
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Histone deacetylases (HDACs), which modulate the expression of genes, are potential therapeutic targets in several cancers. Targeted inhibition of HDAC prevents the expression of oncogenes thereby help in the treatment of cancers. Hence, several pharmaceutical companies developed inhibitors of HDAC and tested them in preclinical models and in clinical trials. SAHA (suberanilohydroxamic acid) is one such HDAC inhibitor developed for treating breast and colorectal carcinomas. However, due to poor efficacy in clinical trials the utility of SAHA for treating cancers was discouraged. Similarly another HDAC inhibitor Trichostatin-A (TSA) also showed promising results in clinical trials but exhibited severe adverse effects, which dampened the interest of using this molecule for cancer treatment. Therefore, search for developing a potent HDAC inhibitor with minimal side effects still continues. Hence, in this study we have screened benzoic acid and benzoic acid derivatives with hydroxylic (-OH) groups and methoxy (-OCH3) groups for their efficacy to bind to the TSA binding site of HDAC using molecular docking studies. Molecules that showed much stronger affinity (than TSA) to HDAC were tested for inhibiting HDAC expressing cultured cancer cells. DHBA but not Dimethoxy Benzoic Acid (DMBA) inhibited HDAC activity, leading to cancer cell growth inhibition through the induction of ROS and cellular apoptosis mediated by Caspase-3. In addition, DHBA arrested cells in G2/M phase of the cell cycle and elevated the levels of sub-G0-G1 cell population. In summary, results of this study report that DHBA could be a strong HDAC inhibitor and inhibit cancer cell growth more effectively.
Abbreviations
| BA | = | Benzoic acid |
| BCA | = | Bicinchoninic acid |
| DHBA | = | Dihydroxy benzoic acid |
| DMBA | = | Dimethoxy methoxy benzoic acid |
| DMEM | = | Dulbecco's Modified Eagle's Medium |
| DMSO | = | Dimethyl sulfoxide |
| EDTA | = | Ethylene diamine tetra acetic acid |
| EGTA | = | Ethylene glycol-bis(2-aminoethylether)- tetra acetic acid |
| FBS | = | Fetal bovine serum |
| HAT | = | Histone acetyl transferase |
| HDAC | = | Histone deacetylase |
| HEPES | = | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| LDH | = | Lactate dehydrogenase |
| MHDMBA | = | Monohydroxy dimethoxy benzoic acid |
| MHMMBA | = | Monohydroxy monomethoxy benzoic acid |
| MgCl2 | = | Magnesium chloride |
| NaCl | = | Sodium chloride |
| NP- 40 | = | Nonidet P-40 |
| PDB | = | Protein data bank |
| PI | = | Propidium iodide |
| RCSB | = | Research Collaboratory for structural bioinformatics |
| ROS | = | Reactive oxygen species |
| SAHA | = | Suberanilohydroxamic acid |
| SRB | = | Sulforhodamine b |
| TCA | = | Trichloro acetic acid |
| TSA | = | Trichostatin A |
Introduction
Acetylation is one of the major post translational modification of histones controlled by histone acetyl transferases (HAT) and histone deacetylases (HDAC).Citation1 Expression of genes is in part regulated by a fine balance between HAT and HDAC activities.Citation1 Aberrations in the expression of HAT and HDACs can (a) transform a normal cell in to cancerous one; (b) make a cell resistant to chemotherapeutic agents; (c) promote the migration of cells thereby induce metastasis and (d) deregulate the normal cellular apoptotic machinery.Citation2 Studies have demonstrated that increase in HAT activity promote apoptosis in cells, whereas elevated HDAC can promote cancer pathogenesis by silencing tumor regulatory genes.Citation2 In addition to deacetylating histone proteins, HDACs can target proteins that are involved in cell proliferation, cell cycle control, angiogenesis and invasion.3Several cancer cell lines and cancer tissues were shown to exhibit higher levels of HDACs, which made this family of proteins as possible therapeutic targets.Citation4 Thus, identification of potent and safe HDAC inhibitors is a viable strategy to inhibit cancer cell progression. Currently SAHA, Belinostat and Romidepsin are the 3 FDA approved HDAC inhibitors available for treating cancers.Citation3 Other than these synthetic inhibitors, few naturally occurring HDAC inhibitors were also reported for treating carcinomas of breast, prostate and colon and rectum.Citation5 Trichostatin A (TSA) a hydroxamate isolated from actinomycetes Streptomyces hygroscopicus is a potent HDAC inhibitor with IC50 lesser than 10 nM.Citation5 In general, HDAC inhibitors promote cancer cell death through the induction of ROS levels and by inhibiting cell cycle progression and by triggering apoptosis either by intrinsic or extrinsic pathways.Citation6,7 Lower efficacy of SAHA in clinical trials, and adverse effects associated with TSA, observed during phase II trials, emphasizes the need for identification of potent HDAC inhibitors with lesser adverse effects.Citation8,9 Thus identifying naturally occurring HDAC inhibitors could be a promising approach to treat cancers.
Phenolic acids are naturally occurring phytochemicals found abundantly in fruits and vegetables.Citation10 Based on their structure they are classified into simple and complex phenolic acids.Citation11 Benzoic acid and their derivatives are a class of simple phenolic acids with known pharmacological properties. Citation11 Gallic acid, a trihydroxylated benzoic acid derivative is known to retard cancer cell growth by inhibiting angiogenesis and invasion and by inducing apoptosis in cervical cancer cell lines.Citation12 Another benzoic acid derivative, ie., protocatechuic acid also inhibited the growth of breast cancer cells.Citation13 Although several reports on their anticancer activities are available, much is not known about their effect on tumor promoting HDACs. In addition, it is also not fully understood about the key structural requirements of benzoic acids to exhibit potent HDAC inhibition. Therefore in the current study, first, we have tested the ability of benzoic acid and its derivatives for binding to TSA binding site of HDAC using in silico modeling. Since, Trichostatin A ((2E,4E,6R)-7-[4-{Dimethylamino}phenyl]-N-hydroxy-4,5-dimethyl-7-oxo-2,4-heptadienamide, is a potent and selective inhibitor of histone deacetylase with Ki value of 3.4 nM, TSA binding region of HDAC was selected for identifying potent hydroxy benzoic acid derivatives.Citation14 Next, the potent compound exhibiting stronger binding to HDAC was evaluated for its ability to inhibit HDACs present in the nuclear extracts of HeLa. Our studies have identified DHBA as the potent HDAC inhibitor, hence, it was tested for its potential to retard cancer cell growth. In addition, the mechanisms of action of DHBA for inhibiting cancer cell growth were determined by measuring the levels of apoptosis using acridine orange and ethidium bromide staining, as well as by assessing the levels of caspase-3 expression.
