
ABSTRACT
It remains uncertain whether there is an correlation between clinical pharmacokinetic parameters and outcomes for metastatic colorectal cancer especially with UGT1A1 *28 and *6 wild type (*1/*1-*1/*1) for serious events associated with Irinotecan are largely excluded. This study retrospectively analyzed the relationship between Irinotecan metabolite levels and outcomes of UGT1A1 *1/*1-*1/*1 genotype arrangement. Blood samples (n = 244) were collected for analysis of plasma DPD activity (before first chemotherapy) and SN-38 levels (1.5 and 49 hour after CPT-11 administration). Clinical variables such as toxicity and outcomes were then assessed. Of the *1/*1 -*1/*1 genotype combination, the median progression free survival of the CSN-38 1.5 h > 50.24 ng/ml subset was remarkably longer than that of the CSN-38 1.5 h ≤ 50.24 ng/ml subset. However, there were no differences between the CSN-38 49 h > 15.25 ng/ml subgroup and the ≤ 15.25 ng/ml group. It was lower DPD activity that responsible for the relatively higher incidence of bone marrow hypocellular, diarrhea, and oral mucositis in the CSN-38 1.5 h > 50.24 ng/ml and CSN-38 49 h > 15.25 ng/ml subsets. Therefore, plasma SN-38 levels is related to outcomes for UGT1A1 *1/*1-*1/*1 genotype, to improve efficacy, patients with CSN-38 1.5 h lower than 50.24 ng/ml, CPT-11 dosage could be added in next chemmotherapy on SN-38 plasma level monitoring. Additionally, in patients with DPD activity below 3.18 before treatment, appropriate reduction of 5-FU dose could be considered to minimize the incidence of adverse events.
Abbreviations
| 5-FU | = | fluorouracil |
| AJCC | = | American Joint Committee on Cancer |
| BSA | = | body surface area |
| CI | = | confidence interval |
| CSN-38 1.5 h | = | plasma SN-38 concentration 1.5 hours after CPT-11 infusion |
| CSN-38 49 h | = | plasma SN-38 concentration 49 hours after CPT-11 infusion |
| CPT-11 | = | Irinotecan |
| CTCAE | = | National Cancer Institute common terminology criteria for adverse events |
| DHFU | = | Dihydrofluorouracil |
| DPD | = | dihydropyrimidine dehydrogenase |
| Dntp | = | deoxy-ribonucleotide triphosphate |
| ECOG | = | Eastern Cooperative Oncology Group |
| EDTA | = | Ethylenediaminetetraacetic acid |
| IS | = | internal standard |
| HPLC | = | high performance liquid chromatography |
| mOS | = | median OS |
| mPFS | = | median PFS |
| mCRC | = | metastatic colorectal cancer |
| MTD | = | maximum tolerable dosage |
| OS | = | overall survival |
| PD | = | progression of disease |
| PFS | = | progression free survival |
| RECIST | = | the Response Evaluation Criteria in Solid Tumors |
| SAP | = | Shrimp Alkaline Phosphatase |
| SN-38 | = | 7-ethyl-10-hydroxycampothecin |
| SN-38G | = | SN-38 glucuronide |
| SNP | = | Single Nucleotide Polymorphism |
| U | = | Uracil |
| UGT1A1 | = | uridine diphosphate glucuronosyltransferase 1A1 |
| UH2 | = | dihydrouracil |
Introduction
The treatment of metastatic colorectal cancer (mCRC) has marched forward to a new age of targeted treatment (noted by Heinemann et al.Citation1 and Elez et al.Citation2), however, cytotoxic anticancer drugs remain the cornerstone of cancer therapy. Clinical effects and treatment-associated toxicities may vary widely in different populations and individuals as a result of changes in the pharmacokinetics and pharmacodynamics of therapeutics. This is caused by genetic variations of drug metabolizing enzymes, drug carriers, and/or targets (according to Snozek et al.Citation3). Therefore, many researchers are exploring new methods of administration according to different targets in drug metabolic pathways, as opposed to the traditional dosing mode calculated by body surface area (BSA) (according to Sawyer et al.Citation4). These new methodologies should improve patient outcomes and prevent severe side effects by choosing a better dosage for patients.
The single nucleotide polymorphisms (SNPs) of drug metabolic enzymes, especially for rate-limiting enzymes that play a key role in drug metabolism, are also major causes of variations in drug efficacy and toxicities. For some drugs with relatively clear metabolic pathways, such as irinotecan (CPT-11), it is feasible to accomplish personalized administration by making use of rate-limiting enzyme SNPs and pharmacokinetics.
More than 60 variants in the coding and promoter regions of the uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) gene have been identified to date. Mutations located in the promoter of UGT1A1*28 have been identified in up to 35% of Caucasians (noted by Deeken et al.Citation5), while mutations in the coding region of UGT1A1*6 are mainly found in Asian populations. A number of studies have shown that homozygous mutations of UGT1A1*6 alone or in combination with UGT1A1*28 were associated with severe hematologic toxicity (according to Han et al.,Citation6 Ando et al.,Citation7 Yamasaki et al.Citation8 and Onoue et al.Citation9). However, considering the fact that the total percentage of UGT1A1*28 and *6 homozygous genotype is below 5%, another aim of personalized treatment using CPT-11 is to improve its therapeutic efficacy in accordance with 7-ethyl-10-hydroxycampothecin (SN-38) pharmacokinetics once any associated serious side effects have been basically precluded. This is considered at least for wild type cases, since UGT1A1 activity is within or beyond the normal range.
