
ABSTRACT
The TP53 gene is distinguished as the most frequently mutated gene in cancer. Unlike most cancer-relevant genes, the TP53 gene is also distinguished by the existence of coding region polymorphisms that alter p53 sequence, and in some cases, also alter p53 function. A common coding region variant at amino acid 72 of p53 encodes either proline (P72) or arginine (R72). P72 is the ancestral variant and is most common in populations near the equator. The frequency of the R72 variant increases in a linear manner with latitude. To date, why the R72 variant arose in humans and was possibly selected for has remained unclear. Here-in we show that this single nucleotide polymorphism (SNP) influences the phosphorylation of p53 and the transactivation of the key p53 target CDKN1A (p21) specifically in response to nutrient deprivation, but not in response to conventional cytotoxic agents. Following activation of the kinase AMPK, R72 cells show increased phosphorylation on serine-15 and increased transactivation of the cyclin-dependent kinase inhibitor CDKN1A (p21) and the metabolic response genes PPARGC1B (PGC-1β) and PRKAB2 (AMPK-β2). This is accompanied by increased growth arrest and decreased apoptosis in R72 cells compared with P72 cells. The combined data fit best with the hypothesis that the R72 polymorphism confers increased cell survival in response to nutrient deprivation. This differential response to nutrient deprivation may explain part of selection for this SNP at northern latitudes, where nutrient deprivation might have been more frequent.
Introduction
The p53 tumor suppressor protein is a critical player in human cancer, and the TP53 gene continues to be distinguished as the most frequently mutated gene in human cancer.Citation1 In addition to mutation, several coding region polymorphisms in TP53, and in genes that regulate p53, function to dampen this pathway and contribute to cancer (seeCitation2,3 for review). For example, a single nucleotide polymorphism in the promoter of MDM2, which negatively regulates p53, increases MDM2 level and consequently decreases p53 activity, leading to earlier age of onset for many cancers.Citation4,5 Additionally, a coding region SNP in TP53 at amino acid 47 (Pro47Ser) leads to decreased ability of p53 to induce ferroptosis, coupled with increased cancer risk in mice and humans.Citation6-8 In contrast to these examples, a common coding region SNP in p53 at codon 72 has failed to show compelling impact on cancer risk, despite the fact that this SNP is predicted to alter p53 structure,Citation9 and data from numerous groups that this SNP alters p53 function.Citation9–15 An intriguing yet poorly understood fact is that this SNP shows a linear association with latitude: specifically, the arginine 72 (R72) variant shows a linear increase in frequency in populations away from the equator, whereas the proline 72 variant (P72) is most common in populations near the equator.Citation16,17
To date genome-wide association studies have failed to find an association between the p53 codon 72 SNP and cancer. In contrast, significant associations with body mass index, waist circumference, and type 2 diabetes have been found for this SNP. Specifically, the R72 variant is associated with increased weight and type 2 diabetes risk.Citation18–21 To elucidate a possible mechanism for this, we previously created a mouse model for the codon 72 variants, in the background of a Humanized p53 knock-in (Hupki) allele.Citation11,22 We exposed these mice to a diet-induced obesity regimen by feeding them a high fat diet (HFD), and found that mice with the arginine 72 (R72) variant of p53 showed increased fat accumulation on a HFD, compared with mice with the proline 72 variant (P72), leading to increased diabetic phenotypes. Mechanistically we found that this increased fat accumulation was due to increased transactivation by the R72 variant of 2 genes, Tnf and Npc1l1, following exposure to a high fat diet.Citation12 The combined data from this previous report suggested that the possible selection for the R72 variant in populations in Northern latitudes might be due to the differences in function of this SNP in response to nutrient excess. In this work, we sought to test the impact of the codon 72 SNP of p53 on the response to nutrient deprivation.
Results
In an effort to probe the influence of the codon 72 polymorphism of p53 to nutrient deprivation, we sought to determine the p53 response in tumor and normal cells to the activation of AMP-dependent protein kinase (AMPK), a key sensor and regulator of the nutrient deprivation response. Toward this end, we activated AMPK in cultured cells by incubating them with the AMP analog 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR). Previous studies indicated that following AICAR treatment p53 is phosphorylated on serine 15 and stabilized, leading to increased transactivation of CDKN1A (hereafter p21) and subsequent growth arrest (see schematic ).Citation23,24 Consistent with this, treatment of Hct116 (p53 WT) and Hct116 p53 −/− cells with different doses of AICAR for 24 hours led to marked induction of p53 and serine 15 phosphorylation of p53 (a marker of p53 activation), along with increased levels of the p53 target proteins MDM2 and p21 in Hct116 cells but not Hct116 p53 −/− cells (). We next tested these same concentrations of AICAR in normal human fibroblasts (NHF) containing WT p53, compared with the same cell line stably-infected with a lentivirus encoding a short hairpin for p53 (shp53). This experiment revealed that treatment of NHF led to stabilization of p53 and induction of p21, and that 2 mM AICAR led to the maximal response (); therefore a 2 mM concentration was selected for further analysis.