Results
Docking studies comparing the efficacy of BA derivatives for binding to TSA-binding site of Human HDAC identified DHBA as the most potent inhibitor of HDAC
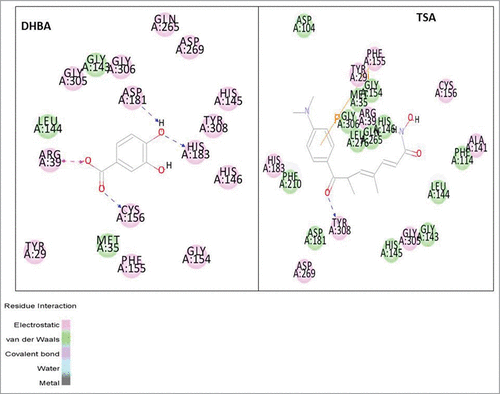
Inorder to identify the most potent benzoic acid derivative among BA, HBA, DHBA, and methylated versions MMBA, MHMMBA, DMBA, DHMMBA, TMBA, the binding efficacy was determined by assessing the ability to interact strongly with TSA-binding site of HDAC (See for structures). First, the X-ray crystal structure of HDAC (PDB ID: 3 MAX) with good resolution (2.05 Å) and Ramachandran plot properties was retrieved from protein data bank () and docked with BA, HBA, DHBA, MHMMBA, DMBA, MHDMBA and TMBA at trichostatin A (TSA) binding active sites and the c-docker energy and molecular interactions calculated (). Among BA derivatives tested, DHBA exhibited stronger interactions with HDAC as evidenced by lower C-docker energy (−30.06 kcal·mol−1) compared with even the positive control TSA, which has −8.2 kcal·mol−1. Even the C-docker interaction of DHBA (−30.05 kcal·mol−1) was comparatively higher than that of TSA (−42.2 kcal·mol−1). More over, DHBA is involved in the formation of 4 hydrogen bonds with HDAC, compared with BA, which did not exhibit any hydrogen bonding to HDAC. Since hydrogen bonds play an important role in the stability of ligand-protein (receptor ie., HDAC) complex, DHBA, compared with BA, was considered as more potent HDAC binding molecule. Molecularly, DHBA interacted with Arginine (A: 39) and the cysteine (A: 156) residues by forming electrostatic and hydrophilic bonds respectively () while TSA formed covalent bonds with phenylalanine (A: 115) and hydrophilic bonds with tyrosine (A: 308) ().
Table 1. Structure, IUPAC- and Common Name of Benzoic acid derivatives.
Figure 1a. The X-ray crystallographic structure of human HDAC enzyme complexed with an N-(2-aminophenyl) benzamide was retrieved from Research Collaboratory for Structural Bioinformatics (RCSB) Protein data Bank (PDB, ID: 3MAX) (http://www.rcsb.org/pdb)
Table 2. C-docker energy and C-docker intercations of BA derivatives with HDAC.
Figure 1b. Trichostatin A, a well-known HDAC inhibitor, binds to HDAC by interacting with key amino acid residues of the protein and forming electrostatic and hydrophilic bonds with higher docker energy.
DHBA inhibited HDAC activity ex vivo and in vitro
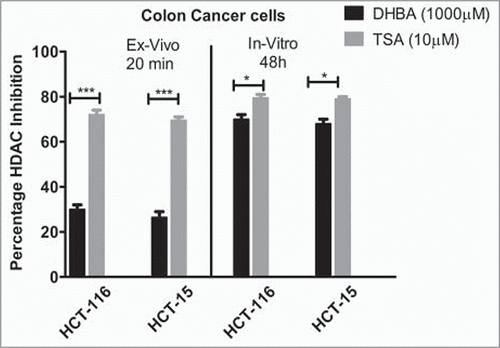
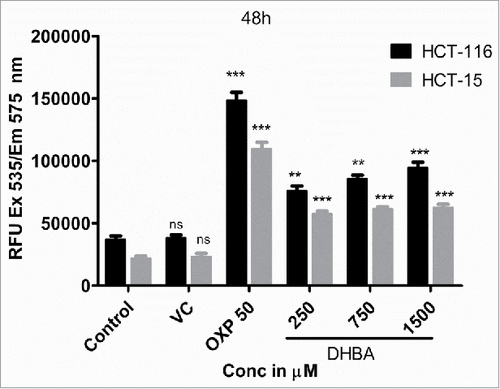
To check whether the in silico data, which identified DHBA as a potent inhibitor of HDAC, also replicate in ex vivo and in vitro studies, the HDAC inhibitory potential of BA derivatives was determined using fluorometric assay as described in methods section. In brief, the nuclear extract of HeLa cells, which contains HDAC enzyme, was incubated with increasing concentrations of DHBA and corresponding –OCH3 derivative and percentage inhibition compared with untreated controls calculated. Analysis of data showed that compounds with –OCH3 were less potent compared to corresponding –OH version (). For example, at 1000 μM concentration DHBA exhibited 22.8% HDAC inhibition while DMBA, which showed only 8.33% inhibition (). However at lower concentration (500 μM) both DHBA and DMBA did not show any inhibition. Positive control Trichostatin-A (10 μM) inhibited HDAC activity by 75% (). Since DHBA was found to be a potent inhibitor of HeLa cells' HDAC, next, the efficacy was also tested using nuclear extracts (containing high HDAC activity) collected from HCT116 and HCT15 colorectal carcinoma cells (). In addition, HCT116 and HCT-15 cells were treated 1000 µM of DHBA for 48 h and HDAC activity in nuclear extracts compared with untreated, and vehicle DMSO treated cells ().Citation15 DHBA inhibited the HDAC enzyme collected from HCT116 and HCT15, respectively, by 30% and 24%. Interestingly, analysis of HDAC activity present in the nuclear extracts of cells treated with DHBA or untreated and DMSO treated cells showed a much higher inhibition (). About 70% and 68% inhibition was noticed with DHBA treatment in HCT-116 and HCT-15 cell lines respectively. Trichostatin A, at 10 µM consistently showed higher HDAC inhibition both ex vivo and in vitro more than 65% inhibition ().
Figure 2a. DHBA identified as a potent inhibitor in the docking studies, and DMBA – methoxylated version of DHBA were incubated with HDAC enzyme (isolated from HeLa cells) for 20 minutes and the percentage inhibition determined. In vitro data showed better inhibition of HDAC compared with methoxylated version.
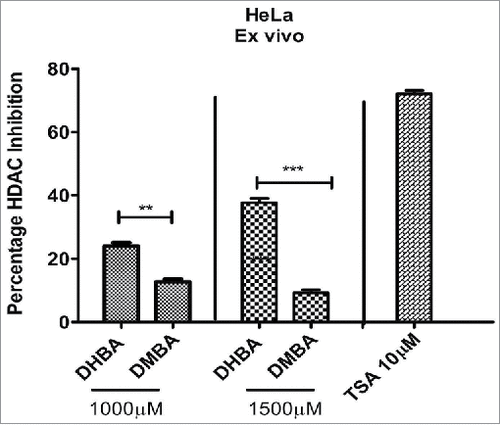
Figure 2b. DHBA inhibited HDAC activity both ex vivo and in vitro in colon cancer cell lines further confirming its potential to bind even when it is present in cells. The inhibitory effect was found to be more potential in vitro.
HDAC inhibitor DHBA retarded the growth of carcinoma cells representing cervix and colon and rectum
Although DHBA exhibited potent inhibitory activity against HDAC enzyme present in cervical and colon cancer cell lines, it is not known whether the concentrations used for HDAC inhibition also good enough to inhibit cell viability. Therefore a dose and time dependent cytotoxicity assay was performed on cell lines representing cervical (HeLa and SiHa) and colorectal (HCT-116 and HCT-15) cancers (). Analysis of the data demonstrated a time- and dose dependent cytotoxic effect on cervical and colon cancer cells with colon cancer cells being more sensitive compared with cervical cancer cells (). For example, at 1.25 mM, DHBA inhibited the growth of 48% HCT-116 cells at 48 h, however, only 23% inhibition was observed when tested on HeLa cells (). Although normal colon epithelial cells such as CCD18Co or FHC (CRL-1831) would have been the right control cells for this study to determine the therapeutic efficacy of DHBA, these cell lines were currently not available, hence, compound selectivity could not be tested. However, RAW 264.7 (mouse macrophage cell line) cell line was used to determine the selectivity. DHBA inhibited the growth of RAW264.7 cells at doses higher than 3000 µM (). Supporting the compounds cytotoxicity on cancer cell line, analysis of lactate dehydrogenase released in to the cell culture media, an indicator of cell death, also showed that DHBA treatment induced death in colorectal cancer cells ().