Dihydropyrimidine dehydrogenase (DPD) is a key regulatory enzyme for fluorouracil (5-FU) that inactivates 80–85% of 5-FU into the inactive metabolite dihydrofluorouracil (DHFU) via the liver (noted by Ezzeldin et al.Citation10). Since 5-FU is commonly used in combination with CPT-11 and has similar adverse reactions, the occurrence of toxicities such as bone marrow hypocellular or diarrhea could mistakenly be attributed to CPT-11 when 5-FU plasma clearance is inhibited with a prolonged half-life due to a drop in DPD activity.
This study, therefore, retrospectively analyzed 244 cases with UGT1A1 *6 and *28 wild type (*1/*1-*1/*1) to determine whether there is a relationship between clinical parameters (such as DPD activity before chemotherapy, SN-38 levels after CPT-11 administration) and outcomes (efficacy or toxicities). This study will serve as a reference for prospective studies on individualized administration of CPT-11 and 5-FU for mCRC with the assessed genotype arrangements.
Results
Distribution of UGT1A1*28 and/or *6 SNPs and comparison of adverse reactions between UGT1A1*28 and/or *6 wild type cases
The sequencing results of UGT1A1 *28 and *6 SNPs are shown in , and the distribution of different genotype combinations is listed in . In addition to 244 cases of *1/*1-*1/*1 genotype arrangement, there were 10 cases of *1/*1-*6/*6, 8 cases of *28/*28-*1/*1, 2 cases of *1/*28-*6/*6, and 1 case of *28/*28-*1/*6 genotype combinations, which could be identified as wild type or heterozygous if only one locus was focused on. Moreover, the incidence of bone marrow hypocellular, diarrhea, and increased alanine aminotransferase was significantly lower in UGT1A1*6 and *28 wild type arrangements than in UGT1A1*6 or *28 wild type alone (shown in ).
Figure 1. Sequence analysis for UGT1A1 *28 and *6 and distribution of UGT1A1*28 and/or *6 genotype among all screened cases (n = 499). Sequencing results for UGT1A1 *28 and *6 using the FinchTV software were: (A) *1/*1 genotype; (B) *1/*28 genotype; (C) *28/*28 genotype; (D) *1/*1 genotype; (E) *1/*6 genotype; and (F) *6/*6 genotype. Distribution of UGT1A1*28 and/or *6 SNPs: the overall distribution of UGT1A1*28 and *6 SNPs by separate or synchronous identification is shown in (G). It should be noted that there were 10 cases of *1/*1-*6/*6, 8 cases of *28/*28-*1/*1, 2 cases of *1/*28-*6/*6, and 1 case of *28/*28-*1/*6 genotype combination in (H) and (I), which could be identified as wild type or heterozygous by one locus detection.
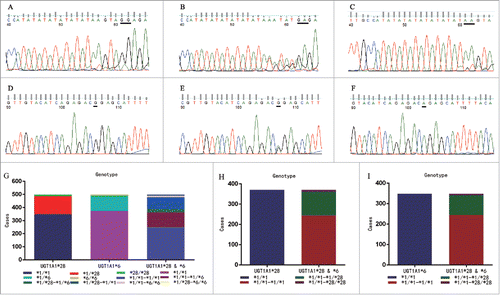
Figure 2. Comparison of adverse reactions between UGT1A1*28 and/or *6 wild type and in different CSN-38 1.5 h and CSN-38 49 h subsets. Comparison of adverse reactions in UGT1A1*28 and/or *6 wild type: 244, 370 and 348cases were identified with *1/*1-*1/*1, *1/*1 (UGT1A1*28) and *1/*1 UGT1A1*6) genotype from all the 499 cases, the incidence of bone marrow hypocellular, diarrhea, and increased alanine aminotransferase was significantly lower in the UGT1A1*6 and *28 wild type arrangement than in UGT1A1*6 (A) *1/*1-*1/*1 genotype vs. *1/*1 genotype: p = 0.004 for bone marrow hypocellular, p = 0.004 for diarrhea, and p = 0.01 for increased alanine aminotransferase) or *28 wild type (B) *1/*1-*1/*1 genotype genotype vs.*1/*1: p = 0.003 for bone marrow hypocellular, and p = 0.04 for diarrhea). Comparison of adverse reactions in the CSN-38 1.5 h > 50.24 ng/ml, ≤ 50.24 ng/ml, CSN-38 49 h > 15.25 ng/m and ≤ 15.25 ng/ml subsets for UGT1A1*28 and *6 wild type arrangement: the incidence of side effects was significantly higher in the CSN-38 1.5 h > 50.24 ng/ml subgroup than in the ≤ 50.24 ng/ml group (C) p < 0.001 for bone marrow hypocellular, p = 0.003 for diarrhea, p = 0.009 for nausea, p < 0.001 for oral mucositis, and p = 0.02 for fatigue). Similar trends were observed in the CSN-38 49 h > 15.25 ng/m subset and the ≤ 15.25 ng/ml subset (D) p < 0.001 for bone marrow hypocellular, p < 0.001 for diarrhea, p = 0.02 for increased alanine aminotransferase, p < 0.001 for nausea, p < 0.001 for oral mucositis, and p < 0.001 for fatigue). Note: nausea and fatigue are scored as 0–3 grades.
