
ABSTRACT
The cancer-predisposing Lynch Syndrome (LS) arises from germline mutations in DNA mismatch repair (MMR) genes, predominantly MLH1, MSH2, MSH6, and PMS2. A major challenge for clinical diagnosis of LS is the frequent identification of variants of uncertain significance (VUS) in these genes, as it is often difficult to determine variant pathogenicity, particularly for missense variants. Generic programs such as SIFT and PolyPhen-2, and MMR gene-specific programs such as PON-MMR and MAPP-MMR, are often used to predict deleterious or neutral effects of VUS in MMR genes. We evaluated the performance of multiple predictive programs in the context of functional biologic data for 15 VUS in MLH1, MSH2, and PMS2. Using cell line models, we characterized VUS predicted to range from neutral to pathogenic on mRNA and protein expression, basal cellular viability, viability following treatment with a panel of DNA-damaging agents, and functionality in DNA damage response (DDR) signaling, benchmarking to wild-type MMR proteins. Our results suggest that the MMR gene-specific classifiers do not always align with the experimental phenotypes related to DDR. Our study highlights the importance of complementary experimental and computational assessment to develop future predictors for the assessment of VUS.
| Abbreviations | ||
| ATM | = | Ataxia telangiectasia mutated |
| ATR | = | ATM- and Rad3-related |
| CDDP | = | cisplatin; also known as cis-Diammineplatinum(II) dichloride |
| DDR | = | DNA damage response |
| HCPS | = | hereditary cancer predisposing syndrome |
| LS | = | Lynch syndrome |
| MMR | = | mismatch repair |
| MMS | = | methyl methane sulfonate |
| VUS | = | variants of uncertain significance |
| wt | = | wild-type |
| 5-FU | = | 5-Flourouracil |
Introduction
Inherited mutations in DNA mismatch repair (MMR) genes result in autosomal-dominant inheritance of hereditary nonpolyposis colorectal cancer (HNPCC) or Lynch Syndrome (LS). These mutations also strongly predispose individuals to endometrial cancer (EDC), colorectal cancer (CRC), and other tumors, forming a spectrum of hereditary cancer-predisposing syndromes (HCPS). These predispositions are largely due to mutations in 4 MMR genes, MSH2, MSH6, MLH1, and PMS2.Citation1-3 MMR is a post-replicative DNA repair mechanism in which 2 protein complexes, MutSα (formed of a heterodimer of MSH2 and MSH6) and MutSβ (a heterodimer of MSH2 and MSH3) wrap around replicated DNA, scan the sequence, and recognize errors in DNA replication. MutSα recognizes mismatched bases and small loops, while MutSβ recognizes both small and large loops (∼10 nucleotides), associated with insertions and deletions.Citation4 When a MutS complex (either α or β) recognizes a nucleic aberration(s), it recruits an MLH1-PMS2 heterodimer (MutLα), a weak endonuclease that introduces nicks in the damaged DNA.Citation5,6 Subsequent recruitment of exonuclease 1 (Exo1) excises mismatched single-stranded DNA from the newly synthesized strand. Finally, the DNA replication machinery resynthesizes the DNA and fills the gap.Citation4 Importantly, MMR proteins signal to induce cell cycle arrest, providing time to complete DNA repairs, or alternatively signal to induce apoptosis in the context of DNA damage too extensive to be repaired.Citation7
Testing for mutations in MMR genes is routinely considered in clinic in suspected cases of LS. The current National Comprehensive Cancer Network (NCCN) guidelines for gastrointestinal cancers indicate that a pathogenic MSH2 or MLH1 mutation confers a 52–82% lifetime risk of CRC 52–82% and 25–60% for EDC. Pathogenic MSH6 mutations result in lifetime risk of 10–22% for CRC and 16–26% for EDC. Pathogenic PMS2 mutations are associated with risks of 15–20% for CRC and 15% for EDC.Citation8 Patients with such mutations receive guidance and preventive care, and are advised to undergo intensive screening to consider prophylactic surgery. Complicating diagnosis and genetic counseling, ∼20–30% of MMR variants identified in high-throughput genomic studies, or in panel testing of individuals without access to family pedigree analysis, are rare variants of uncertain significance (VUS). Most of these cause single amino acid changes which may or may not impact protein function, and which have not yet been functionally linked to cancer risk.Citation9 Understanding the significance of MMR variants would not only enhance risk assessment for families and individuals, but may also impact treatment decisions.Citation10 Recent studies have shown that MMR-defective tumors are particularly susceptible to immunotherapy drugs targeting the programmed death 1 (PD-1) (Le, et al., 2015) and its ligand (PD-L1).Citation11
Current methods for assessment of VUS rely on epidemiological and pathological data when available, but otherwise rely on a variety of computational tools.Citation12 Generic mutation phenotype prediction servers, such as SIFT,Citation13 PolyPhen-2,Citation14 and MutationAssessor,Citation15 are often used (reviewed in Citation16). Such generic predictors make phenotypic predictions for missense variants in any protein. Their accuracies on independent benchmarks depend strongly on the nature of the testing set and its overlap with the training set.Citation17 In an effort to increase the accuracy of phenotypic prediction for potential LS patients, several specific structural predictors have been developed for MMR proteins. MAPP-MMRCitation18 makes predictions for MLH1 and MSH2, CoDPCitation19 makes predictions for MSH6, and PON-MMR220 makes predictions for MLH1, PMS2, MSH2, and MSH6. These computational tools are primarily based on analyses of sequence conservation and the severity of the substitution based on the range of variation at the assessed position.Citation12