Figure 3a. DHBA treatment induced death of HCT-116 and HCT-15 (colorectal cancer cell lines), and HeLa and SiHa (cervical cancer cell lines) cells in a time and dose dependent manner. Colon cancer cells were found to be more sensitive to cervical cancer cells at all time points tested.
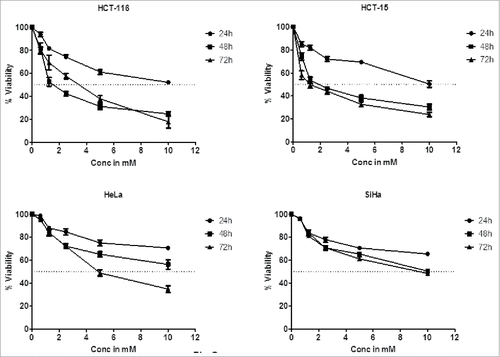
Figure 3b. DHBA was less toxic to mouse macrophage cell line RAW 264.7 at 24 and 48 h indicating that these compounds were safe. DHBA at 3000 µm exhibited 84% viable cells even after 48 h of exposure.
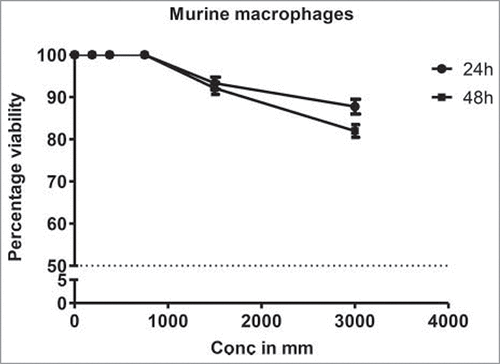
Figure 3c. DHBA treatment, for 48 h, enhanced the release of lactate dehydrogenase from HCT-116 and HCT-15 cells indicating the induction of cancer cells death which was found to be dose dependent.
DHBA elevated reactive oxygen species generation and arrested cells in G2/M phase of the cell cycle
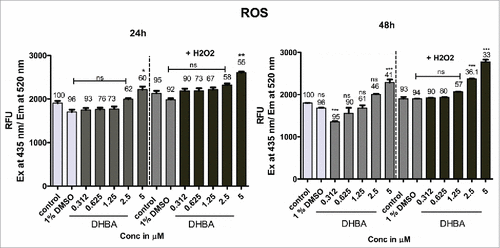
Since many studies have shown induction of reactive oxygen species when HDAC was inhibited, we have estimated the levels of ROS in the cells treated with DHBA. As predicted, treatment of HCT-116 cells with increasing concentrations of DHBA demonstrated a significant increase in reactive oxygen species at 24 h and 48 h (). Furthermore, addition of H2O2 to DHBA treated cells much more elevated ROS levels thereby enhanced cellular damage ().
Figure 4a. HDAC inhibitor DHBA increased ROS levels in a dose and time dependent manner to induce cell death. Further, in the presence of an external ROS generator like H2O2, the production of ROS was much higher compared with H2O2 and DHBA alone, indicating that these molecules cause cell death through ROS induction. Percentage of cell viability was determined using SRB assay simultaneously in both DHBA alone and DHBA with H2O2 treatment (indicated above bars). Although the viable cells reduced with treatment, ROS production increased indicating that compounds could potentially elevate the ROS levels.
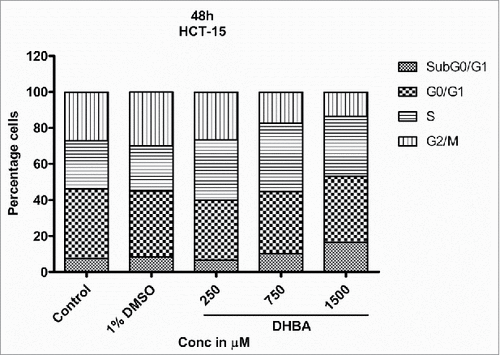
Cell cycle is a well-orchestrated and tightly regulated phase of a proliferating cell. Pharmacological agents that can halt the progression of cell cycle are potential anti-cancer agents. Hence, the effect of DHBA on cell cycle progression was assessed by treating HCT-116 () and HCT-15 () cells with DHBA for 48 h and by staining with propidium iodide followed by analyzing the stained cells using FACS. DHBA induced the levels of Sub G0/G1 phase, an indicator of apoptotic cells, and arrested cells in S and G2/M transition phases.Citation16,17 For example, treatment of HCT116 cells with DHBA increased the percentage sub-G0-G1 (1.18% in untreated control to 2.94% in 750 µM DHBA treated cells), S-phase (11.5% in untreated control to 15.8% in 1500 µM DHBA treated cells) and G2/M population (15.95% in untreated cells to 21.62% in 1500 µM DHBA treated cells), with a decrease in G0-G1 phase cells (71.43% in untreated control to 61.65% in 1500 µM DHBA treated cells) (). Oxaliplatin (50 µM), a positive control used in this study, arrested HCT116 cells in G2-M phase (15.95% in untreated control to 49.81% in oxaliplatin treated cells). Treatment of HCT15 cells with DHBA increased the sub-G0-G1 level from 7.6% in control untreated cells to 16.6% in 1500 µM treated cells (). In addition, a significant increase in the S-phase population with a simultaneous decrease in the G0-G1 and G2/M population was also observed when HCT15 cells were exposed to increasing concentration of DHBA ().
Figure 4b. HCT-116 cell lines were treated with DHBA for 48 h and stained with propidium iodide and analyzed using FACS. Analysis of HCT-116 cells treated with DHBA for 48 h showed induction of apoptotic sub-G0-G1 population and arrest of cells in S- and G2/M phase.
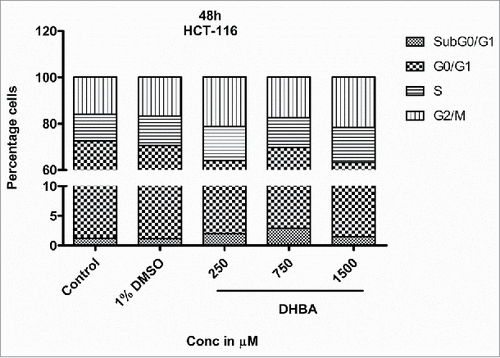
Figure 4c. Exposure of HCT-15 cells with DHBA for 48 h arrested the cells in S phase and also induced apoptosis as evident by accumulation of cells in Sub-G0-G1 phase similar to HCT-116 cell lines.