Pertinence analysis of CSN-38 1.5 h and CSN-38 49 h outcome or toxicity
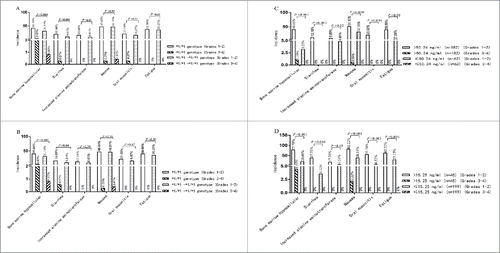
Plasma SN-38 concentration 1.5 hours and 49 hours after CPT-11 infusion (CSN-38 1.5 h, CSN-38 49 h) and DPD activity served as dependent variables, whereas gender, age, Eastern Cooperative Oncology Group (ECOG) score, TNM staging, metastases, CPT-11 starting dosage, short-term response, progression free survival (PFS), and side effects were independent variables. Stepwise regression analysis suggested that CSN-38 1.5 h was related to PFS, while CSN-38 49 h and DPD activity were linked to bone marrow hypocellular (). Pearson correlation analysis also revealed that the relativity of CSN-38 1.5 h to PFS was well above that of CSN-38 49 h to PFS (Pearson correlation coefficient: 0.77 vs. 0.35). Subsequently, 244 cases with *1/*1-*1/*1 genotype combination were allocated to 2 groups according to the adjusted predicted value and standard deviation of CSN-38 1.5 h and CSN-38 49 h based on the stepwise regression analysis (60.84 ± 10.60 ng/ml and 11.13 ± 4.12 ng/ml): the > 50.24 ng/ml and ≤ 50.24 ng/ml subgroup for CSN-38 1.5 h and the > 15.25 ng/ml and ≤ 15.25 ng/ml subgroup for CSN-38 49 h. Compared to the CSN-38 1.5 h ≤ 50.24 ng/ml subset, the CSN-38 1.5 h > 50.24 ng/ml group achieved a longer mPFS. However, no significant differences were found between the CSN-38 49 h > 15.25 ng/ml and ≤ 15.25 ng/ml subgroups with regards to median PFS (mPFS) (see ). Furthermore, cases with CSN-38 1.5 h ≤ 50.24 ng/ml made up 25.41% of the total *1/*1-*1/*1 genotype combination (see ).
Figure 3. Comparison of mPFS in different CSN-38 1.5 h and CSN-38 49 h subsets for the UGT1A1*28 and *6 wild type. The CSN-38 1.5 h > 50.24 ng/ml subgroup achieved a better mPFS than the CSN-38 1.5 h ≤ 50.24 ng/ml group (6.77 ± 0.09 months vs. 5.50 ± 0.41 months, p < 0.001). However, no statistically significant differences were observed between CSN-38 49 h > 15.25 ng/ml and ≤ 15.25 ng/ml subgroups in terms of mPFS (6.73 ± 0.11 months vs. 6.70 ± 0.14 months, p = 0.42). Similarly, there was no significant difference in mPFS between DPD activity in the > 3.18 and ≤ 3.18 subgroups (6.73 ± 0.13 months vs. 6.83 ± 0.12 months, p = 0.35).
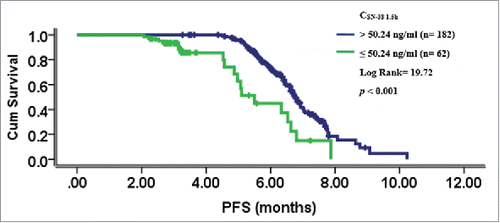
Figure 4. CSN-38 1.5 h distribution of the total *1/*1-*1/*1 genotype and DPD activity comparison between the CSN-38 1.5 h > 50.24 ng/ml and CSN-38 49 h >15.25 ng/ml subgroups with or without adverse effects. Distribution of CSN-38 1.5 h after CPT-11 infusion: the CSN-38 1.5 h for 62 cases was below the cut-off point of 50.24 ng/ml, which made up approximately 25% of the total 244 cases from the waterfall plot (A) DPD activity comparison between different CSN-38 1.5 h and CSN-38 49 h subsets with or without adverse effects: the levels of DPD activity in patients with side effects were statistically lower than those in patients without adverse reactions in CSN-38 1.5 h > 50.24 ng/ml (B) p < 0.001 for bone marrow hypocellular, p < 0.001 for diarrhea and p < 0.001 for oral mucositis) and CSN-38 49 h > 15.25 ng/ml subgroups (C) p = 0.005 for bone marrow hypocellular, p = 0.001 for diarrhea, and p = 0.002 for oral mucositis).
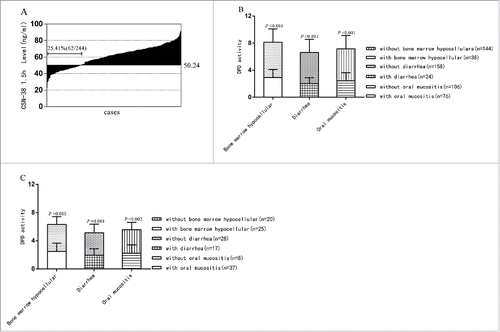
The relationship between clinical efficacy, side effects, and DPD activity in the CSN-38 1.5 h > 50.24 ng/ml and CSN-38 49 h > 15.25 ng/ml subgroups
demonstrate that the incidence of adverse reactions in the CSN-38 1.5 h > 50.24 ng/ml and CSN-38 49 h > 15.25 ng/ml subsets was higher than in the CSN-38 1.5 h ≤ 50.24 ng/ml and CSN-38 49 h ≤ 15.25 ng/ml subsets. However, the adverse effects were limited to grades 1–2. We next assessed whether there were differences in DPD activity between cases with bone marrow hypocellular, diarrhea, and oral mucositis, and those without these associated toxicities in CSN-38 1.5 h > 50.24 ng/ml and CSN-38 49 h > 15.25 ng/ml subgroups. The levels of DPD activity in patients with side effects were statistically lower than in cases without adverse reactions (). Additionally, to determine whether clinical outcome is affected by downregulation of 5-FU dose due to lower DPD activity, the *1/*1-*1/*1 genotype group was also separated into 2 subgroups based on the adjusted predicted value and standard deviation after stepwise regression (4.58 ± 1.40): DPD activity > 3.18 and ≤ 3.18 subset. The results showed that no significant difference was detected between the 2 subgroups in terms of mPFS.