Figure 1. AICAR induces p53 activation. (A) p53 is involved in the metabolic stress response pathway to regulate cell fate. (B) Hct116 cells with or without (p53−/−) functional p53 were treated with AICAR for 24 hours. Expression levels of p53 and p53 target proteins (MDM2 and p21) were detected by western blot analysis. Actin is included as the loading control. (C) Normal human fibroblast cells (NHF 6113 cell line) with WT p53 or treated with a short hairpin RNA to p53 (shp53) were treated with AICAR at the concentrations depicted for 24 hours. Expression levels of p53 and p21 were detected by western blot analysis. GAPDH is included as the loading control.
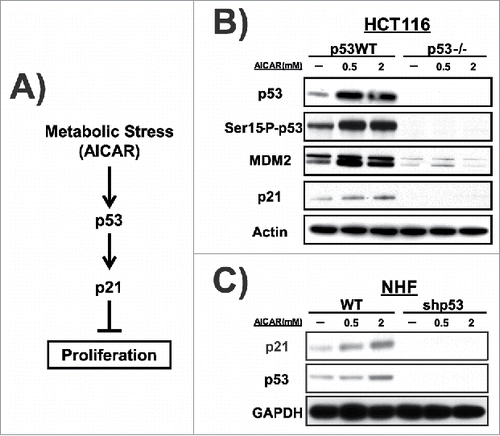
We next sought to test for an impact of the codon 72 polymorphism of p53 on the response to AMPK activation. We performed a time course of p53 induction and serine 15 phosphorylation of p53 in NHF that are homozygous for the proline 72 (P72) and arginine 72 (R72) polymorphic variants of p53. To ensure rigor we used 2 different sets of NHF that were homozygous for P72 and R72, and obtained consistent results (see Materials and Methods). Following treatment with 2 mM AICAR for 1, 6 and 24 hours, p53 level was increased in both cell lines, and serine-15 phosphorylation was also increased; consistently however the increase in both was greater in cells encoding the R72 variant compared with P72 (). Densitometric quantification of the averaged results from 3 independent experiments supported the conclusion that there was a significant increase in the phosphorylation of the R72 variant following AICAR, compared with P72, leading to an increase in steady-state levels of p53 protein in R72 cells (). This led to a significant increase in the mRNA level of p21 in R72 cells following AICAR, as assessed by quantitative reverse transcription polymerase chain reaction analysis (qRT-PCR) ().
Figure 2. AMPK activation induces p53 with distinct kinetics in cells that differ in the codon 72 polymorphism. (A) P72 and R72 NHF were treated with AICAR (2 mM) for 1, 6, or 24 hr. Expression levels of p53 and serine-15 phosphorylated p53 were detected by western blot analysis. Actin is included as the loading control. (B) Levels of p53 and serine-15 phosphorylated p53 were quantified after 6 hours of AICAR treatment. Error bars represent standard error. The values depicted are the average quantification from 2 independent experiments. (C) qRT-PCR assessment of the mRNA level of the p53 target gene CDKN1A (p21) in NHF cells treated with AICAR for 6 hr. The data represent the average from 3 independent experiments, error bars mark standard error. The asterisk denotes p-value <0.05. (D) Human lung cancer cell line H1299 expressing doxycycline-inducible P72 or R72 versions of p53 were treated with doxycycline plus etoposide. Expression levels of p53, serine-15 phosphorylated p53 and CDKN1A (p21) were detected by western blot analysis. GAPDH is the loading control. (E) Human lung cancer cell line H1299 expressing doxycycline-inducible P72 or R72 versions of p53 were treated with doxycycline plus AICAR, as indicated in the figure. Expression levels of p53, serine-15 phosphorylated p53 and CDKN1A (p21) were detected by western blot analysis. GAPDH is the loading control. (F) P72 and R72 NHFs were deprived of glucose for 0, 5, or 24 hr and probed by western blotting for the proteins indicated. The results depicted are representative of 3 independent experiments. (G) P72 and R72 NHFs were treated with Metformin (5 mM) for 6 hr. Western blot analyses were performed using antisera against the proteins indicated. The results depicted are representative of 2 independent experiments; densitometry results are normalized to GAPDH control.