DHBA induced apoptosis in colon cancer cells by triggering caspase-3 levels
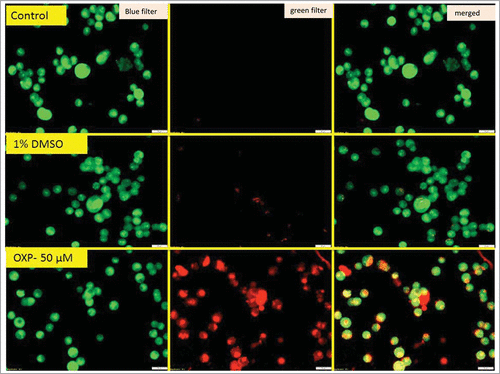
Since, an about 3-fold increase was observed in the sub-G0-G1 population when HCT-116 cells were treated with 750 µM DHBA, next, we have determined the levels of DHBA induced apoptotic cell death by staining the cells with acridine orange and ethidium bromide and also by measuring the levels of caspase-3 (). Experimentally, DHBA treated () and untreated HCT-116 () cells (48 h) were stained with ethidium bromide and acridine orange to evaluate apoptosis. In principle, the cells undergoing apoptosis exhibit condensed chromatin and show orange color compared with the live cells that appear green when observed under fluorescence microscope. Analysis of photomicrographs showed significantly high orange-stained cells when treated with DHBA (). But, no such stained cells were observed in untreated or vehicle 1% DMSO treated cells (). The number of orange stained cells increased with increasing DHBA concentration.
Figure 5a. Acridine orange and ethidium bromide staining of colon cancer cells indicated that DHBA induced apoptosis as evidenced by the presence of condensed chromatin (orange stained cells).
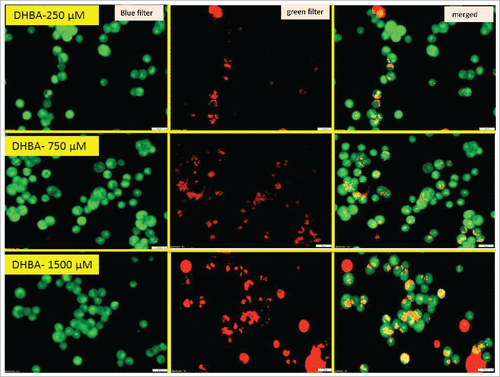
Figure 5b. Untreated and vehicle treated cells appeared green without orange stains indicating all live cells, however treatment with oxaliplatin induced apoptosis, as evidenced by increased orange cells.
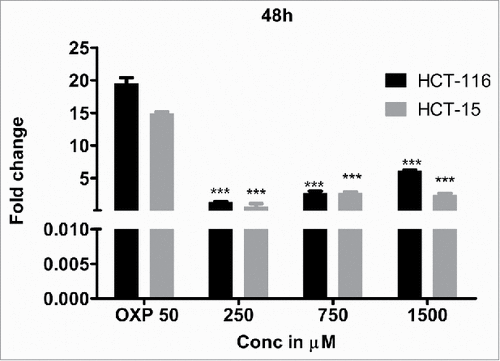
Next, to determine whether the apoptosis induction is due to caspase-3 expression, the levels of caspase-3 in the DHBA treated cells was compared with untreated and 1% DMSO vehicle treated cells (). A dose dependent increase in caspase-3 expression was observed when HCT-15 and HCT-116 cells were treated with increasing concentration of DHBA (). For example, a significant 2.5 to 5.5-fold high caspase-3 compared with DMSO treated cells was observed, respectively, when HCT-15 and HCT-116 cells were exposed to 1500 µM DHBA (). In total, our data demonstrated that DHBA induced apoptosis was mediated by caspase-3 expression in colorectal carcinoma cell lines.
Figure 5c. DHBA treated colon cancer cells expressed high levels of caspase-3, an apoptotic marker, compared with vehicle treated cells, confirming the induction of apoptosis.
Discussion
Transformation of normal cells to cancerous cells involves changes in gene expression controlled by HDAC/HAT enzymes.Citation18 Appropriate control over their expression and activity is very much required to regulate timely expression of genes.Citation19 Defects in this control promote the transformation of a normal cell in to cancerous one.Citation20 Several studies have shown elevated HDAC activity resulting in the suppression of tumor suppressor proteins in cancers.Citation21 Hence, targeted inhibition of HDAC activity is a viable strategy to halt the proliferation of cancer cells and to sensitize them to chemo- and radio-therapies. Several HDAC inhibitors such as SAHA and derivatives of SAHA, and TSA have been developed and tested for their safety and efficacy in vitro and in vivo. However, these HDAC inhibitors found successful only with minimal efficacy when administered alone. Higher doses could not be tested due to adverse side effects.Citation22 Even, other HDAC inhibitors triggered adverse effects such as thrombocytopenia, fatigue, diarrhea and nausea.Citation9 Hence, identification of new HDAC inhibitors with lesser side effects is essential. Dietary phenolic compounds that include derivatives of benzoic acid are known anticancer agents with minimal side effects.Citation11 Although these molecules are known to inhibit cancer cell proliferation, not much is known about their effect on HDAC inhibition. Moreover, it is also not known whether substitution of benzene ring with hydroxylic groups enhance the HDAC inhibitory activity of these molecules. Since substitution of benzene ring with hydroxylic groups has been shown to enhance the anti-oxidant and anti-inflammatory properties and likely to promote anti-cancer activity, we have hypothesized that the benzoic acid derivatives with 2 or 3 hydroxylic groups exhibit better HDAC inhibitory activity thereby induce cancer cell death.
Molecular docking is one of the recent developments helping to quickly screen pharmacological agents in silico and understand the mode of action.Citation23 Since TSA is a potent inhibitor of HDAC and for which the binding sites are well known, first, we have assessed the binding strength of BA and BA derivatives to HDAC using Discovery Studio software. Among the BA derivatives tested for docking energy and strength, the dihydroxylated version (DHBA) showed better interaction with HDAC compared with corresponding methoxylated derivatives and even the positive control TSA, indicating that DHBA could be a potent inhibitor of HDAC. Further analysis determining the key residues involved in HDAC binding pointed that the para hydroxyl group of 3,4 DHBA formed a hydrophilic bond with His (A: 183) and Asp (A: 181) while the carboxylic group interacted with Cys (A: 156) and Arg (A:39). These interactions promoted the binding strength of DHBA to HDAC. Even the in vitro and in vivo results replicated the docking data further confirming that DHBA could be a good inhibitor of HDAC. Further analysis of key structural requirements of benzoic acids to exhibit potent HDAC inhibition showed better HDAC inhibition with benzoic acid derivatives containing more number of free hydroxylic groups. For example, 3,4-dihydroxybenzoic acid (DHBA) compared with monohydroxy-, monohydroxymonomethoxy- benzoic acid derivatives showed better HDAC inhibition.
In colorectal carcinomas, the elevated HDAC expression and activity promoted cell growth and made the cells resistant to various therapeutic agents.Citation24,25 Targeted inhibition of HDAC using siRNA or pharmacological agents retarded the growth of colon cancer cells in vitro and in vivo.Citation26 In this study, we have demonstrated that treating HCT-116 and HCT-15 cell lines with DHBA reduced the HDAC activity by 70 and 68%, respectively, and retarded cell growth by 50% to 60%. A prior study by Khaopha, S., et al., 2015 also showed better HDAC inhibitory activity of pea-nuts testa extracts that contain DHBA and MHDMBA.Citation27 Other than this, no other report attributed the HDAC inhibitory property to BA derivatives. Hence, to the best of our knowledge, this is the first study where a comparative assessment of BA derivatives for inhibiting HDAC activity is reported.Citation28
Like many other HDAC inhibitors, even DHBA inhibited the cell growth by inducing cellular oxidative stress and triggering caspase-3 mediated apoptosis.Citation29 Since induction of ROS promotes the double stranded DNA breaks there by trigger cell death, compounds, like DHBA, elevating cellular ROS are good anti-cancer agents.Citation3,30–32 In addition, DHBA sensitized colorectal carcinoma cells to H2O2, which could be due to unusually high levels of ROS produced by DHBA and H2O2 combination.