Discussion
Dose individualization of a cytotoxic drug makes it feasible to optimize the outcome on the condition that the information about the tolerance of a given dose is available before or after treatment (such as the activity or SNPs of drug-associated metabolic enzymes or pharmacokinetic properties). As for UGT1A1, the rate-limiting enzyme associated with CPT-11 inactivation, many studies focused on the UGT1A1 *28 and the UGT1A1 *6 alleles (according to Strassburg,Citation11 Sugatani,Citation12 Horsfall et al.,Citation13 and Gagné et al.Citation14), and the risk of severe adverse events (noted by Onoue et al.,Citation9 Marcuello et al.,Citation15 Paoluzzi et al.Citation16 and Takano et al.Citation17). However, the occurrence of homozygotes for UGT1A1*28 or *6 is about 3% in Asians (according to Takano et al.,Citation17 Fukui et al.Citation18 and Glimelius et al.Citation19) as well as in China (according to Wang et al.Citation20), but considering the frequencies of allelic variation among different populations, we synchronously determined the SNP of both loci. Compared to 2-site synchronous detection (as seen from ), the incidence of bone marrow hypocellular and diarrhea significantly increased with single-locus identification. Therefore, 2-point simultaneous determination can improve the accuracy of UGT1A1*28 or *6 homozygote identification and provide the *1/*1-*1/*1 genotype with security for CPT-11 dose escalation.
For most wild type or heterozygous types of UGT1A1*28 and/ or *6 in Chinese population, it is important to improve outcome than to reduce or avoid toxicities in CPT-11 individualized administration because of the normal or near-normal activity of UGT1A1. A previous study demonstrated that the maximum tolerable dosage (MTD) of CPT-11 for UGT1A1*28 heterozygous and wild type were 450 mg/m2 and 390 mg/m2, respectively, which are far greater than the recommended dosage of 180 mg/m2 (noted by Marcuello et al.Citation21). Therefore, there is a great range of CPT-11 dose increasing for UGT1A1*28 and *6 wild type arrangement. While determination of optimal dosage for CPT-11 based on UGT1A1 genotypes is still accomplished by dose-escalation (noted by Freyer et al.,Citation22 Kim et al.,Citation23 Lu et al.Citation24 and Innocenti et al.Citation25), this approach can be affected by dose-increasing extent, and can result in patients receiving sub-clinical doses. Furthermore, estimation of the optimum dose may also be limited to calculating the area under the curve of CPT-11, SN-38, or SN-38 glucuronide (SN-38G) owing to some factors such as numerous sampling, high testing costs, time, and poor compliance (according to Innocenti et al.,Citation25 Satoh et al.Citation26 and Lee et al.Citation27).
Considering 0.5– 3.75 hour of peak-reaching time for SN-38 after intravenous administration, 10– 20 h of elimination of half-life, and a linearly increasing relationship between absorption area and CPT-11 doses within the range of 50–350 mg/m2, CSN-38 1.5 h and CSN-38 49 h were selected as appropriate detection points for understanding the pharmacokinetics of SN-38 and outcomes. Our results showed that the mPFS of the CSN-38 1.5 h > 50.24 ng/ml subset was longer than that of the ≤ 50.24 ng/ml group. Furthermore, CSN-38 49 h had little effect on efficacy, suggesting that insufficient dosage was unfavorable for the treatment outcome. Cases with CSN-38 1.5 h ≤ 50.24 ng/ml was around 25% of the total, which was similar to the results of our previous study investigating only UGT1A1*28 (noted by Cai et al.Citation28). These results also serve as a reference for further prospective investigation of CPT-11 dosage individualization by CSN-38 1.5 h.
From a standpoint of toxicities, indicated that higher CSN-38 1.5 h and CSN-38 49 h subsets demonstrated relatively high incidence of toxicities than the lower ones. Since previous studies demonstrated that the application of 5-FU did not change CPT-11 metabolism within a certain range in vivo (according to Ducreux et al.Citation29 and Saltz et al.Citation30), and the side effects caused by 5-FU are similar to those induced by CPT-11, so we tested whether 5-FU could affect the statistical analysis of CPT-11 associated toxicities between subgroups. We compared DPD activity between patients with and without bone marrow hypocellular, diarrhea, and oral mucositis in the higher CSN-38 1.5 h and CSN-38 49 h subsets, the result showed that DPD activity in the toxicity group was substantially below that in the non-toxicity group, respectively. Moreover, cases of *1/*1-*1/*1 genotype were also allocated to > 3.18 and ≤ 3.18 groups according to the adjusted predicted value and standard deviation by stepwise regression, there were no significant differences between these 2 groups in terms of mPFS. All these results suggested that the increased incidence of adverse reactions might result from decreased DPD activity leading to enhanced cytotoxicity, but without influencing the outcome.Therefore, prior dosage reduction on DPD activity and subsequent regulation of 5-FU by plasma level monitoring may reduce 5-FU associated side reactions.
In summary, by retrospective analysis, we found that simultaneous determination of UGT1A1 *6 and *28 may improve the accuracy of homozygous genotype identification; for cases with DPD activity below 3.18 before treatment, appropriate reduction of 5-FU dose according to DPD activity could be taken to minimize the incidence of adverse reactions; while for those of UGT1A1 *6 and *28 wild type arrangement with CSN-38 1.5 h lower than 50.24 ng/ml, next CPT-11 dosage could be added in the premise of CSN-38 1.5 h monitoring. However, further studies are needed to put personalized administration of CPT-11 into effect for UGT1A1 *6 and *28 wild type such as prospective designed researches according to the effective range of plasma SN-38 levels and DPD activity from the retrospective study. In addition, other factors might influence CPT-11 distribution in vivo after CPT-11 infusion including competitive inactivation via the CYP3A4 pathway (noted by Goey et al.Citation31), the effect on SN-38 levels by SNPs of organic anion-transporting polypeptide 1B (noted by Teft et al.Citation32), and ATP-Binding Cassette Subfamily C, all of which need to be further assessed in clinical practice.