Importantly, if a VUS is predicted to have a moderate or minimal effect on protein structure, but disrupts critical protein interactions that influence specific functions, the resulting biologic functionality or disease penetrance may be complex. For example, a specific VUS might impede apoptotic signaling, while leaving MMR fully functional.Citation21,22 However, in contrast to the common use of a growing number of computational prediction methods, direct experimental approaches to assess protein function are rarely used to explore and validate predictions, although some recent studies have suggested the value of this approach.Citation23,24 In this study, we have integrated phenotype predictions and functional biologic assessment for 15 missense VUS in 3 MMR genes (MSH2, MLH1, and PMS2). As part of this work, we have analyzed our results in the context of predictions made by 8 commonly used generic and specific computational predictors. Based on this work, we propose that a panel of cell-based in vitro methods can contribute to the assessment of the impact of VUS on DDR functionality.
Materials & methods (See also Supplementary Methods)
Computational analysis of VUS, protein, and mRNA structure
MMR variants were analyzed by using multiple computational tools for assessment of SNP impact on protein function. We used several generic predictors such as: PolyPhen-2.1Citation14 http://genetics.bwh.harvard.edu/pph2/, SIFTCitation13 http://sift.jcvi.org/, PROVEANCitation25 http://provean.jcvi.org/index.php, MutationAssessorCitation15 http://mutationassessor.org/r3/ (see supplementary methods for extended predictor and computational methods). Additionally, we analyzed each variant with the Biological Assembly Support Vector Machine (BA-SVM),Citation26 a generic phenotype predictor developed by our group which incorporates sequence conservation, structural disorder, physiochemical properties, and interface data from the structures of oligomeric protein complexes (biologic assemblies) into a support vector machine. We also used 2 MMR-specific predictors: MAPP-MMRCitation18 (http://mappmmr.blueankh.com) and PON-MMR2 Citation20 (http://structure.bmc.lu.se/PON-MMR2). All the predictive tools provide a score from which the prediction on the protein variant can be categorized as benign/neutral, pathogenic/disease-causing, or intermediate/uncertain. For each variant, we also calculated the DDG of the mutation using FoldXCitation27 to evaluate protein stability. Variants were also submitted to the Human Splicing Finder (HSF),Citation28 SNPfold,Citation29 and MfoldCitation30 servers to evaluate potential alterations in mRNA splicing or structure.
Molecular modeling
A complete 3-dimensional model of the MutSα heterodimer was constructed from the crystal structure of the human MSH2/MSH6 heterodimer (PDB: 2O8BCitation31) by building loops with the program YASARACitation32 for several loop regions missing coordinates in the experimental structure. The MutLα C-terminal heterodimer model was created with HHpred,Citation33 using the human C-terminal MLH1 crystal structure (PDB: 3RBN (Dombrovsky et al., unpublished, DOI: 10.2210/pdb3rbn/pdb) and S. cerevisiae MutLα C-terminal heterodimer crystal structure (PDB 4FMOCitation34) as templates. Energy minimization was performed with YASARA, and side chains were rebuilt with SCWRL4.Citation35 The N-terminal heterodimer was constructed by superposing the human N-terminal domain crystal structures of PMS2 (PDB: 1EA636) and MLH1 (PDB 4P7A,Citation37 using the MLH1 homodimer structure (PDB: 4P7A) as a template.Citation37
Results
MMR variants and computational predictions of pathogenicity
outlines the activity of the MutSα/β and MutLα complexes during MMR repair. The MMR variants in this study () were selected based on the following criteria: 1) Identified in patients with LS and/or HCPS; 2) Rare, missense germline variants; 3) Clinically classified as VUS; 4) Not previously analyzed. Five VUS each in MSH2, MLH1, and PMS2 () that met the above criteria were analyzed using 8 generic and 2 MMR protein-specific predictors (Tables 2 and S2). The scores from each predictor were translated into a 3-tiered classification system (Neutral (N), Moderate (M), Deleterious (D)) to facilitate comparison between predictor programs (Table S1). Results in Tables 2 and S2 highlight the variability in the results of these computational programs. While there is some consensus for 6 variants (PMS2: V475E, I853M; MLH1: R423T, H264L, V213L; MSH2: R534P, for the remaining 9 VUS there was significant disagreement among the predictions. We also incorporated analysis of codon usage bias in the initial assessment, which exhibited differences that correlated with the observed phenotype of some of the variants as discussed below (summarized in Table S2).