Supporting the anti-cancer activity of DHBA, a study by Yin, M.C. et al., in 2009 also reported the growth inhibitory properties of cervical, breast, lung, liver and prostate cancers.Citation33 Cell growth inhibition is further evidenced by the release of cytosolic enzyme lactate dehydrogenase (LDH).Citation34 Furthermore, analysis of DHBA treated cells by FACS showed elevated sub-G0-G1 population (an indicator of apoptosis) and S and G2/M arrest.Citation35 Similar to DHBA, a recent study by Du L et al., 2014 and Karthik et al., 2014 showed G1/S or G2/M phase arrest when cancer cells were exposed to HDAC inhibitors such as romidepsin, vorinostatin.Citation16,17,36 The apoptosis induction is usually mediated by the activation of caspases in cells.Citation37,38 Analysis of cells treated with DHBA also showed a significant induction in caspase-3 indicating the anti-cancer potential of DHBA.Citation39 In conclusion, DHBA retarded cancer cell growth by binding and inhibiting HDAC, which induced cellular ROS and caspase-3 to trigger apoptosis in cells.
Conclusion
In summary, analysis of in silico docking studies, in vitro and ex vivo studies identified DHBA as a potent HDAC inhibitor compared with benzoic acid or other benzoic acid derivatives. Mechanistically, DHBA induced cancer cell death is mediated by (a) elevated caspase-3 and reactive oxygen species; (b) G2/M phase cell cycle arrest culminating in to increased sub-Go-G1cell population. Hence, DHBA could be a potent anti-cancer agent, which requires further validation in animal models for safety and efficacy.
Materials and methods
Materials
Benzoic acid (BA), para-hydroxybenzoic acid (HBA), 3,4-dihydroxybenzoic acid (DHBA), 4-hydroxy, 3-methoxy benzoic acid (MHMMBA), 3,4-dimethoxybenzoic acid (DMBA), 4-hydroxy, 3,5-dimethoxybenzoic acid (MHDMBA) were purchased from Sigma Aldrich Chemical Company.The structure, name of the compound given by International Union of Pure and Applied Chemistry (IUPAC), common name and molecular weight are shown in .
Cancer cell lines representing carcinomas of cervix (HeLa and SiHa), and colon and rectum (HCT-116 and HCT-15) were procured from NCCS, Pune, Maharashtra, India. Cell culture reagents and disposables for cell culture were procured from Life Technologies and Tarson's India Pvt Ltd. Histone deacetylase fluorometric (HDAC) assay kit was from Abcam, [cat#: ab156064], Pierce BCA protein estimation kit was from ThermoFischer Scientific, [cat#: 23227], Lactate dehydrogenase activity assay kit was from Biovision [cat#: K730–500]. Caspase-3/CPP32 colorimetric assay kit was from Biovision [cat#: K106–100]. Acridine orange, ethidium bromide, propidium iodide were from SRL.
Accelrys Discovery Studio 3.5 software is from Biovia.(Available at Sri Padmavati Mahila Visvavidyalayam (Women's University).
Methods
Comparative assessment of the binding potential of benzoic acid and benzoic acid derivatives to Trichostatin-A binding site of HDAC using Discovery Studio 3.5
Selection of HDAC structure from Protein Data Bank
The X-ray structures of human HDAC complexed with N-(2-aminophenyl) benzamide was retrieved from Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB, ID: 3MAX) (http://www.rcsb.org/pdb). The PDB file was cleaned and the hetero-atoms (HETATM) of the receptors were removed manually as they were the non-standard residues of proteins.Citation40
Generation of ligand data set
Ligand structures and the properties (hydrogen donors, acceptors, Log-P values, refractivity, pH and molecular weight) of each ligand were obtained from PubChem compound database (https://pubchem.ncbi.nlm.nih.gov/). Before docking, the ligands were prepared using the “prepare ligand” module (available in Discovery studio 3.5) for removing duplicates, enumerating isomers and tautomers, and generating 3D conformations.
Active site analysis of 3MAX structure
Possible binding sites of HDAC were identified by first defining the receptor molecule using binding site tools of Discovery Studio. In brief, active sites were selected according to PDB site records. A total of 5 active sites were evaluated for their binding to ligands, which include the control Trichostatin A, with lowest C Dock energies. Active site-4 (with XYZ attributes 65.913, 30.249,1.149.) was chosen for further evaluation to identify potent inhibitors of HDAC.
Molecular Docking using Discovery Studio 3.5
The docking of ligands in the receptor-binding site was performed using C-docker module available in Discovery studio 3.5. C-docker docks ligands using the validated CDOCKER algorithm, a grid-based molecular docking method that uses CHARMm with CDOCKER. Experimentally, first, initial ligand conformations were sampled using high temperature molecular dynamics and allowed to flex during the refinement (via simulated annealing MD). Next, the conformations were translated into binding site and candidate poses created using random rigid-body rotations followed by simulated annealing. A final minimization is then used to refine the ligand poses. The binding mode for all ligands to human HDAC (PDB ID:3 MAX) was investigated by CDOCKER protocol which had been incorporated into Discovery Studio 3.5.Citation41 Different poses of protein-ligand complex were obtained after docking process with their specific CDOCKER energy and CDOCKER interaction scores displayed in output file. The best ligand was chosen on the basis of their highly interacting amino acid residues and lower C docker energy score.
Assessment of HDAC inhibitory potential of benzoic acid and benzoic acid derivatives
Histone deacetylase (HDAC) inhibitory potential of few BA and BA-derivatives was determined using the HeLa cells nuclear extract supplied in the kit (Ex vivo analysis; Histone deacetylase fluorometric (HDAC) assay kit (ab156064) from Abcam, Cambridge, UK) as well as the nuclear extract collected from the HCT116 cells upon treatment with BA and BA derivatives (In vitro analysis). Details of the procedure can be obtained from the following web link: [http://www.abcam.com/ps/products/156/ab156064/documents/ab156064%20HDAC%20Activity%20Assay%20Kit%20v3%20(website).pdf]
Isolation of cellular nuclei and collection of nuclear extract
Isolation of nuclear fraction was performed as described in the kit protocol. In brief, 1 × 106 cells from control untreated, vehicle DMSO treated and BA and BA-derivatives exposed flasks were collected by trypsinization, followed by centrifugation at 3000 rpm for 5 minutes. The pelleted cells were washed twice with PBS, and resuspended in 1.0 mL lysis buffer containing 10 mM Tris HCl (pH 7.5), 10 mM NaCl, 15 mM MgCl2, 250 mM Sucrose, 0.5% NP-40 and 0.1 mM EGTA. After vortexing for 15.0 seconds the cell suspension was incubated on ice for 15.0 minutes, centrifuged at 10,000 rpm for 10.0 minutes at 4°C with 4.0 mL of sucrose cushion. The pellet collected was washed using cold 10.0 mM Tris HCl (pH 7.5) and 10.0 mM NaCl to obtain a fraction rich in nuclei.