Patients and methods
Patients
A total of 499 patients from Zhongshan hospital (104 cases), Shanghai Cancer Center (98 cases), Ruijin hospital (85cases), Renji hospital (77 cases), Shanghai General hospital (69 cases), and Shanghai Tenth People's Hospital (66 cases), diagnosed with local advanced or mCRC with pathological and imaging information were screened from December 2012 to May 2014. Patient identified as *1/*1-*1/*1 genotype, with measurable lesions untreated, Eastern Cooperative Oncology Group (ECOG) Performance Status grades 0–2, and life expectancy of more than 3 months were qualified for the study (244 cases, see ). These patients were received the FOLFIRI regimen for at least 3 cycles as first-line chemotherapy and had at least one evaluation; plasma bilirubin and transaminase levels did not exceed 1.5 and 5 times of the normal upper limit for patients with liver metastases. The exclusion criteria were as follows: pregnant or lactating women, patients complicated with complete or incomplete intestinal obstruction; patients with a history of chronic enteritis, extensive bowel resection; patients with a history of being seriously allergic to CPT-11; evaluable lesions having received radiotherapy or other treatments; vital organ dysfunction; or patients with other malignancies; and patients demonstrating poor compliance.
Table 1. Disposition and baseline characteristics of mCRC patients with *1/*1-*1/*1 genotype who participated in this study.
Table 2. Stepwise regression of CSN-38 1.5 h, CSN-38 49 h, and DPD activity with clinical parameters in the *1/*1-*1/*1 genotype combination.
Methods
All patients gave their informed consent before entering the study, which was approved by the bioethics committee of all hospitals participated in the study. Peripheral blood samples (anticoagulated with Ethylenediaminetetraacetic acid (EDTA)) were collected 3 d before first chemotherapy for UGT1A1*28 and *6 genotype identification and detection of DPD activity. The eligible patients were then accepted into FOLFIRI protocol, which included 1.5 hour of CPT-11 infusion (180 mg/m2) and 2 hours drip of leucovorin (400 mg/m2), followed by 5-FU bolus (400 mg/m2) on day 1 and a 46 hours continuous infusion (2,400 mg/m2) for each cycle. This process was repeated every other week. Another blood sample was obtained 1.5 hour and 49 hours after CPT-11 infusion for plasma SN-38 level testing (CSN-38 1.5 h and CSN-38 49 h) by high performance liquid chromatography (HPLC).
UGT1A1*28 and *6 SNP analysis
Genomic DNA was extracted using a DNA extraction kit (Qiagen, 51304) for PCR amplification. PCR primer sequences designed for the target DNA fragment of UGT1A1 gene (including *28 and *6) were as follows: forward: 5′-TCCCTGCTACCTTTGTGGAC-3′, reverse: 5′-AGCAGGCCCAGGACAAGT-3′. The PCR reaction system contained 2 μl of 10× PCR buffer (15 mM magnesium chloride), 2 μl of deoxy-ribonucleotide triphosphate (dNTP) (2.5 mM), 1 μl of primers (10 μm), 1 μl of DNA templates, 0.2 μl of DNA Taq polymerase (5 U/μl), and 18.8 μl of double distilled water. Amplification conditions were as follows: an initial denaturation step at 94°C for 5 min, followed by 40 cycles of denaturation (94°C for 15 s), then an annealing (55°C for 25 s) step, an extension (72°C for 50 s) step, followed by another extension (72°C for 7 min) step. Having verified by electrophoresis, 5 μl of eligible PCR specimen was mixed with 2 μl Shrimp Alkaline Phosphatase (SAP) and incubated at 37°C for 60 min, followed by an additional incubation step at 80°C for 15 min. The mixture was then stored at 4°C. Approximately 3 μl of positive PCR hydrolysates were mixed with 1 μl sequencing reagent (BigDye®) and 2 μl sequencing primer (*28 prime: forward 5′-CAGCCTCAAGACCCCACA-3′, reverse 5′-TGCTCCTGCCAGAGGTTC-3′; *6 prime: forward 5′-TCCCTGCTACCTTTGTGGA-3′, reverse 5′- AGGAAAGGGTCCGTCAGC-3′) for PCR amplification: initial denaturation at 96°C for 1 min, followed by 25 cycles of denaturation (96°C for 10 s), annealing (50°C for 5 s), and extension (60°C for 4 min), then storage at 4°C. Eventually, the purified products were tested using a DNA sequencer (ABI-373), and the results were analyzed and displayed using the FinchTV® software.
DPD activity detection (UH2/U ratio) (according to Gamelin E et al.Citation33)
Plasma (300 μl) mixed with internal standard (IS) (2 μg/ml of bromouracil) and 1.5 ml of extraction reagent (n-propyl alcohol: tert butyl methyl ether = 25:75, v/v) was oscillated for 2 min, and centrifuged at 2, 250 g for 5 min. The organic phase was then removed, and the above process was repeated. The aqueous phase was dried with nitrogen and re-dissolved with 50 μl H2O and 20 μl of dichloromethane after eddy mixing for 30 s and 5 s consecutively. This was followed by centrifugation at 2, 250 g for 10 min. The supernatant (10 μl) was then drained for analysis. Chromatographic conditions were as follows: shim-pack CLC-ODS column (6 mm ×150 mm, 5 μm), sodium biphosphate buffer (0.05 mol/L, pH 4.0) as the mobile phase, 260 nm wavelength detection for Uracil (U) and 205 nm for dihydrouracil (UH2), and column temperature at 35°C. A standard curve was drawn by taking the U and UH2 concentrations as abscissa and the peak height ratio of the sample and IS as ordinate. The standard and quality controls were prepared using 3% bovine serum albumin.