Figure 1. (A) Basic schematic of the MMR pathway in replication-coupled repair. During DNA replication, a misincorporation of nucleotide(s) results in the creation of mismatched base(s) in the daughter strand of the DNA. If DNA polymerase fails to recognize and correct the misincorporated base pair(s), the post -replicative MMR pathway is activated. A MutS heterodimer, incorporating MSH2 with MSH6 (MutSα) or MSH3 (MutSβ), recognizes the mismatch, and recruits MutLα (a MLH1 heterodimer PMS2) to nick the daughter strand at either the 3′- or -5′ end, depending on the location of the mismatch. Nicks in DNA are extended by Exo1, after which the DNA replication machinery fills in the correct base(s) and DNA ligase connects the new sections of the DNA. (B) Mapping of variants/mutants on MMR protein structures. Panels: A. Cartoon representation of MutSα. Purple: MSH2. Orange: MSH6. Magenta spheres: mutation sites. Cyan: dsDNA. Gray sphere and blue sticks: Mg2+ and ADP. (B) Cartoon representation of MutLα, with N-terminal domain on top and C-terminal domain below. Unstructured linker arms are represented as dashed lines and are not to scale. Green: MLH1. Cyan: PMS2. Magenta spheres: mutation sites. Cyan: dsDNA. Gray sphere and orange sticks in N-terminal domain: Mg2+ and ADP. Orange sticks on C-terminal domain: EXO1 fragment. C-G. Zoomed-in MSH2 mutations. H-K. Zoomed-in MLH1 mutations. L-O. Zoomed-in PMS2 mutations.
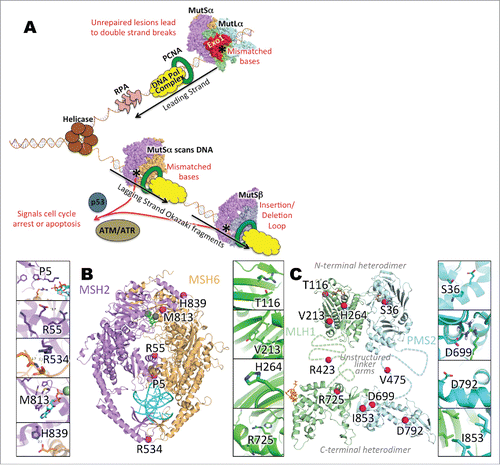
Table 1. Variant information, clinical classification and ExAC summary. Key: AA, amino acid; CMMR-D, constitutional MMR deficiency; CRC, colorectal cancer; HCPS, Hereditary Cancer Predisposing Syndrome; Hx, History; LS, Lynch Syndrome; MMR, Mismatch Repair; MSS, Microsatellite Stable; NF, Not Found; Pt, Patient. ExAC estimates were based on May 2016 data release (http://exac.broadinstitute.org/about).
Table 2. Summary of generic and gene-specific computational predictors. Key: AA, mutant amino acid; PP2, PolyPhen-2 score; MUTA, MutationAssessor score; BA, Biological Assembly SVM score; PROV, Provean score; N, neutral; M, moderate; D, damaging; n/a, not available. These predictive tools provide a variety of scores from which protein variants can be categorized as benign/neutral, pathogenic/disease causing, or intermediate /uncertain. For comparative purposes, raw prediction scores were translated into 3 classes (neutral, moderate, damaging) using the expanded criteria given in Table S2.