To confirm these data in an independent setting, we analyzed H1299 cells containing doxycycline-inducible versions of p53 encoding the P72 and R72 variants. To establish the importance of AICAR in the differential response of P72 and R72, we analyzed these lines treated with etoposide, which induces DNA damage, compared with AICAR. In response to doxycycline combined with etoposide, the P72 and R72 variants responded similarly to each other; specifically, there was equivalent serine 15 phosphorylation, p53 stabilization, and p21 induction ()wh. In contrast, following doxycycline plus AICAR treatment the R72 variant showed increased serine 15 phosphorylation, p53 level, and p21 induction (). Therefore, in 2 independent experimental systems, the response to AICAR was enhanced in R72 cells. This increased response of the R72 variant was not specific to AMPK activation by AICAR, as it was also observed in R72 NHF treated with glucose deprivation () and metformin, which like AICAR leads to AMPK activation (). The combined data support the premise that cells containing the R72 variant of p53 show a more robust response to nutrient deprivation.
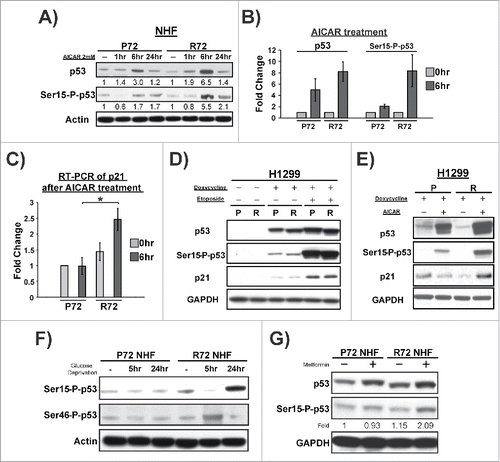
We next sought to probe the growth arrest and cell death response following AICAR treatment in P72 and R72 NHFs, and in H1299 doxycycline-inducible cells. Analysis of S phase cells using BrdU staining in P72 and R72 NHF 24 hours following AICAR treatment revealed a consistent increase in growth arrest in R72 cells (). This finding was corroborated using propidium iodide staining in NHFs, which showed a marked increase in G1 phase in R72 cells following AICAR treatment compared with P72 (); in this assay, the lack of G1-arrested P72 cells appeared to reflect an increased apoptotic response of cells with this SNP. To test this possibility further we used Annexin V staining to assess the accumulation of apoptotic cells. This analysis revealed a statistically significant increase in apoptosis in P72 cells treated with AICAR, compared with R72 (p<0.005, ). This increased apoptotic response of P72 was also evident in the doxycycline-inducible H1299 cells as well (p<0.05, ). The combined data support the premise that growth arrest versus cell death decision of cells following AICAR treatment is influenced by the codon 72 polymorphism. Specifically, P72 cells show increased apoptosis compared with R72, while R72 cells show increased growth arrest.
Figure 3. AICAR differentially induces cell cycle arrest and apoptosis in P72 and R72 cells. (A) Quantification of BrdU staining to detect S phase cells in P72 and R72 NHFs treated with AICAR (2 mM) for 24 hr. (B) Cell cycle analysis using propidium iodide staining was used to determine the percent of G1 phase cells in P72 and R72 NHFs treated with AICAR (2 mM) for 24 hr. The data represent the average from 4 independent experiments; error bars mark standard error. (C) Annexin V staining was used to assess apoptosis in P72 and R72 NHFs treated with AICAR (2 mM) for 24 hr. The data represent the average from 3 independent experiments; error bars mark standard error. The double asterisk denotes p-value <0.005. (D) H1299 Tet-inducible p53 cells were treated with doxycycline along with AICAR (2 mM) for 24 hr and subjected to Annexin V staining to determine level of apoptosis. The data depicted represent 3 independent experiments; error bars mark standard error. The single asterisk denotes p-value <0.05.