SN-38 plasma level detection (according to Hirose et al.Citation34 and Schoemaker et al.Citation35)
SN-38 and IS (acetone) were dissolved with dimethyl sulfoxide into 1.0 mg/ml of storage solution, and stored at −80°C. Approximately 2– 500 ng/ml of SN-38 plasma control was prepared with blank plasma. Then, 200 μl of plasma sample was vortexed with 200 μl of acetonitrile (100 ng/ml) for 1 min and centrifuged at 1, 400 g for 10 min. The supernatant (200 μl) was then extracted and mixed with 100 μl of HCl (1 mol/L). The mixture was then analyzed after being vortexed with 100 μl of hydrochloric acid (1 mol/L) for 30 s. The standard curve and linear regression were determined by considering the peak area ratio of SN-38 and IS as the vertical axis. The chromatographic condition used was the Waters® Nova-Pak C18 Guard column (20 mm × 3.9 mm, 5 μm of particle diameter), and the mobile phase was 0.05 mol/L disodium hydrogen phosphate (pH 4.0, including 0.05 mol/L sodium 1-heptanesulfonate) and acetonitrile (75: 25, v/v). Testing conditions included flow rate of 1 ml/min, column temperature at 25°C, 370 nm wavelength of excitation, and 470 nm wavelength of emission.
Evaluation criteria and follow-up
The first efficacy follow-up was performed using CT or MRI scans after 3 cycles for all qualified cases. Partial remission was reconfirmed 4 weeks after the initial assessment based on the Response Evaluation Criteria in Solid Tumors (RECIST, edition 1.1). Subsequent evaluation was done after every 3 cycles until disease progression (PD). The median follow-up was 16 months (8–25 months). Toxicity was graded according to National Cancer Institute common terminology criteria for adverse events (CTCAE, version 4.0).
Statistical analyses
Count data are expressed as mean ± standard deviation. mPFS was calculated using the Kaplan-Meier survival function, and survival differences were analyzed by the log rank test. The Chi-square test was used to compare count data. Differences in toxicity, plasma concentration, and efficacy between subgroups were assessed by one way-ANOVA (Bonferroni option was used for post-hoc test). A Pearson correlation test was applied to evaluate the relationship between variables. All values were 2-sided, and statistical significance was accepted at the p < 0.05 level. The SPSS® (version 19.0) software was used for all statistical analyses.
Disclosure of potential conflicts of interest
We declare that we have no financial or personal relationships with other people or organizations that could inappropriately influence this study. There is no professional or other personal interest of any nature or kind in any product, service, and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled “Correlative analysis of plasma SN-38 levels and DPD activity with outcomes of FOLFIRI regimen for metastatic colorectal cancer with UGT1A1 *28 and *6 wild type and its implication for indidualized chemotherapy.”
Patients were admitted to the study providing that they were exposed to the principle of “Ethics, consent, and permissions.” Before the plasma specimen was collected, patients were fully informed of the purpose and methodology of the study, as well as the role of the plasma specimen in the study design. They were also informed that participation in the study would not increase their medical costs or pain, and the materials and results of the study would be used for the purposes of scientific research without any conflicts of interest. Any report and publication of individual patient data (in the form of images, videos, voice recordings, etc.) required the approval of the patient enrolled in the study.
Acknowledgments
The authors thank the phase I clinical research group at Shanghai General Hospital for DPD activity testing and the Shanghai Yuanqi Biomedical Science & Technology Co. for gene polymorphism testing of UGT1A1 *28 and *6. We also thank Dr Siyu Shao at the Epidemic Prevention branch of the Disease Control Center in Putuo District for assistance with statistical analysis. This work was supported by the Science and Technology Foundation of the Health and Family Planning Commission of Guizhou province, China (gzwjkj2016–1–028).
Disclosure statement
The design and plan of the study were prepared with full consideration of the principles of safety and fairness; therefore, the study was harmless and risk-free to patients. This article does not contain any experiments with human participants, performed by any of the authors. Patients that provided plasma specimens did so on their own will and gave informed consent for its use. The investigator protected the patients' rights and privacy to the maximum extent and ensured that there were no conflicts of interest between the contents and the results of the study. The ethics committee of the Shanghai General Hospital agreed that the study would proceed as scheduled after deliberation.
