSE_TO_DNA_DAMAGE_STIMULUS while (D) represented how many downregulated genes in PLASMA_MEMBRANE.
Since cells in LSCC samples appeared with the characteristic of keratosis and intercellular bridge, including spindle cell carcinoma, pathways associated with the structure of cell membrane could become abnormal. Growth and division of cells could be constant and hereditary substance became variated after malignant transformation process, which could stimulate the overexpression of suppressor genes in cells.
Co-expression of DEIs and TSG or oncogenes
Co-efficient of up-or-downregulated lncRNA with TSGs or oncogene was calculated out respectively by R function. The thresholds of 0.5and −0.5 was used to screen out the co-expression pair of lncRNAs-genes.SFTA1P appeared to be the most co-expressed gene between downregulated lncRNAs and oncogenes (). There were 7 co-expression genes, that werePTPRB, SLC34A2, FGR,ROS1,CSF3R, CXCL2, RND1. CACNA1Dwas found to be the most co-expression oncogeneswith lncRNAs, while all these co-expression oncogenes were downregulated.There were 2 co-expressed genes, KCNH8 and SOX2, between upregulated lncRNAs and oncogenes and MIAT was the one with the most co-expression. PABPC1L was found to be the oncogene co-expressing with more than one lncRNAs.
Table 4. Co-expression pairs of lncRNA and oncogenes.
There were 3 genes with most co-expression between downregulated lncRNAs and TSGs, SFTA1P with the 16 co-expression genes, while C10orf95 with 8 co-expression genes and CASC2 with 7 co-expression genes (). All the TSGs, that is,GADD45B, CSRNP1, RHOBTB2, DOK2 were all downregulated.Four co-expression genes in DLEU1, 5 co-expression genes in GAS5 and 4 co-expression genes in SNHG12. The 3 TSGs, RPL15, SELT, RBX1, were all overexpressed.
Table 5. Co-expression pairs between lncRNA and TSGs.
Discussion
Long non-coding RNA (lncRNA), is a kind of RNAs whose length larger than 200 bases with biologic functions. LncRNAs can suppress translation by biding to their corresponding mRNAs and that's the main reason why they have an essential role in gene regulation. It's involved in the growth and pathological process by the way of chromatin reprogramming, cis regulation at enhancers or post-trtanscriptional regulation, which was similar with way of the encodinggenes. Since the close relation between gene expression patterns and tumor subtypes was essential in understanding the molecular basis of tumorigenesis,Citation23, 24 the 2 differentially expressed IncRNAs between normal samples and patients with SCLC we identified in this study, SFTA1P and CASC2, may of much value in clinical management of the disease.
There were reports about abnormal expression of some IncRNA in the occurrence, invasion and development in many types of tumor tissues, indicating their association with the suppression of cancer. The expression degree of these IncRNAs may reflect the degree of prognosis process. Given their specialty in cancer tissues, these IncRNAs were regarded as prognostic biomarkers of cancer disease.
In this study, spermatogenesis was the most enriched GO terms in both up and downregulated IncRNAs. We speculated that there was much difference of differentially expressed IncRNAs between normal samples and cancer specimens, which thus affected the regulation of reproduction and inheritance process. Besides, the results of pathway analysis revealed that these IncRNAs were also involved in the pathways of cell cycle regulation and alexin.
Functional analysis of oncogenes revealed that they had large effect in signaling pathways associated with cytokine and DNA damage, indicating that there was large variation in genetic material of NSCC and activation of signaling pathways led to the deterioration of the cancer.
Two IncRNA, SFTA1P and CASC2 were found to be associated with most of tumor supressor genes and oncogenes and had an great effect on the regulation of LSCC. It was reported that SFTA1P was downregulated in NSCC and its main function was lying on epidermal growth, cell attachment and response to DNA damage.Citation25 The other IncRNA, CASC2, was reported to be inhibited by miR-21 in gliomas and,Citation26, 27 which could be the ceRNA of other oncogenes.
It was reported that lncRNAs could in cis regulate the expression of their neighboring genes,Citation4, 28 we suspected the effect of these 2 genes may be related with this pathway, which needed more relevant research.
In summary, we identify 2 potential biomarkers, that is, SFTA1P and CASC2, associated with the regulation and development of lung squamous cell carcinoma, which could provide more specific and accurateprognostic and predictive indicators to clinical outcome of LSCC patients,implying their application in clinical diagnosis and treatment of this disease.
Conclusions
In summary, we identify 2 potential biomarkers, that is, SFTA1P and CASC2, associated with the regulation and development of lung squamous cell carcinoma, which could provide more specific and accurate prognostic and predictive indicators to clinical outcome of LSCC patients, implying their application in clinical diagnosis and treatment of this disease.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
GQ.H, ZP.K and B.G conceived and designed the experiments; GQ.H, ZP.K performed the experiments; ZP.K and B.G analyzed the data; Y.C contributed analysis tools; GQ.H, ZP.K, B.G and HB.H wrote the paper.
Supplementary_Table_1-3.zip
Download Zip (139.3 KB)References
- Network C. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489(7417): 519-25; PMID:22960745; http://dx.doi.org/10.1038/nature11404
- Chiba I, Takahashi T, Nau MM, D'Amico D, Curiel DT, Mitsudomi T, Buchhagen DL, Carbone D, Piantadosi S, Koga H, et al. Mutations in the p53 gene are frequent in primary, resected non-small cell lung cancer. lung cancer study group. Oncogene 1990; 5(10): 1603-10; PMID:1979160.