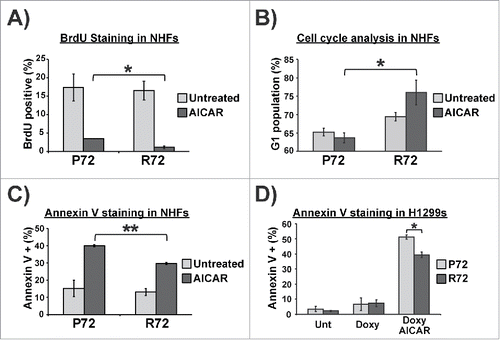
To address the potential mechanism underlying the cell fate differences following AICAR treatment between P72 and R72 cells, we performed qRT-PCR analysis for p53 target genes in these cells. For this analysis we focused on p53 target genes involved in growth arrest and metabolism (including p21, PPARGC1B (PGC-1β), and PRKAB2 (AMPK-β), as well as genes with roles in apoptosis (BBC3 (PUMA), PMAIP1 (NOXA) and DRAM1). The majority of other p53 target genes that we analyzed showed no differences between P72 and R72 (data not shown; see details in Materials and Methods). In NHF, we found significant increases in the mRNA levels of genes associated with growth arrest and metabolism in R72 cells, compared with P72 (); the increased induction of p21 is consistent with the increased growth arrest in R72 cells. Conversely, we saw consistent increases in genes involved in apoptosis in P72 cells treated with AICAR, relative to R72 (). Importantly, these data were recapitulated in doxycycline-inducible P72 and R72 cells (). We next tested the hypothesis that the increased transactivation of p21 in R72 cells, and NOXA in P72 cells, was mediated by altered recruitment of these p53 variants to the promoters of these genes. Toward this end we performed chromatin immunoprecipitation in H1299 cells with doxycycline-inducible P72 and R72 in the absence and presence of doxycycline and AICAR. This analysis revealed a marked increase in the R72 protein bound to the major p53 binding site in the p21 promoter, compared with P72. Conversely there was a marked decrease in the binding of R72 protein to the p53 binding site in the NOXA promoter (). The combined data support the premise that the altered transcriptional profiles in P72 and R72 cells appear to be mediated by altered promoter binding of these variants following AICAR treatment.
Figure 4. Distinctive gene expression profiles in P72 and R72 cells following AICAR treatment. (A) P72 and R72 NHFs were treated with 2 mM AICAR for 6 hr and the level of the p53-target genes indicated was assessed by qRT-PCR; the data depicted represent the averages from 3 independent experiments. (B) H1299 cells containing doxycycline-inducible P72 or R72 were treated with doxycycline and AICAR for 6 hr. Total RNA were extracted and subjected to qRT-PCR; the data depicted represent the averages from 3 independent experiments. (C) Chromatin immunoprecipitation (ChIP) analysis data from H1299 cells with tetracycline-inducible P72 and R72 following treatment of cells with doxycycline and AICAR after 6 hours. The percent of DNA bound normalized to input for IgG and p53 antibody from 3 independent experiments is depicted for the binding sites of CDKN1A (p21) and PMAIP1 (NOXA).
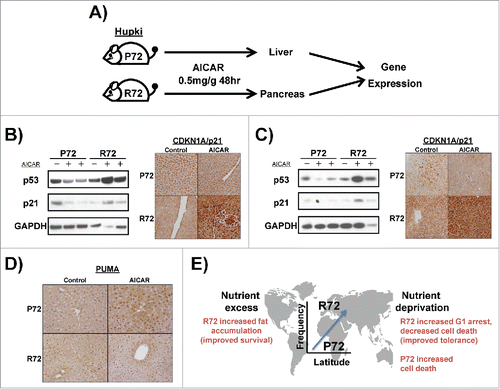
We next sought to recapitulate our findings about the differential response of P72 and R72 to AICAR treatment in animals. Toward this end, we treated P72 and R72 mice, which we previously established and characterized,Citation11,22 with AICAR. P72 and R72 mice were treated with a single intra-peritoneal dose of AICAR at 0.5 mg/kg, and the liver and pancreas were analyzed for p53 and p21 level by western blot, as well as p21 and PUMA by immunostaining analyses, after 48 hours (). Consistent with the data obtained in cell lines, we noted increased p53 and p21 protein level in the pancreas and liver of AICAR-treated R72 mice, relative to P72 ( and ). In contrast, we noted increased PUMA induction in the livers of P72 mice compared with R72 (). In sum, in normal and transformed cell lines and in mice, the R72 variant consistently shows increased induction of p53 and p21 in response to nutrient deprivation, while the P72 variant shows increased induction of apoptotic markers like PUMA.
Figure 5. Differential regulation of CDKN1A (p21) in P72 and R72 mice following AICAR treatment. (A) Design of experiment: P72 and R72 mice were injected with AICAR (2 mg/g) or dilution vehicle and the liver and pancreas were collected after 48 hours. Total protein was extracted and subjected to western blotting (n = 2 mice per genotype per treatment). Additionally, tissues were fixed, sectioned and subjected to staining (n = 2 mice per genotype per treatment). (B) CDKN1A (p21) protein expression in the pancreas from P72 and R72 mice treated with AICAR was assessed by western blotting and immunohistochemical staining. (C) CDKN1A (p21) protein expression in the liver from P72 and R72 mice treated with AICAR was assessed by western blotting and immunohistochemical staining. (D) PUMA protein expression in the liver from P72 and R72 mice treated with AICAR was assessed by immunohistochemical staining. E. Model for the relationship between the codon 72 SNP of p53 with latitude and metabolic stress.