References
- Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol 2014; 15:1065-75; PMID:25088940; http://dx.doi.org/10.1016/S1470-2045(14)70330-4
- Elez E, Argilés G, Tabernero J. First-line treatment of metastatic colorectal cancer: Interpreting FIRE-3, PEAK, and CALGB/SWOG 80405. Curr Treat Options Oncol 2015; 16:52; PMID:26374340; http://dx.doi.org/10.1007/s11864-015-0369-x
- Snozek CL, O'Kane DJ, Algeciras-Schimnich A. Pharmacogenetics of solid tumors: Directed therapy in breast, lung, and colorectal cancer: A paper from the 2008 william beaumont hospital symposium on molecular pathology. J Mol Diagn 2009; 11:381-9; PMID:19644023; http://dx.doi.org/10.2353/jmoldx.2009.090003
- Sawyer M, Ratain MJ. Body surface area as a determinant of pharmacokinetics and drug dosing. Invest New Drugs 2001; 19:171-7; PMID:11392451; http://dx.doi.org/10.1023/A:1010639201787
- Deeken JF, Figg WD, Bates SE, Sparreboom A. Toward individualized treatment: Prediction of anticancer drug disposition and toxicity with pharmacogenetics. Anticancer Drugs 2007; 18:111-26; PMID:17159598; http://dx.doi.org/10.1097/CAD.0b013e3280109411
- Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, Jang IJ, Lee DH, Lee JS. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol 2006; 24:2237-44; PMID:16636344; http://dx.doi.org/10.1200/JCO.2005.03.0239
- Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, Yokoyama A, Saitoh S, Shimokata K, Hasegawa Y. Polymorphisms of UDPglucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res 2000; 60:6921-26; PMID:11156391
- Yamasaki S, Tanimoto K, Kohno K, Kadowaki M, Takase K, Kondo S, Kubota A, Takeshita M, Okamura S. UGT1A1 *6 polymorphism predicts outcome in elderly patients with relapsed or refractory diffuse large B-cell lymphoma treated with carboplatin, dexamethasone, etoposide and irinotecan. Ann Hematol 2015; 94:65-9; PMID:25055799; http://dx.doi.org/10.1007/s00277-014-2170-5
- Onoue M, Terada T, Kobayashi M, Katsura T, Matsumoto S, Yanagihara K, Nishimura T, Kanai M, Teramukai S, Shimizu A, et al. UGT1A1*6 polymorphism is most predictive of severe neutropenia induced by irinotecan in Japanese cancer patients. Int J Clin Oncol 2009; 14:136-42; PMID:19390945; http://dx.doi.org/10.1007/s10147-008-0821-z
- Ezzeldin H, Diasio R. Dihydropyrimidine dehydrogenase deficiency, a pharmacogenetic syndrome associated with potentially life-threatening toxicity following 5-fluorouracil administration. Clin Colorectal Cancer 2004; 4:181-9; PMID:15377401; http://dx.doi.org/10.3816/CCC.2004.n.018
- Strassburg CP. Gilbert-Meulengracht’ s syndrome and pharmacogenetics: Is jaundice just the tip of the iceberg?. Drug Metab Rev 2010; 42:168-81; PMID:20070246; http://dx.doi.org/10.3109/03602530903209429
- Sugatani J. Function, genetic polymorphism, and transcriptional regulation of human UDP -glucuronosyltransferase (UGT) 1A1. Drug Metab Pharmacokinet 2013; 28:83-92; PMID:23089802; http://dx.doi.org/10.2133/dmpk.DMPK-12-RV-096
- Horsfall LJ, Zeitlyn D, Tarekegn A, Bekele E, Thomas MG, Bradman N, Swallow DM. Prevalence of clinically relevant UGT1A alleles and haplotypes in African populations. Ann Hum Genet 2011; 75:236-46; PMID:21309756; http://dx.doi.org/10.1111/j.1469-1809.2010.00638.x
- Gagné JF, Montminy V, Belanger P, Journault K, Gaucher G, Guillemette C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38). Mol Pharmacol 2002; 62:608-17; PMID:12181437; http://dx.doi.org/10.1124/mol.62.3.608
- Marcuello E, Altés A, Menoyo A, Del Rio E, Gómez-Pardo M, Baiget M. UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br J Cancer 2004; 91:678-82; PMID:15280927; http://dx.doi.org/10.1038/sj.bjc.6602042
- Paoluzzi L, Singh AS, Price DK, Danesi R, Mathijssen RH, Verweij J, Figg WD, Sparreboom A. Influence of genetic variants in UGT1A1 and UGT1A9 on the in vivo glucuronidation of SN-38. J Clin Pharmacol 2004; 44:854-60; PMID:15286088; http://dx.doi.org/10.1177/0091270004267159
- Takano M, Kato M, Yoshikawa T, Sasaki N, Hirata J, Furuya K, Takahashi M, Yokota H, Kino N, Horie K, et al. Clinical significance of UDP-glucuronosyl -transferase 1A1* 6 for toxicities of combination chemotherapy with irinotecan and cisplatin in gynecologic cancers: A prospective multi-institutional study. Oncology 2009; 76:315-21; PMID:19299905; http://dx.doi.org/10.1159/000209335
- Fukui T, Mitsufuji H, Kubota M, Inaoka H, Hirose M, Iwabuchi K, Masuda N, Kobayashi H. Prevalence of topoisomerase I genetic mutations and UGT1A1 polymorphisms associated with irinotecan in individuals of Asian descent. Oncol Lett 2011; 2:923-8; PMID:22866151; http://dx.doi.org/10.3892/ol.2011.346
- Glimelius B, Garmo H, Berglund A, Fredriksson LA, Berglund M, Kohnke H, Byström P, Sørbye H, Wadelius M. Prediction of irinotecan and 5-fluorouracil toxicity and response in patients with advanced colorectal cancer. Pharmacogenomics J 2011; 11:61-71; PMID:20177420; http://dx.doi.org/10.1038/tpj.2010.10
- Wang Y, Xu JM, Shen L, Xu N, Wang JW, Jiao SC, Zhang JS, Song ST, Li J, Bao HY, et al. Polymorphisms of UGT1A gene and irinotecan toxicity in Chinese colorectal cancer patients. Zhonghua Zhong Liu Za Zhi 2007; 29:913-6; PMID:18478930