Extraction of nuclei
The fraction rich in nuclei was resuspended in ∼100.0 μL nuclear extraction buffer made up of 50 mM HEPES-KOH (pH 7.5), 420 mM NaCl, 0.5 mM Na2EDTA, 0.1 mM EGTA, 10% Glycerol and vortexed briefly. The mixture was incubated on ice for 30.0 minutes and centrifuged at 20,000 rpm for 10 minutes. The supernatant containing crude nuclear extract was used for determination of HDAC activity. The protein content in the nuclear extract was estimated using BCA kit (ThermoFischer, Waltham, MA, USA) as described below and extracts stored at −80°C until further use.
Estimation of total protein using BCA method
Total protein content in the nuclear extracts was estimated using Pierce BCA kit from ThermoFischer ScientificCitation42 [https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf]. A calibration graph was prepared by incubating 10.0 µL increasing concentration of 25, 125, 250, 500, 750, 1000 and 1500 μg/mL of bovine serum albumin (BSA) with 200 µL BCA reaction mixture made up of 50 parts of reagent A containing 0.8% sodium bicarbonate, 4% bicinchoninic acid and 0.16% sodium tartrate in 0.1M sodium hydroxide and 1 part of reagent B made up of 4% cupric sulfate at 37°C for 30.0 minutes, followed by measuring the absorbance at 562 nm using a multimode plate reader (PerkinElmer, Germany). Appropriately diluted test samples were also processed similarly and concentration determined using the calibration curve.
HDAC inhibitory potential assessment using ex vivo method
HeLa crude nuclear extract provided in the HDAC kit was used for determining the HDAC inhibitory potential of BA and BA-derivatives. Experimentally, first, 5.0 µL assay buffer and 5.0 µL fluoro-substrate were mixed with 5 µL nuclear extracts and the reaction volume made up-to 30.0 µL using distilled water. The reaction mixture was incubated for 20.0 minutes at room temperature. Appropriate experimental controls that include (a) no enzyme control with all components except the nuclear lysates; (b) a developer control consisting of fluro-deacetylated substrate instead of fluoro-substrate; (c) an inhibitor control with 10.0 μM of Tricostatin-A (a known HDAC Inhibitor); and (d) a solvent control were also processed similarly. The reaction was stopped by the addition of 20.0 µL stop solution, and the fluorescence read in a kinetic mode after the addition of 5.0 µL of developer at an excitation of 365 nm and emission of 450 nm.
HDAC inhibitory potential assessment using in vitro method
In vitro HDAC inhibitory potential assessment was performed as described by Senawoong T et al., 2013.Citation43 Experimentally, first, 1 × 106 HCT116 cells were plated in 10.0 mL of DMEM-supplemented with 10% FBS in a 100 mm Petri plate. After 48 h exponentially growing cells were treated with DHBA (1000 µM), and Tricostatin A (10.0 µM) for further 48 h. The cells were trypsinized and nuclear lysate (10.0 µg) used for the determination of HDAC enzyme activity as detailed before.
Determination of the anti-cancer activity of benzoic acid derivatives
The anti-cancer activity of benzoic acid derivatives was measured according to Madhunapantula et al., 2008.Citation44 In brief 0.5 × 104 cells, representing carcinomas of colon and rectum (HCT-116 and HCT-15) and cervix (HeLa and SiHa), and noncancerous murine macrophage cell line (RAW 267.4) in 100 µL DMEM supplemented with 10% FBS were seeded in a 96-well plate and incubated at 37°C in a cell culture incubator maintained at 5% CO2 and 90% relative humidity. After 48 h, the cells were exposed to increasing concentration of benzoic acid derivatives (dissolved in DMSO and diluted in DMEM-10% FBS medium) for 24, 48 and 72 h. Viability of cells was measured using sulforhodamine-B assay and the percentage growth inhibition compared with vehicle DMSO treated cells calculated.
Measurement of cell viability using sulforhodamine-B assay (SRB assay)
SRB assay was performed according to Skehan et al., 1990.Citation45 Experimentally, cells were fixed in 1/4th volume of cold 50% (w/v) TCA for 4°C. After 1 h the media is removed and the wells washed with water (200 µL X 4 times) to remove TCA and serum proteins. The plates were dried, incubated with 100 µL 0.4% SRB for 30.0 minutes to stain the cellular proteins. Unbound SRB was removed by washing quickly with 1% acetic acid (200 µL X 4 times). The bound SRB was solubilized in 10.0 mM Tris base solution (100 µL/well) and the absorbance measured in a multimode plate reader operating at 490 nm. Percentage cell viability calculated using the equation shown below
Determination of cell viability by assessing the levels of lactate dehydrogenase release in to the cell culture media
Picoprobe lactate dehydrogenase activity assay kit from Biovision was used to estimate the amount of LDH in the colon cancer cell lines treated with DHBA. (http://www.biovision.com/manuals/K730.pdf). Lactate dehydrogenase, which catalyzes the conversion of lactate to pyruvate, is commonly used as a marker of tissue damage. The amount of LDH released to the media is directly proportional to the extent of damage caused by the compounds to the cells. 0.3 × 106 HCT-116 and HCT-15 cells in 2.0 mL media/well were plated in a 6-well plate. After 48 h, the exponentially growing cells were treated with 250, 750 and 1500 μM of DHBA for 48 h. Twenty five micro liters media was collected from the treated cells and made up to 50.0 μL with LDH assay buffer. Next, 50.0 μL reaction mixture containing 45.5 μL of LDH assay buffer, 2.5 μL of Picoprobe and 2.0 μL LDH substrate was added. The fluorescence was measured using multimode plate reader at an excitation of 535 nm and emission of 587 nm immediately in a kinetic mode for 10–30 minutes at 37°C. NADH was used as the standard (0–500 pmol) and the time point in the linear range was selected for calculations.
Evaluation of intracellular Reactive Oxygen Species (ROS)
Levels of ROS was estimated according to Shailasree et al., 2015 with minor modifications.Citation46 0.5 × 104 HCT-116 cells were plated in 96 well plates and incubated in a carbon dioxide incubator (maintained at 5% CO2) at 37°C for 48 h. Then the growing cells were first treated with DHBA (0.312, 0.625, 1.25, 2.5, 5 mM) for 24 h and 48 h, and subsequently with 250 μM H2O2 for 1 h. Cells were washed with PBS to remove the traces of media and incubated with 10 µm of 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCFDA, prepared in PBS). The plates were wrapped with sterile aluminum foil and incubated at 37°C in the CO2 incubator for 30 minutes. Amount of fluorescence was measured using a multimode plate reader operating at an excitation of 435 nm and emission of 520 nm.