Specific VUS affect mRNA and protein expression of MMR genes
To establish consequences of analyzed VUS for mRNA and protein expression, providing a baseline for interpreting functional results, we transiently introduced plasmids expressing wild-type (wt) or VUS forms of MSH2, MLH1, or PMS2 into the highly transfectable HEK293 cell line ( and Fig. S1).Citation38 For MSH2, expression of VUS R534P and M813I was equivalent to wt while H839R was variable. Unexpectedly, MSH2 variants P5Q and R55G were not detectable. For MLH1, variants T116R, V213L, H264L, and R423T, were expressed equivalently to wt, while R725H was reduced by ∼60%. For PMS2, all VUS (S36R, V475E, D699H, D792N, and I853M) were expressed equivalently to wt. For each overexpressed protein, we also examined expression of the binding partner in each heterodimer (based on MSH2-MSH6 and MLH1-PMS2 pairing, and ): this was not significantly affected for any VUS ().
Figure 2. Analysis of MMR variant protein expression in HEK293 cell lines. (A-B) represents expression of MSH2 variants (P5Q, R55G, R534P, M813I, H839R) (dark gray bars) with corresponding MSH6 expression (light gray bars). (C-D) represents expression of MLH1 variants ((T116R, V213L, H264L, R423T, R725H) (dark gray bars) with corresponding PMS2 expression (light gray bars). (E-F) represents expression of PMS2 variants ((S36R, V475E, D699H, D792N, I853M) (dark gray bars) with corresponding PMS2 expression (light gray bars). For all experiments, E, empty vector transfection and WT, wild type transfection. (A, C, E) Quantification from 3 independent experiments ± SEM. Statistics were performed using a one-sample T-test, where * represents p < 0.05, ** represents p < 0.01 and *** represents p < 0.001. Protein was analyzed 48 hr post transfection. The data are analyzed relative to wt expression as % expression ± SEM. (B, D, F) Top image represents MSH2/MLH1/PMS2 with GAPDH and bottom image represents corresponding MSH6/PMS2/MLH1 with GAPDH
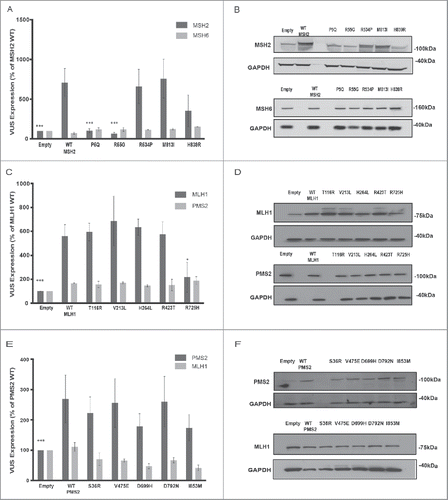
We also assessed mRNA levels for each VUS (Fig. S1). Surprisingly, the mRNA expression of MSH2 VUS P5Q and R55G was barely detectable above the endogenous expression in HEK293 cells, while the level of MSH2 VUS H839R was reduced by ∼77% versus wt (Fig S1A), which was comparable to the observed protein expression differences for these variants (). For MLH1 and PMS2, wt and VUS mRNAs were expressed at equivalent levels with the exception of MLH1 T116R, which exhibited reduced (∼40%) RNA expression (Fig. S1B), even though no differences in steady-state protein expression were noted ().
Impact of MMR VUS on basal cellular viability
The expression of MMR proteins has been associated with the activation of cell cycle checkpoints and apoptosis, especially in response to certain DNA damaging agents.Citation39 Several studies have used in vitro biochemical methods for assessment of VUS.Citation40,41 A limitation of these methods is that they do not fully recapitulate the intracellular environment: they may minimize the impact of VUS that impact protein interactions with diverse partners, or alternatively, fail to capture compensating mechanisms that reduce the impact of some damaging VUS.Citation39 In vivo studies have shown that re-expression of MSH2 or MLH1 can induce apoptosis in both repair-proficient and repair-deficient cells, with loss of such pro-apoptotic activity a reliable indication of damaged MMR signaling function. Hence, several studies have used cell culture-based methods to address MMR functionality in regulating cellular viability and cell death under basal conditions, and during DNA damage response.Citation42-46
Using these approaches, we analyzed the consequences of VUS function in CRC cell line models in which no wt copy of each affected gene was present. We transfected plasmids expressing the MSH2 series VUS into LoVo cells,Citation47 the MLH1 series into HCT116 cells,Citation48 and the PMS2 series into Vaco481 cellsCitation49 (Fig S2 and S3). In the absence of endogenous wt forms of each protein, the protein expression patterns for each VUS were generally similar to those observed in the HEK293 cells (), with the exception that MSH2 H839R and PMS2 I853M were now somewhat reduced compared with wt (∼50%, Fig S2A, B; ∼35%, Fig S2E, F). A complete summary of expression results is presented in .
Table 3. Summary of experimental and structural analysis. AA, amino acid change; +, proficient; -, deficient; +(-), reduced or between empty and wt; wt, as wt; +(-)*; response likely different with different DNA damaging agent.