- Marcuello E, Páez D, Paré L, Salazar J, Sebio A, del Rio E, Baiget M. A genotype-directed phase I-IV dose-finding study of irinotecan in combination with fluorouracil/leucovorin as first-line treatment in advanced colorectal cancer. Br J Cancer 2011; 105:53-7; PMID:21654688; http://dx.doi.org/10.1038/bjc.2011.206
- Freyer G, Duret A, Milano G, Chatelut E, Rebischung C, Delord JP, Merrouche Y, Lledo G, Etienne MC, Falandry C. Pharmacogenetic tailoring of irinotecan-based first-line chemotherapy in metastatic colorectal cancer: Results of a pilot study. Anticancer Res 2011; 31:359-366; PMID:21273624
- Kim KP, Hong YS, Lee JL, Bae KS, Kim HS, Shin JG, Lee JS, Kim TW. A phase I study of UGT1A1 * 28/* 6 genotype-directed dosing of irinotecan (CPT-11) in Korean patients with metastatic colorectal cancer receiving FOLFIRI. Oncology 2015; 88:164-72; PMID:25427841; http://dx.doi.org/10.1159/000368674
- Lu CY, Huang CW, Hu HM, Tsai HL, Huang CM, Yu FJ, Huang MY, Chang SF, Huang ML, Wang JY. Prognostic advantage of irinotecan dose escalation according to uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) genotyping in patients with metastatic colorectal cancer treated with bevacizumab combined with 5-fluorouracil /leucovorin with irinotecan in a first-line setting. Transl Res 2014; 164:169-76; PMID:24462762; http://dx.doi.org/10.1016/j.trsl.2013.12.009
- Innocenti F, Schilsky RL, Ramírez J, Janisch L, Undevia S, House LK, Das S, Wu K, Turcich M, Marsh R, et al. Dose-finding and pharmacokinetic study to optimize the dosing of irinotecan according to the UGT1A1 genotype of patients with cancer. J Clin Oncol 2014; 32:2328-34; PMID:24958824; http://dx.doi.org/10.1200/JCO.2014.55.2307
- Satoh T, Yasui H, Muro K, Komatsu Y, Sameshima S, Yamaguchi K, Sugihara K. Pharmacokinetic assessment of irinotecan, SN-38, and SN-38 glucuronide: A substudy of the FIRIS study. Anticancer Res 2013; 33:3845-53; PMID:24023318
- Lee LS, Seng KY, Wang LZ, Yong WP, Hee KH, Soh TI, Wong A, Cheong PF, Soong R, Sapari NS, et al. Phenotyping of UGT1A1 activity using raltegravir predicts Pharmacokinetics and toxicity of Irinotecan in FOLFIRI. PLoS One 2016; 11:e0147681; PMID:26808671; http://dx.doi.org/10.1371/journal.pone.0147681
- Cai X, Cao W, Ding H, Liu T, Zhou X, Wang M, Zhong M, Zhao Z, Xu Q, Wang L. Analysis of UGT1A1*28 genotype and SN-38 pharmacokinetics for irinotecan-based chemotherapy in patients with advanced colorectal cancer: Results from a multi-center, retrospective study in Shanghai. J Cancer Res Clin Oncol 2013; 139:1579-89; PMID:23892411; http://dx.doi.org/10.1007/s00432-013-1480-7
- Ducreux M, Ychou M, Seitz JF, Bonnay M, Bexon A, Armand JP, Mahjoubi M, Méry-Mignard D, Rougier P. Irinotecan combined with bolus fluorouracil, continuous infusion fluorouracil, and high-dose leucovorin every two weeks (LV5FU2 regimen): A clinical dose-finding and pharmacokinetic study in patients with pretreated metastatic colorectal cancer. J Clin Oncol 1999; 17:2901-8; PMID:10561369; http://dx.doi.org/10.1200/JCO.1999.17.9.2901
- Saltz LB, Kanowitz J, Kemeny NE, Schaaf L, Spriggs D, Staton BA, Berkery R, Steger C, Eng M, Dietz A, et al. Phase I clinical and pharmacokinetic study of irinotecan, fluorouracil, and leucovorin in patients with advanced solid tumors. J Clin Oncol 1996; 14:2959-67; PMID:8918493; http://dx.doi.org/10.1200/JCO.1996.14.11.2959
- Goey AK, Mooiman KD, Beijnen JH, Schellens JH, Meijerman I. Relevance of in vitro and clinical data for predicting CYP3A4-mediated herb-drug interactions in cancer patients. Cancer Treat Rev 2013; 39:773-83; PMID:23394826; http://dx.doi.org/10.1016/j.ctrv.2012.12.008
- Teft WA, Welch S, Lenehan J, Parfitt J, Choi YH, Winquist E, Kim RB. OATP1B1 and tumour OATP1B3 modulate exposure, toxicity, and survival after irinotecan-based chemotherapy. Br J Cancer 2015; 12:857-65; PMID:25611302; http://dx.doi.org/10.1038/bjc.2015.5
- Gamelin E, Boisdron-Celle M, Guérin-Meyer V, Delva R, Lortholary A, Genevieve F, Larra F, Ifrah N, Robert J. Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5-FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: A potential interest for predicting 5-FU toxicity and determining optimal 5-FU dosage. J Clin Oncol 1999; 17:1105-10; PMID:10561167; http://dx.doi.org/10.1200/JCO.1999.17.4.1105
- Hirose K, Kozu C, Yamashita K, Maruo E, Kitamura M, Hasegawa J, Omoda K, Murakami T, Maeda Y. Correlation between plasma concentration ratios of SN-38 glucuronide and SN-38 and neutropenia induction in patients with colorectal cancer and wild-type UGT1A1 gene. Oncol Lett 2012; 3:694-8; PMID:22740978; http://dx.doi.org/10.3892/ol.2011.533
- Schoemaker NE, Rosing H, Jansen S, Schellens JH, Beijnen JH. High-performance liquid chromatographic analysis of the anticancer drug irinotecan (CPT-11) and its active metabolite SN-38 in human plasma. Ther Drug Monit 2003; 25:120-4; PMID:12548157; http://dx.doi.org/10.1097/00007691-200302000-00020