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Research 2012; 22(9): 1775-1789; PMID:22955988; http://dx.doi.org/10.1101/gr.132159.111
- Wang K, Chang H. Molecular mechanisms of long noncoding RNAs. Molecular cell 2011; 43(6): 904-14; PMID:21925379; http://dx.doi.org/10.1016/j.molcel.2011.08.018
- Gupta R, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464(7291): 1071-76; PMID:20393566; http://dx.doi.org/10.1038/nature08975
- Wilusz J, Sunwoo H, Spector D. Long noncoding RNAs: functional surprises from the RNA world. Genes & Development 2009; 23(13): 1494-1504; http://dx.doi.org/10.1101/gad.1800909
- Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 2008; 451(7175): 202-206; PMID:18185590; http://dx.doi.org/10.1038/nature06468
- Wang N, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proceedings of the National Academy of Sciences 2011; 108(43): 17761-66; ; http://dx.doi.org/10.1073/pnas.1114669108
- Guffanti A, et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics 2009; 10(1): 1; PMID:19121221; http://dx.doi.org/10.1186/1471-2164-10-163
- Yamada K, Kano J, Tsunoda H, Yoshikawa H, Okubo C, Ishiyama T, Noguchi M. Phenotypic characterization of endometrial stromal sarcoma of the uterus. Cancer Science 2006; 97(2): 106-12; PMID:16441420; http://dx.doi.org/10.1111/j.1349-7006.2006.00147.x
- Lin R, Maeda S, Liu C, Karin M, Edgington TS. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 2007; 26(6): 851-58; PMID:16878148; http://dx.doi.org/10.1038/sj.onc.1209846
- Luo J, Ren B, Keryanov S, Tseng GC, Rao UN, Monga SP, Strom S, Demetris AJ, Nalesnik M, Yu YP, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 2006; 44(4): 1012-24; PMID:17006932; http://dx.doi.org/10.1002/hep.21328
- He X, et al. Low expression of long noncoding RNA CASC2 indicates a poor prognosis and regulates cell proliferation in non-small cell lung cancer. Tumor Biology 2016; 37(7):9503-9510.
- Du Z, et al. Integrative analyses reveal a long noncoding RNA-mediated sponge regulatory network in prostate cancer. Nature Communications 2016; 7:10982; http://dx.doi.org/10.1038/ncomms10982
- Di Martino M, Guzzi PH, Caracciolo D, Agnelli L, Neri A, Walker BA, Morgan GJ, Cannataro M, Tassone P, Tagliaferri P, et al. Integrated analysis of microRNAs, transcription factors and target genes expression discloses a specific molecular architecture of hyperdiploid multiple myeloma. Oncotarget 2015; 6(22): 19132; PMID:26056083; http://dx.doi.org/10.18632/oncotarget.4302
- Ritchie M, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research 2015; 43(7):e47; http://dx.doi.org/10.1093/nar/gkv007
- Jiang Q, et al. LncRNA2Function: a comprehensive resource for functional investigation of human lncRNAs based on RNA-seq data. BMC Genomics 2015; 16(3): 1. PMID:25553907; http://dx.doi.org/10.1186/1471-2164-16-S3-S2
- Futreal P, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer 2004; 4(3): 177-83; PMID:14993899; http://dx.doi.org/10.1038/nrc1299
- Chen J, Hung WS, Chan HH, Tsai SJ, Sun HS. In silico identification of oncogenic potential of fyn-related kinase in hepatocellular carcinoma. Bioinformatics 2013; 29(4): 420-7; PMID:23267173; http://dx.doi.org/10.1093/bioinformatics/bts715
- Mootha V, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. PGC-1[alpha]-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34(3): 267-73; PMID:12808457; http://dx.doi.org/10.1038/ng1180
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102(43): 15545-50; PMID:16199517; http://dx.doi.org/10.1073/pnas.0506580102
- Yang F, Lyu S, Dong S, Liu Y, Zhang X, Wang O. Expression profile analysis of long noncoding RNA in HER-2-enriched subtype breast cancer by next-generation sequencing and bioinformatics. OncoTargets and Therapy 2016; 9: 761; PMID:26929647; http://dx.doi.org/10.2147/OTT.S97664
- Sørlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Academy of Sciences 2003; 100(14): 8418-23; PMID:12829800; http://dx.doi.org/10.1073/pnas.0932692100
- Sørlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences 2001; 98(19): 10869-74; PMID:11553815; http://dx.doi.org/10.1073/pnas.191367098
- Zhao W, Luo J, Jiao S. Comprehensive characterization of cancer subtype associated long non-coding RNAs and their clinical implications. Scientific Reports 2014; 4: 6591; PMID:25307233; http://dx.doi.org/10.1038/srep06591
- Wang P, Liu YH, Yao YL, Li Z, Li ZQ, Ma J, Xue YX. Long non-coding RNA CASC2 suppresses malignancy in human gliomas by miR-21. Cellular Signalling 2015; 27(2): 275-82; PMID:25446261; http://dx.doi.org/10.1016/j.cellsig.2014.11.011
- Zhang J, Zhu N, Chen X. A novel long noncoding RNA LINC01133 is upregulated in lung squamous cell cancer and predicts survival. Tumor Biology 2015; 36(10): 7465-1; PMID:25908174; http://dx.doi.org/10.1007/s13277-015-3460-9
- Dimitrova N, Zamudio JR, Jong RM, Soukup D, Resnick R, Sarma K, Ward AJ, Raj A, Lee JT, Sharp PA, et al. LincRNA-p21 activates p21 in cis to promote polycomb target gene expression and to enforce the G1/S checkpoint. Molecular Cell 2014; 54(5): 777-90; PMID:24857549; http://dx.doi.org/10.1016/j.molcel.2014.04.025