Discussion
Despite years of work studying the p53 codon 72 polymorphic variants, their precise differences in function, the potential role of this SNP in cancer, and the possible reasons for genetic variation at this locus have been unclear. GWAS and other studies have implicated the R72 SNP in body mass index and diabetes risk.Citation18–21 To explore the mechanism underlying this association, we previously showed that in response to nutrient excess, the R72 variant of p53 shows increased fat accumulation, caused by increased transactivation of 2 p53 target genes Tnf and Npc1l1, which regulate inflammation and cholesterol absorption, respectively. We hypothesized that the R72 variant may have been selected for in environments of northern latitudes due to the increased ability of these individuals to accumulate fat, and consequently survive times of food scarcity and/or colder temperatures. The latter hypothesis is consistent with the finding that the association of the R72 variant with increasing latitude relies more on an association with colder winter temperatures.Citation25
In this work we analyzed the opposing stress on p53, nutrient deprivation. In response to nutrient deprivation, p53 is activated following phosphorylation of serine 15 by the protein kinase AMPK, leading typically to growth arrest.Citation23 Using standard agents that activate AMPK such as the ribonucleoside analog AICAR, the compound metformin or glucose deprivation, we found that the R72 variant of p53 consistently showed heightened stabilization and transactivation of the cdk inhibitor p21, compared with the P72 variant, in 2 different cell line backgrounds and in a mouse model. This led to preferential cell cycle arrest instead of programmed cell death in R72 cells, which would ultimately be predicted to improve cell survival. This might likewise be predicted to improve the survival of individuals moving to northern climates, where food scarcity might lead to occasional periods of nutrient deprivation. The combined data best support a model whereby the migration of individuals from equatorial regions to more northern climates would select for the R72 polymorphism to allow for increased fat accumulation in times of nutrient excess, as well as increased survival following nutrient deprivation (). We propose that this may explain the latitudinal bias of this SNP. Further, the impact of p53 on the control of metabolism may explain why this tumor suppressor gene, unlike so many others, possesses functionally impactful coding region polymorphic variants that differ in frequency in different populations.
It remains unclear how these data might influence our thinking of the contribution of the codon 72 polymorphism with cancer risk and the efficacy of cancer therapy. Despite early reports that this SNP markedly impacted the ability of p53 to induce apoptosis in tumor cell lines, primarily due to increased use of the direct mitochondrial pathway for the R72 variant,Citation10,26 compelling association of this SNP with cancer risk has been lacking.Citation3 Our data suggest that individuals with the R72 variant may be subject to increased weight gain, which might increase cancer risk. Additionally, tumors from these individuals might show evidence for increased survival under periods of nutrient deprivation, which might increase cancer progression. When combined with increased ability of R72 to respond to chemotherapeutic agents, these combined factors may yield no net sum influence on cancer risk. In fact, these opposing cancer risk factors (obesity, survival following nutrient deprivation, and increased therapy response) might account for the numerous conflicting reports, and ultimate lack of a consensus, on the contribution of this SNP to cancer.
A final remaining question regards the underlying mechanism for the increased ability of the R72 variant to transactivate CDKN1A and metabolism genes following AICAR treatment, under conditions where the P72 variant of p53 preferentially transactivates pro-apoptotic genes. Such promoter selectivity governing the cell cycle arrest and apoptotic response of cells has been previously reported.Citation27 We posit that the proline-rich domain of p53, where the codon 72 SNP lies, constitutes a protein-protein interaction domain, and that the codon 72 SNP influences the interaction of a key co-activator for p53. In support of this premise, we previously showed that the SUMO-ligase TRIML2 interacts preferentially with the R72 variant of p53, and modifies p53 with a SUMO2/3 tag, which then regulates the ability of p53 to bind to the promoters of certain genes.Citation14 We propose that a co-activator that is specifically necessary following nutrient deprivation must cooperate with p53 and differentially bind to P72 and R72 variants of this protein.
Materials and methods
Cell lines and reagents
Normal Human Fibroblast (NHF) cells homozygous for p53 variants P72 or R72 (1603 P72, 6113 P72, 5386 R72, and 6555 R72) were purchased from the Coriell Institute for Medical Research (Camden, NJ, USA) and cultured in 15% fetal bovine serum (Gembio), Penicillin (100 I.U./ml)-Streptomycin (100 μg/ml) (Pen/Strep, Cellgro, 30–002-CI), Non-Essential Amino Acids (NEA, Cellgro, 25–030-CI), 2 mM L-glutamine (Cellgro, 25–005-CI) in Dulbecco's Modification of Eagle's Medium (DMEM, Cellgro). H1299 cells containing Tet-inducible variants of p53 P72 and R72 were provided by Steven McMahon (Thomas Jefferson University)Citation11 and cultured in DMEM supplemented with 10% Tet approved system FBS (Clontech, 631106) and Pen/Strep. HCT116 p53 WT and HCT116 p53−/− cells were provided by Bert Vogelstein from Johns Hopkins School of MedicineCitation28 and were cultured in McCoy's 5A medium (Cellgro, 10–050-CV) supplemented with 10% FBS and Pen/Strep. A concentration of 0.75 μg/ml doxycycline (BD Biosciences, 631311) was used to induce gene expression from tetracycline-inducible constructs. Etoposide (Sigma, E1383) was used at a concentration of 100 uM to induce DNA damages. AICAR (5-Aminoimidazole-4-carboxyamide Ribonucleoside; Cell Signaling Technology, 9944) and Metformin (Sigma, 1396309) were used to induce metabolic stresses. Fugene 6 transfection reagent (Promega) was used for all transfection experiments.