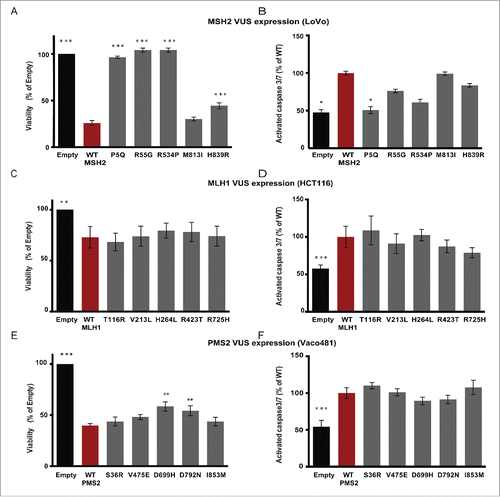
In functional testing, introduction of plasmids for wt MSH2 and its VUS M813I reduced basal LoVo cellular viability by ∼70–75%, while VUS H839R reduced basal viability ∼56%, (), which may reflect the partially reduced expression of this protein. In contrast, the well-expressed variant R534P did not reduce viability, similar to the empty vector control and poorly expressed variants P5Q and R55G. Based on measurement of caspase 3/7 cleavage, the observed reduction of viability approximated the induction of apoptosis by wt MSH2 and MSH2 variants M813I and H839R (). For MLH1, no significant differences were observed between wt and VUS, which each reduced HCT116 viability by ∼25% in comparison to empty vector (), and each increased apoptosis by ∼40% (). Similarly, expression of PMS2 and the VUS series in each case significantly reduced viability of Vaco481 cells (), with wt and VUS S36R, V475E, and I853M reducing viability slightly more than by ∼52–61%, but PMS2 VUS D669H and D792N. These results paralleled capacity to induce apoptosis, with wt and VUS S36R, V475E, and I853M exhibiting slightly higher caspase3/7 cleavage than D669H and D792N ().
Figure 3. Expression of wt and MMR variants impacts cellular viability. LoVo (A, B), HCT116 (C, D), or Vaco481 (E, F) CRC cells were transfected with Empty vector (black bars) or wt (red bars) or VUS (dark gray bars) for MSH2 (A, B), MLH1 (C, D), or PMS2 (E, F). Cell viability (A, C, and E) was determined 72 hours post-transfection by CellTiter-Blue (CTB) assay. The data are represented as % viability relative to E and are from 3 independent experiments ± SEM. The statistical comparisons are with wt. Statistical analysis was performed using a generalized linear model, where * represents p < 0.05. B, D, and F) Post transfection (72 h) cells were assayed for caspase-3/7 activity using the Apo-ONE Homogeneous Caspase 3/7-activity kit according the manufacturer's protocols. Measurements were taken using a Perkin Elmer Envision Plate Reader and plotted as % of wt (red bars) from 3 independent experiments ± SEM. Statistics were performed using a generalized linear model, where * represents p < 0.05, ** represents p < 0.01 and *** represents p < 0.001.
Impact of VUS on viability in response to DNA-damaging treatments
Loss of MMR leads to paradoxical resistance to chemotherapeutic/DNA-damaging agents such as platinum-based compounds (e.g. cisplatin (CDDP)), anti-metabolites such as 5-flourouracil (5-FU)), and methylating agents such as methyl methanesulfonate (MMS)),Citation39, 50, 51 as MMR-deficient cells are able to tolerate moderate DNA damage, whereas MMR responses induces cytostasis and apoptosis. To specifically evaluate the impact of the VUS on response to DNA-damaging agents, we analyzed viability of CRC cell lines expressing wt or variant forms of MSH2, MLH1, or PMS2 72 hours after treatment with a concentration series of CDDP, 5-FU, or MMS (, and extended data in Table S3 with red text highlighting significant dose response and significant dose response relative to wt). Platinum agents and 5-FU are used clinically in the treatment and management of CRC.Citation52
Figure 4. Impact of VUS on IC50 for DNA damaging drugs. LoVo (A-C), HCT116 (C-E), and Vaco481 (F-H) CRC cells were transfected with empty vector (E, black curves) or plasmid expressing wt (red curves) or VUS (gray curves) MSH2 (A-C), MLH1 (D-F), or PMS2 (G-I). 48 hours after transfection cells were treated with vehicle (V), methyl methanesulphonate (MMS), or cisplatin (CDDP) or 5-Flurouracil (5-FU) for 2 h at the concentrations indicated. Cell viability was measured by short-term CellTiter-Blue (CTB) assay (72 hours after drug addition). Data are represented as % viability and are from 3 independent experiments ± SEM. For extended p-values for log dose effect (slope) and log dose slope compared with wt see Table S3 (p-values hihglighted in red are significant).