Cell cycle analysis using propidium iodide staining
Cell cycle analysis was performed according to Hrgovic et al., 2016Citation47 [http://www.abcam.com/protocols/flow-cytometric-analysis-of-cell-cycle-with-propidium-iodide-dna-staining]. 0.3 × 106 HCT-116 and HCT-15 cells were plated in a 6-well plate and allowed to reach ∼70% confluence. Growing cells were treated with DHBA at 250, 750, 1500 μM concentrations for 48 h. Cells treated with 50 µM oxaliplatin, for 48 h, were used as positive control. Control and treated cells were collected by trypsinization, washed thrice with PBS and fixed with chilled 70% ethanol for half an hour at −20°C. The samples were then centrifuged to remove ethanol and washed with twice PBS. The pelleted cells were incubated with 400 μL propidium iodide (50 μg/mL) for 15 minutes and analyzed using BD-FACS caliber
Detection of apoptosis by acridine orange and ethidium bromide staining
Apoptosis detection using acridine orange and ethidium bromide staining method was performed as described by Shailasree et al., 2015.Citation46 In brief, 0.3 × 106 cells were plated in 6-well plates and after ∼36 h exposed to increasing concentrations of DHBA (250, 750, 1500 μM) and oxaliplatin (50 μM) for about 48 h. The control and treated cells were trypsinized and mixed thoroughly to obtain a single cell suspension. Trypsin was neutralized by the addition of complete medium and 20.0 μL cell suspension was incubated with 10.0 μL 100.0 µg/mL ethidium bromide and 10.0 μL of 100.0 µg/mL acridine orange mixture for 5.0 minutes. The cells were imaged using the fluorescence microscope using TRITC and FITC filters. The images obtained using 2 different channels were later merged to obtain a combined image, which emitted green and orange cells.Citation46
Confirmation of apoptosis by measuring caspase-3 activity using a chromogen-coupled DEVD substrate
Activation of caspase-3 is an indicator of apoptosis induction in mammalian cells.Citation48 The caspase-3/CPP32 colorimetric assay kit was used to measure the caspase-3 activity [http://www.biovision.com/manuals/K106.pdf]. Experimentally, first, 0.5 × 106 HCT-116 cells were treated with DHBA (250, 750, 1500 μM) for 48. While cells treated with 50.0 µM Oxaliplatin served as positive control the cells exposed to 1% DMSO served as vehicle control. Cell lysates from control and treated cells were collected using 100 μL of lysis buffer provided in the kit. Total protein content in the cell lysates was estimated using BCA method as described earlier. Caspase-3 activity was determined by incubating 100.0 μg of total protein in a total volume of 50.0 μL cell lysis buffer was incubated at 37°C for 3 h with 50 μL of 2X reaction buffer containing 10.0 mM DTT and 5.0 μL 4.0 mM DEVD-pNA substrate (200.0 μm final concentration). The developed color was read at 405 nm using a multimode plate reader (PerkinElmer). The fold change with respect to vehicle control (1% DMSO) was calculated and plotted against compound concentration.
Statistical analysis
All experiments were conducted in multiple replicates and the results were expressed as mean ±SEM. The results were subjected to one way ANOVA, followed by Tukey's post hoc test to analyze difference between DHBA and controls/positive controls. A value of < 0.05 was considered to be significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
PGA has performed the experiments, compiled the data, prepared tables and graphs. BDR has performed the in silico work and interpreted the data. MPA, MGV and SVM have assisted in designing, drafting and editorial corrections of the manuscript.
Acknowledgments
PGA would like to acknowledge the award of UGC Maulana Azad National Senior Research Fellowship (MAN-SRF). PGA would also like to acknowledge Department of Science and Technology (DST)-Fund for the Improvement of Science and Technology infrastructure (FIST) supported Center of Excellence in Molecular Biology and Regenerative Medicine (CEMR) laboratory, Department of Biochemistry, JSS Medical College, JSS University for allowing me to carry out research studies.
Funding
PGA is supported by University Grants Commission, Government of India by Maulana Azad National Fellowship (F117.1/201415/MANF201415JAIKAR34099).
References
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Ann Rev Biochem 2001; 70:81-120; PMID: 11395403; https://doi.org/10.1146/annurev.biochem.70.1.81
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst 2000; 92(15):1210-6; PMID: 10922406; https://doi.org/10.1093/jnci/92.15.1210
- Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015; 20(3):3898-941; PMID: 25738536; https://doi.org/10.3390/molecules20033898
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5(9):769-84; PMID: 16955068; https://doi.org/10.1038/nrd2133
- Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol 2013; 9(5):319-25; PMID: 23524983; https://doi.org/10.1038/nchembio.1223
- Zhang ZH, Hao CL, Liu P, Tian X, Wang LH, Zhao L, Zhu CM. Valproic acid inhibits tumor angiogenesis in mice transplanted with Kasumi1 leukemia cells. Mol Med Rep 2014; 9(2):443-9; PMID: 24297248; https://doi.org/10.3892/mmr.2013.1834
- Stankov MV, El Khatib M, Kumar Thakur B, Heitmann K, Panayotova-Dimitrova D, Schoening J, Bourquin JP, Schweitzer N, Leverkus M, Welte K, et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014; 28(3):577-88; PMID: 24080946; https://doi.org/10.1038/leu.2013.264
- Zheng L, Fu Y, Zhuang L, Gai R, Ma J, Lou J, Zhu H, He Q, Yang B. Simultaneous NF-kappaB inhibition and E-cadherin upregulation mediate mutually synergistic anticancer activity of celastrol and SAHA In vitro and in vivo. Int J Cancer 2014; 135(7):1721-32; PMID: 24615207; https://doi.org/10.1002/ijc.28810
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007; 109(1):31-9; PMID: 16960145; https://doi.org/10.1182/blood-2006-06-025999
- Mandal SM, Chakraborty D, Dey S. Phenolic acids act as signaling molecules in plant-microbe symbioses. Plant Signal Behavior 2010; 5(4):359-68; PMID:20400851; https://doi.org/10.4161/psb.5.4.10871
- Anantharaju PG, Gowda PC, Vimalambike MG, Madhunapantula SV. An overview on the role of dietary phenolics for the treatment of cancers. Nutr J. 2016; 15(1):99; PMID: 27903278; https://doi.org/10.1186/s12937-016-0217-2
- Zhao B, Hu M. Gallic acid reduces cell viability, proliferation, invasion and angiogenesis in human cervical cancer cells. Oncol Lett 2013; 6(6):1749-55; PMID: 24843386; https://doi.org/10.3892/ol.2013.1632
- Lin HH, Chen JH, Chou FP, Wang CJ. Protocatechuic acid inhibits cancer cell metastasis involving the down-regulation of Ras/Akt/NF-kappaB pathway and MMP-2 production by targeting RhoB activation. Br J Pharmacol 2011; 162(1):237-54; PMID: 20840540; https://doi.org/10.1111/j.1476-5381.2010.01022.x
- Zhang Y, Xu Q, Liu G, Huang H, Lin W, Huang Y, Chi Z, Chen S, Lan K, Lin J, et al. Effect of histone deacetylase on prostate carcinoma. Int J Clin Exp Pathol 2015; 8(11):15030-4; PMID: 26823840
- Lutz L, Fitzner IC, Ahrens T, Geissler AL, Makowiec F, Hopt UT, Bogatyreva L, Hauschke D, Werner M, Lassmann S. Histone modifiers and marks define heterogeneous groups of colorectal carcinomas and affect responses to HDAC inhibitors In vitro. Am J Cancer Res 2016; 6(3):664-76; PMID: 27152243