Human p53 knock-in (Hupki) mice
Hupki P72 and R72 mice in C57/BL6 background were generated as described previously.Citation11,22 All studies with mice complied with all federal and institutional guidelines as per IACUC protocols. Mice were housed in plastic cages with ad libitum diet and maintained at 22°C with a 12-hour dark/12- hour light cycle. Six- to 8-week old male Hupki mice were treated with AICAR (0.5 mg/g body weight) through intra-peritoneal injection. Forty-eight hours post-injection, mice were killed by carbon dioxide inhalation and tissues (liver and pancreas) were harvested. Harvested tissues were either formalin fixed and analyzed by immunohistochemistry using antisera for p21 (Ab6, Calbiochem, OP79) or PUMA (Abcam ab9643), or were homogenized in TissueLyser II (Qiagen) with radio-immunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% sodium dodecyl sulfate [SDS], 0.1% deoxycholic acid) supplemented with 10 mM sodium fluoride, 10 mM sodium orthovanadate (Na3VO4), and protease inhibitor cocktail (Roche). Lysates were clarified and the protein concentration was determined using DC protein assay system (Bio-Rad).
Western blot analysis
Cell lysates were extracted from cells at ∼90% confluence. Cell were washed with cold phosphate-buffered saline (PBS, Gibco), scrape harvested in cold PBS, centrifuged at 1000 × g for 5 min, and lysed with RIPA buffer supplemented with 10 mM NaF, 10 mM Na3VO4, and protease inhibitor cocktail. Lysates were clarified and the protein concentration was determined using DC protein assay system (Bio-Rad). Equal amount of protein was resolved over sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using pre-cast NuPAGE® Bis-Tris gels (Life Technologies) and transferred onto PVDF membranes (Bio-Rad). Primary antibodies used in this study include p53 (Ab6, Calbiochem, OP43; CM5, Leica Biosystems), p53 Ser-15-P (Cell Signaling, 9284), p53 Ser-46-P (Cell Signaling, 2521), GAPDH (14C10, Cell Signaling, 2118), Actin (AC-15, Sigma, A5441), p21 (Ab6, Calbiochem, OP79), MDM2 (Ab1-OP46, Ab2-OP115, Calbiochem), and PUMA. Secondary antibodies conjugated to Horseradish peroxidase were used at a dilution of 1:10,000 (Jackson Immunochemicals). ECL (Amersham, RPN2232) was then applied to blots and protein levels were detected using autoradiography. Densitometry quantification of protein signals was performed using ImageJ software (NIH).
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cells using RNeasy Mini kit (Qiagen, 74104) including on-column DNase digestion (Qiagen, 79254) following the manufacturer's protocol. High Capacity cDNA Reverse Transcription Kit (Applied Biosciences, 4368814) was used to transcribe RNA to cDNA. Quantitative PCR (qPCR) was performed using Brilliant III Ultra Fast SYBR® Green QPCR Mix kit (Agilent Technologies, 600882) on the Stratagene Mx3005P device (Agilent Technologies), and data analysis was performed using the MxPro program (Stratagene). mRNA expression levels were normalized to cyclophilin A. Primers used in this study include (5′; to 3′;) p21: GGAAGACCATGTGGACCTGT and GGATTAGGGCTTCCTCTTGG; PGC-1β: CCGAGCTCTTCCAGTTGAC and CGAAGCTGAGGTGCATGATA; AMPK-β2: GTGTTCAGCCTCCCTGACTC and AAGACCTCCTTGCCTCCTTC; PUMA: GTCCCCTGCCAGATTTGTG and AGAGGCCGCAGGACACTG; NOXA: CCAGTTGGAGGCTGAGGTT and TTCTGCCGGAAGTTCAGTTT; DRAM: CTTGGATTGGTGGGATGTT and CAGACAAAGGCCAAAAGAGC.