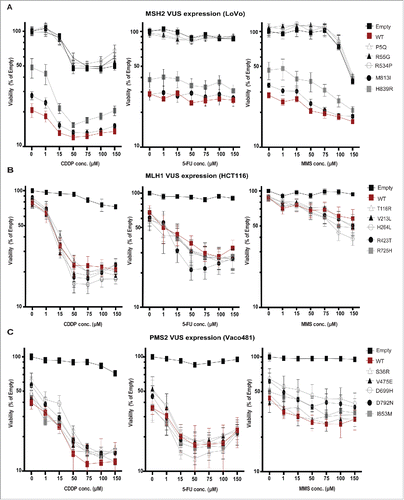
For MSH2 in LoVo cells, the 2 poorly expressed VUS (P5Q and R55G) and VUS R534P had IC50 curves for all 3 drugs similar to cells transfected with empty vector (). Strikingly, VUS M813I and H839R showed more subtle alterations in patterns of response, referenced to wt MSH2. For example, while expression of wt MSH2 had the greatest impact in viability in the absence of drug, IC50 curves were relatively flat as concentration of 5-FU was increased, indicating limited dose responsiveness. However increasing concentrations of CDDP and MMS indicated significantly increased dose responsiveness of wt MSH2 ( and Table S3). In contrast, VUS H839R, expressed at a lower but detectable level had a weaker effect on basal viability, but was associated with a proportionally greater loss of cell viability following treatment with these drugs, while VUS M813I had an intermediate effect compared with wt.
For MLH1/HCT116 and PMS2/Vaco481, cells transfected with empty vector had almost no reduction in viability following treatment with any of the drugs, whereas introduction of either wt or VUS forms of MLH1 or PMS2 resulted in a dose-dependent loss of viability. Surprisingly, even though the expression of MLH1 R725H was significantly reduced compared with wt (Fig. S2C and D), this difference did not impair drug response phenotype (). Also surprisingly, several MLH1 VUS had a non-statistically significant trend toward greater drug-induced loss of viability than did wt (). For PMS2, I853M CDDP showed a significant dose response difference from wt, while VUS D699H and D792N had a near-significant difference from wt specifically following CDDP and MMS treatment ().
Expression of VUS impacts DDR signaling
The kinases ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) are the key regulators of DNA damage response (DDR) signaling,Citation53 activated by MMR-dependent detection of specific DNA lesions.Citation54 MSH2 directly recruits ATR to the sites of DNA damage,Citation54 while ATM activation may stabilize the MutL complex in response to DNA damage.Citation55 ATM-dependent phosphorylation of histone H2AX phosphorylation at Ser139 (gH2AX) in the presence of DNA damage marks chromatin for recruitment of DNA repair proteins.Citation56 Certain MSH2 missense mutations have been suggested to uncouple repair and DDR signaling functions,Citation21,22 making it of interest to assess how the VUS affect ATM/ATR signaling activation and other indicators of DDR signaling.
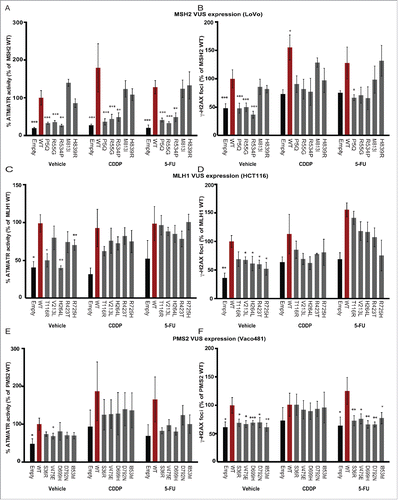
We used antibodies directed at canonical ATM/ATR substrate phosphorylation sites (phospho-S*/T*Q),Citation57,58 and monitored appearance of gH2AX foci (as in Citation23) in cells overexpressing wt or VUS proteins, either under basal conditions or 16 hours after treatment with 15mM of CDDP or 5-FU (determined as responsive dose in ) (, and Table S4 (red text highlights significance)). Overall, VUS phenotypes paralleled those obtained with viability and apoptosis assays, with VUS capable of reducing viability and inducing apoptosis performing similarly to wt MMR proteins in inducing ATM/ATR phosphorylation and gH2AX induction under both basal and drug treatment conditions, with some interesting exceptions. Comparably increased ATM/ATR-dependent and gH2AX phosphorylation () was induced by MSH2 wt, VUS M813I and H839R, but not by VUS P5Q, R55G and R534P. In contrast, several of the MLH1 VUS and all of the PMS2 VUS showed reduced induction of ATM/ATR-dependent and gH2AX phosphorylation compared with wt MLH1 and PMS2 ().