- Du L, Risinger AL, King JB, Powell DR, Cichewicz RH. A potent HDAC inhibitor, 1-alaninechlamydocin, from a Tolypocladium sp. induces G2/M cell cycle arrest and apoptosis in MIA PaCa-2 cells. J Nat Prod 2014; 77(7):1753-7; PMID: 24999749; https://doi.org/10.1021/np500387h
- Karthik S, Sankar R, Varunkumar K, Ravikumar V. Romidepsin induces cell cycle arrest, apoptosis, histone hyperacetylation and reduces matrix metalloproteinases 2 and 9 expression in bortezomib sensitized non-small cell lung cancer cells. Biomed Pharmacother 2014; 68(3):327-34; PMID:24485799; https://doi.org/10.1016/j.biopha.2014.01.002
- Moreira JM, Scheipers P, Sorensen P. The histone deacetylase inhibitor Trichostatin A modulates CD4+ T cell responses. BMC Cancer 2003; 3:30; PMID: 14606959; https://doi.org/10.1186/1471-2407-3-30
- Chen Y, Wang H, Yoon SO, Xu X, Hottiger MO, Svaren J, Nave KA, Kim HA, Olson EN, Lu QR. HDAC-mediated deacetylation of NF-kappaB is critical for Schwann cell myelination. Nat Neurosci 2011; 14(4):437-41; PMID: 21423191; https://doi.org/10.1038/nn.2780
- Chen HP, Zhao YT, Zhao TC. Histone deacetylases and mechanisms of regulation of gene expression. Crit Rev Oncog 2015; 20(1-2):35-47; PMID: 25746103; https://doi.org/10.1615/CritRevOncog.2015012997
- Singh BN, Zhou H, Li J, Tipton T, Wang B, Shao G, Gilbert EN, Li Q, Jiang SW. Preclinical studies on histone deacetylase inhibitors as therapeutic reagents for endometrial and ovarian cancers. Future Oncol 2011; 7(12):1415-28; PMID: 22112317; https://doi.org/10.2217/fon.11.124
- Ma X, Ezzeldin HH, Diasio RB. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs 2009; 69(14):1911-34; PMID: 19747008; https://doi.org/10.2165/11315680-000000000-00000
- Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol 2007; 152(1):9-20; PMID: 17549047; https://doi.org/10.1038/sj.bjp.0707305
- Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol 2011; 7(2):263-83; PMID: 21345145; https://doi.org/10.2217/fon.11.2
- Wilson AJ, Byun DS, Nasser S, Murray LB, Ayyanar K, Arango D, Figueroa M, Melnick A, Kao GD, Augenlicht LH, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell 2008; 19(10):4062-75; PMID: 18632985; https://doi.org/10.1091/mbc.E08-02-0139
- Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang XF, Counter CM, Yao TP. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res 2008; 68(18):7561-9; PMID: 18794144; https://doi.org/10.1158/0008-5472.CAN-08-0188
- Somprasong Saenglee SJ, Patanathai Aran, Senawong Thanaset. Cytotoxic effects of peanut phenolic compounds possessing histone deacetylase inhibitory activity on human colon cancer cell lines. Turk J Biol 2016; 40:1258-71; https://doi.org/10.3906/biy-1601-23
- Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem 2008; 19(9):587-93; PMID: 18061431; https://doi.org/10.1016/j.jnutbio.2007.08.002
- Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A 2004; 101(52):18030-5; PMID: 15596714; https://doi.org/10.1073/pnas.0408345102
- Karthik S, Sankar R, Varunkumar K, Anusha C, Ravikumar V. Blocking NF-kappaB sensitizes non-small cell lung cancer cells to histone deacetylase inhibitor induced extrinsic apoptosis through generation of reactive oxygen species. Biomed Pharmacother 2015; 69:337-44; PMID:25661379; https://doi.org/10.1016/j.biopha.2014.12.023
- Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A 2010; 107(33):14639-44; PMID: 20679231; https://doi.org/10.1073/pnas.1008522107
- Rosato RR, Almenara JA, Maggio SC, Coe S, Atadja P, Dent P, Grant S. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther 2008; 7(10):3285-97; PMID: 18852132; https://doi.org/10.1158/1535-7163.MCT-08-0385
- Yin MC, Lin CC, Wu HC, Tsao SM, Hsu CK. Apoptotic effects of protocatechuic acid in human breast, lung, liver, cervix, and prostate cancer cells: potential mechanisms of action. J Agr Food Chem 2009; 57(14):6468-73; PMID: 19601677; https://doi.org/10.1021/jf9004466
- Chan FK, Moriwaki K, De Rosa MJ. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol Biol 2013; 979:65-70; PMID: 23397389; https://doi.org/10.1007/978-1-62703-290-2_7
- Urrego D, Tomczak AP, Zahed F, Stuhmer W, Pardo LA. Potassium channels in cell cycle and cell proliferation. Philos Trans R Soc Lond B Biol Sci 2014; 369(1638):20130094; PMID: 24493742; https://doi.org/10.1098/rstb.2013.0094
- Sellers WR, Fisher DE. Apoptosis and cancer drug targeting. J Clin Invest 1999; 104(12):1655-61; PMID: 10606616; https://doi.org/10.1172/JCI9053
- Shi Y. Caspase activation, inhibition, and reactivation: a mechanistic view. Protein Sci 2004; 13(8):1979-87; PMID: 15273300; https://doi.org/10.1110/ps.04789804
- McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 2015; 7(4); PMID: 25833847; https://doi.org/10.1101/cshperspect.a026716
- Silva G, Cardoso BA, Belo H, Almeida AM. Vorinostat induces apoptosis and differentiation in myeloid malignancies: genetic and molecular mechanisms. PloS One 2013; 8(1):e53766; PMID: 23320102; https://doi.org/10.1371/journal.pone.0053766
- Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, O'Connell S, Grimshaw CE, Navre M, Gangloff AR. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Med Chem Lett 2010; 20(10):3142-5; PMID: 20392638; https://doi.org/10.1016/j.bmcl.2010.03.091
- Wu G, Robertson DH, Brooks CL, 3rd, Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J Compu Chem 2003; 24(13):1549-62; PMID: 12925999; https://doi.org/10.1002/jcc.10306
- Brown RE, Jarvis KL, Hyland KJ. Protein measurement using bicinchoninic acid: elimination of interfering substances. Anal Biochem 1989; 180(1):136-9; PMID: 2817336; https://doi.org/10.1016/0003-2697(89)90101-2
- Senawong T, Misuna S, Khaopha S, Nuchadomrong S, Sawatsitang P, Phaosiri C, Surapaitoon A, Sripa B. Histone deacetylase (HDAC) inhibitory and antiproliferative activities of phenolic-rich extracts derived from the rhizome of Hydnophytum formicarum Jack.: sinapinic acid acts as HDAC inhibitor. BMC Complement Altern Med 2013; 13:232; PMID: 24053181; https://doi.org/10.1186/1472-6882-13-232
- Madhunapantula SV, Desai D, Sharma A, Huh SJ, Amin S, Robertson GP. PBISe, a novel selenium-containing drug for the treatment of malignant melanoma. Mol Cancer Ther 2008; 7(5):1297-308; PMID: 18483317; https://doi.org/10.1158/1535-7163.MCT-07-2267
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82(13):1107-12; PMID: 2359136; https://doi.org/10.1093/jnci/82.13.1107
- Shailasree S, Venkataramana M, Niranjana SR, Prakash HS. Cytotoxic effect of p-Coumaric acid on neuroblastoma, N2a cell via generation of reactive oxygen species leading to dysfunction of mitochondria inducing apoptosis and autophagy. Mol Neurobiol 2015; 51(1):119-30; PMID: 24760364; https://doi.org/10.1007/s12035-014-8700-2
- Hrgovic I, Doll M, Kleemann J, Wang XF, Zoeller N, Pinter A, Kippenberger S, Kaufmann R, Meissner M. The histone deacetylase inhibitor trichostatin a decreases lymphangiogenesis by inducing apoptosis and cell cycle arrest via p21-dependent pathways. BMC Cancer 2016; 16(1):763; PMID: 27716272; https://doi.org/10.1186/s12885-016-2807-y
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35(4):495-516; PMID: 17562483; https://doi.org/10.1080/01926230701320337