Chromatin immunoprecipitation
Chromatin immunoprecipitation analysis was performed according to the manufacturer's protocol (EZ-ChIP; Millipore). Briefly, cells were cultivated to 90% confluence, harvested, then fixed for 10 min in freshly made fixation solution (1% formaldehyde, 10 mM NaCl, 0.1 mM EDTA, 5 mM HEPES, pH7.9). Fixation was quenched by 0.125 M Glycine, followed by PBS wash, and cells were lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH8.1) containing protease inhibitors. Chromatin was sheared by sonication to an average size of 200 to 1000 bp, clarified, and precleared for 1 h at 4°C with salmon sperm DNA-saturated protein A-Sepharose beads (Millipore, 16–157). The supernatant was incubated with normal mouse immunoglobulin G or anti-p53 (Ab6, Calbiochem, OP43) and rotated overnight at 4°C. Lysates were immunoprecipitated with salmon sperm DNA-saturated protein A-Sepharose beads for 1 h at 4°C and washed extensively. Input and immunoprecipitated protein/DNA complexes were eluted at room temperature, and the cross-linking was reversed overnight at 65°C in the presence of 200 mM NaCl, before being treated with RNase A (37°C for 30 min) and proteinase K (45°C for 2 h). Sample DNAs were purified through DNA columns and subjected to Quantitative PCR using Brilliant III Ultra Fast SYBR® Green QPCR Mix kit on the Stratagene Mx3005P device. Primers used for ChIP analysis include (5′; to 3′;) p21: GTGGCTCTGATTGGCTTTCTG and CTGAAAACAGGCAGCCCAAG; NOXA: CAGCGTTTGCAGATGGTCAA and CCCCGAAATTACTTCCTTACAAAA.
BrdU staining, Annexin V and cell cycle analyses, statistical analysis
Cells were grown on coverslips before fixed for immunofluorescence. Detection of BrdU incorporation was performed by using the BrdU detection kit I (Sigma, 11296736001) according to the manufacturer's protocol. For each cell line, at least 6 separate fields and over 300 cells were counted. Annexin V staining to detect apoptotic cells and cell cycle analysis were performed using Guava Nexin Reagent (Millipore, 4500–0450) and Propidium iodide staining on the Guava easyCyte HT system according to the manufacturer's protocols, respectively. Statistical analyses were performed using 2-sided unpaired Student's t test. Data were considered significant if p<0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
Conception and Design: C.P.K. and M.E.M.
Data Analysis: all
Drafting and approval of manuscript: all
Acknowledgments
The authors declare no conflicts of interest. This work was supported by R01s CA201430 and CA102184 (M.E.M.). The authors would like to acknowledge the Histotechnology, Laboratory Animal and Imaging facilities at The Wistar Institute. Support for Core Facilities used in this study was provided by Cancer Center Support Grant (CCSG) CA010815 to The Wistar Institute.
Funding
This work was supported by the NIH under grants R01CA102184 and CA201430 (M.E.M).
References
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Na Rev Cancer 2009; 9:749-58; PMID:19776744; https://doi.org/10.1038/nrc2723
- Murphy ME. Polymorphic variants in the p53 pathway. Cell Death Differ 2006; 13:916-20; PMID:16557270; https://doi.org/10.1038/sj.cdd.4401907
- Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Na Rev Cancer 2009; 9:95-107; PMID:19165225; https://doi.org/10.1038/nrc2584
- Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, Bartel F, Taubert H, Wuerl P, Hait W, et al. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res 2006; 66:5104-10; PMID:16707433; https://doi.org/10.1158/0008-5472.CAN-06-0180
- Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004; 119:591-602; PMID:15550242; https://doi.org/10.1016/j.cell.2004.11.022
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 2016; 30:918-30; PMID:27034505; https://doi.org/10.1101/gad.275891.115
- Basu S, Barnoud T, Kung CP, Reiss M, Murphy ME. The African-specific S47 polymorphism of p53 alters chemosensitivity. Cell cycle 2016; 15:2557-60; PMID:27484708; https://doi.org/10.1080/15384101.2016.1215390
- Murphy ME, Liu S, Yao S, Huo D, et al. A functionally significant SNP in TP53 and breast cancer risk in African American women. NPJ Breast Cancer 2017; 3:5; PMID:28232952; https://doi.org/:10.1038/s41523-017-0007-9
- Thomas M, Kalita A, Labrecque S, Pim D, Banks L, Matlashewski G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol Cell Biol 1999; 19:1092-100; PMID:9891044; https://doi.org/10.1128/MCB.19.2.1092
- Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 2003; 33:357-65; PMID:12567188; https://doi.org/10.1038/ng1093
- Frank AK, Leu JI, Zhou Y, Devarajan K, Nedelko T, Klein-Szanto A, Hollstein M, Murphy ME. The codon 72 polymorphism of p53 regulates interaction with NF-{kappa}B and transactivation of genes involved in immunity and inflammation. Mol Cell Biol 2011; 31:1201-13; PMID:21245379; https://doi.org/10.1128/MCB.01136-10
- Kung CP, Leu JI, Basu S, Khaku S, Anokye-Danso F, Liu Q, George DL, Ahima RS, Murphy ME. The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell Rep 2016; 14:2413-25; PMID:26947067; https://doi.org/10.1016/j.celrep.2016.02.037
- Leu JI, Murphy ME, George DL. The p53 Codon 72 polymorphism modifies the cellular response to inflammatory challenge in the liver. J Liver 2013; 2:117; PMID:23991369; https://doi.org/10.4172/2167-0889.1000117
- Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME. Identification of TRIML2, a Novel p53 target, that enhances p53-SUMOylation and regulates the transactivation of pro-apoptotic genes. Mol Cancer Res 2015 Feb; 13(2):250-62; PMID:25256710; https://doi.org/10.1158/1541-7786.MCR-14-0385
- Feng Z, Zhang C, Kang HJ, Sun Y, Wang H, Naqvi A, Frank AK, Rosenwaks Z, Murphy ME, Levine AJ, et al. Regulation of female reproduction by p53 and its family members. FASEB J 2011; 25:2245-55; PMID:21402718; https://doi.org/10.1096/fj.10-180166
- Sjalander A, Birgander R, Kivela A, Beckman G. p53 polymorphisms and haplotypes in different ethnic groups. Hum Hered 1995; 45:144-9; PMID:7615299; https://doi.org/10.1159/000154275
- Beckman G, Birgander R, Sjalander A, Saha N, Holmberg PA, Kivela A, Beckman L. Is p53 polymorphism maintained by natural selection? Hum Hered 1994; 44:266-70; PMID:7927355; https://doi.org/10.1159/000154228
- Burgdorf KS, Grarup N, Justesen JM, Harder MN, Witte DR, Jorgensen T, Sandbæk A, Lauritzen T, Madsbad S, Hansen T, et al. Studies of the association of Arg72Pro of tumor suppressor protein p53 with type 2 diabetes in a combined analysis of 55,521 Europeans. PloS one 2011; 6:e15813; PMID:21283750; https://doi.org/10.1371/journal.pone.0015813
- Gaulton KJ, Willer CJ, Li Y, Scott LJ, Conneely KN, Jackson AU, Duren WL, Chines PS, Narisu N, Bonnycastle LL, et al. Comprehensive association study of type 2 diabetes and related quantitative traits with 222 candidate genes. Diabetes 2008; 57:3136-44; PMID:18678618; https://doi.org/10.2337/db07-1731
- Gloria-Bottini F, Banci M, Saccucci P, Magrini A, Bottini E. Is there a role of p53 codon 72 polymorphism in the susceptibility to type 2 diabetes in overweight subjects? A study in patients with cardiovascular diseases. Diabetes Res Clin Pract 2011; 91:e64-7; PMID:21146884; https://doi.org/10.1016/j.diabres.2010.11.031
- Reiling E, Lyssenko V, Boer JM, Imholz S, Verschuren WM, Isomaa B, Tuomi T, Groop L, Dollé ME. Codon 72 polymorphism (rs1042522) of TP53 is associated with changes in diastolic blood pressure over time. Eur J Hum Genet 2012; 20:696-700; PMID:22189267; https://doi.org/10.1038/ejhg.2011.240
- Reinbold M, Luo JL, Nedelko T, Jerchow B, Murphy ME, Whibley C, Wei Q, Hollstein M. Common tumour p53 mutations in immortalized cells from Hupki mice heterozygous at codon 72. Oncogene 2008; 27:2788-94; PMID:17998932; https://doi.org/10.1038/sj.onc.1210932
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18:283-93; PMID:15866171; https://doi.org/10.1016/j.molcel.2005.03.027
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 2007; 67:6745-52; PMID:17638885; https://doi.org/10.1158/0008-5472.CAN-06-4447
- Shi H, Tan SJ, Zhong H, Hu W, Levine A, Xiao CJ, Peng Y, Qi XB, Shou WH, Ma RL, et al. Winter temperature and UV are tightly linked to genetic changes in the p53 tumor suppressor pathway in Eastern Asia. Am J Hum Genet 2009; 84:534-41; PMID:19344876; https://doi.org/10.1016/j.ajhg.2009.03.009
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004; 6:443-50; PMID:15077116; https://doi.org/10.1038/ncb1123
- Carvajal LA, Manfredi JJ. Another fork in the road–life or death decisions by the tumour suppressor p53. EMBO Rep 2013; 14:414-21; PMID:23588418; https://doi.org/10.1038/embor.2013.25
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497-501; PMID:9822382; https://doi.org/10.1126/science.282.5393.1497