Figure 5. Impact of VUS on wt DNA damage signaling responses. ImageExpress automated quantitation of signal for ATM/ATR substrate phosphorylation (A, C, E.) or gH2AX (B, D, F) in Lovo (A, B), HCT116 (C, D) or Vaco481 (E, F) CRC cells. Imaging is performed 16 hours after drug treatment with vehicle (V), with 15 mM cisplatin (CDDP) or 5-flourouracil (5-FU) of cells transfected with empty vector (E, black bars) or plasmids expressing wt (red bars) or VUS for indicated proteins. Numbers reflect mean positive fluorescence (for ATM/ATR phosphorylation) or mean foci count (for gH2AX). Data are represented as % positive mean relative to wt and are from 3/4 independent experiments ± SEM. Statistical analysis was performed using a generalized linear model, where where * represents p < 0.05, ** represents p < 0.01 and *** represents p < 0.001.
Structural and computational analysis of VUS
Based on all the data described above, several VUS reduce or eliminate mRNA expression (MSH2-P5Q, R55G, H839R; MLH1-T116R, H264L) and/or protein expression (MSH2-P5Q, R55G, H839R; MLH1-R725H; PMS2-I853M), while several VUS impair function but not expression (MSH2-R534P; MLH1-V213L, R423T, R725H; PMS2-V475E, D699H, D792N). The experimental results may be compared with the output of programs designed to predict changes in protein stability or function or association with disease .13-15,18-20 For 2 mutations with consistent experimental functionality, MSH2-R534P with impaired function and PMS2-I853M with wt functionality, the computational predictions indicate agreement with the experimental results (6/7 deleterious (D) and 5/6 neutral (N) respectively; see ). In 5 cases, there was strong consensus among the predictors: MSH2-H839R (5/7 N), MLH1-V213L (7/7 N), MLH1-H264L (6/7 D), MLH1-R423T (5/6 N), and PMS2-V475E (6/6 N), while the experimental results indicated some wt functionality and some impaired functionality (columns 4–6, ). For the remaining 8 cases, the predictors produced mixed results. Of these, 2 mutations were completely defective due to lack of mRNA expression (MSH2-P5Q and MSH2-R55G), which the predictors are generally not designed to detect since they are based on protein sequence and structural information. Two mutations were similar to wild type in all 3 assays (MSH2-M183I and PMS2-S36R), while the remaining 4 were moderately function, having impaired function in one or 2 of the 3 assays (MLH1-T116R, MLH1-R725H, PMS2-D699H, PMS2-D792N)
To further guide interpretation of biologic data, we mapped each VUS onto the coordinates of the 3-dimensional structures of the MMR heterodimers to examine local interactions for each affected residue. We also calculated the DDG of mutation with the program FoldX ,27 and examined potential alterations to transcription and translation using online prediction tools. Here we highlight the most relevant results for several variants ( and ), with additional text and data for the entire test set noted in supplementary text and Table S1 respectively.
Neutral variants
Three variants were found to be experimentally functional in all 3 assays compared with wt: MSH2-M813I, PMS2-S36R, PMS2-I853M. The conservative, hydrophobic mutations MSH2-M813I and PMS2-I853M are both in heterodimeric interfaces (with MSH2 and MLH1 respectively) but molecular modeling suggested no significant steric conflicts upon mutation and preservation of hydrophobic contacts. PMS2 S36R is solvent exposed on the N-terminal domain of PMS2, easily accommodating a larger, polar side chain at the same site. The computational results are further detailed in supplementary text.
Interface-disrupting VUS
Several more drastic variants were located at the interfaces of biologic assemblies, and have the potential to disrupt dimerization events. Here we highlight VUS MSH2-R534P from , while details on MSH2-H839R, MLH1-T116R, MLH1-R725H and MLH1-D699H are in supplementary text. Functional assays showed the greatest impairment of activity for a VUS affecting MSH2-R534, which is at the MutSα heterodimer interface. The R534 side chain is in close proximity to backbone carbonyls of R988 and N989 on MSH6, with N-O distances of 3.1 and 3.7 Angstroms, respectively. Additionally, R534 backbone amide hydrogen stabilizes a β turn of residues 532–535 through a hydrogen bond to the backbone carbonyl oxygen of residue 535. The R534P mutation would likely disrupt the β turn at the interface by eliminating the amide hydrogen (due to proline ring closure) and the backbone hydrogen bond. Additionally, any favorable intermolecular electrostatic interactions to residues 988 and 989 of MSH6 would be lost.
Ambiguous variants
The effects for some variants were more difficult to rationalize with a high level of confidence from a structural perspective. Both MLH1-R423 and PMS2-V475 are located in the disordered linker regions between the N- and C-terminal domains of MLH1 and PMS2, respectively (). Although neither of these sites were included in the critical linker arm regions identified by Plys et al.Citation59 the biophysical consequences of mutations in this region are difficult to predict. Other ambiguous variants such as PMS2-D792N, MLH1 V213L and H264L are summarized in supplementary text.
RNA expression-deficient variants
Five variants showed partial or severe deficiencies in RNA expression: MSH2-P5Q (severe), MSH2-R55G (severe), MSH2-H839R (partial), MLH1-T116R (partial), and MLH1-H264L (partial). Alternative splicing leading to premature truncation of protein coding capacity can trigger nonsense-mediated decay,Citation60 providing one possible explanation for reduced steady-state mRNA levels. The Human Splicing Finder (HSF) serverCitation28 suggests that mRNA splicing can potentially be altered through several mechanisms, including the creation of an exonic splicing silencer (ESS) site, creation of a new donor or acceptor site, or alteration of an exonic splicing enhancer (ESE) site. For the variants affecting RNA expression, HSF predicts the following, MSH2-P5Q (ESS), MSH2-R55G (ESS, donor), MLH1-T116R (ESS, ESE, and acceptor), and MLH1-H264L (ESS). Since we have transfected DNA sequences without introns, these changes are not necessarily relevant to the expression of RNAs unless they create new splice sites. Of the remaining 11 VUS, 7 of them also have predicted changes by the Human Splicing Finder (Table S2). For individuals carrying a VUS in an MMR gene, changes in RNA splicing may be a mechanism for downregulating expression of the gene product, even though this was not assessed in our experiments.
Alternatively, gross changes in mRNA secondary structure can also destabilize gene transcripts.Citation61 SNPfoldCitation29 predicted statistically significant structural alterations to RNA structure for MSH2-P5Q (p = 0.0226) and MLH1-T116R (p = 0.0115), which showed impaired RNA expression, and for MLH1-V213L (p = 0.0106) and PMS2-V475E (p = 0.0436) which did not. MfoldCitation30 was used to generate the predicted mRNA 2° structures of the reference and variant sequences (Fig. S4). Mfold predicts that the P5Q RNA structure gains a circular loop in the middle of the stem (Figs. S4A, B). The R55G RNA structure was predicted to form a circular loop instead of 2 short arms (Figs. S4C, D). H839R is predicted to eliminate a significant loop in the RNA structure. Such changes, if validated, could significantly affect mRNA stability.
Discussion
This study examined the relation between computational prediction of VUS severity and MMR functionality. We systematically assessed the role of 15 rare germline VUS in MSH2, MLH1 and PMS2 ( and ), using multiple assays to address the consequences for RNA and protein expression, viability, apoptosis induction, and DNA damage signaling. Of the VUS analyzed, most were detected either in patients with features selective of LS or HCPS, or with definitive LS, while some of the VUS (MSH2-R534P, MSH2-H839R, MLH1-R725H, and PMS2-D699H) () were also detected in patients with CRC. Major conclusions of the study were: 1) existing generic and gene-specific predictor programs failed to correctly classify many novel VUS, and care should be taken when using these methods in a clinical setting; 2) for at least 3 VUS, single residue changes were associated with significant changes at the steady-state mRNA level, which may reflect differences in mRNA secondary structure; 3) the use of multiple function-testing assays was useful to distinguish different categories of potential defect associated with individual VUS; and 4) mapping the variants onto protein structure provided additional insights suggesting the mechanistic basis for their effect, with many VUS localizing in important functional domains ( and ).
Among the VUS assessed, most were expressed at normal protein levels, but several exhibited moderate impairment of activity in one or more assays, with a small number associated with more profound defects. Most of the MLH1 and PMS2 variants were classified as moderately functional (). Interestingly, VUS affecting these genes sometimes varied significantly in their consequences for viability and DDR signaling in response to cisplatin and 5-FU DNA damaging treatments (, , and Table S4). Three of 5 MSH2 variants assessed had moderate to severe defects in activity. For 2, P5Q and R55G, MSH2 bearing these variants were detectable neither at the mRNA nor the protein level ( and Figs. S1 and S2), which may reflect impact of the variants at the level of RNA structure, with variant-associated changes potentially influencing interaction of affected transcripts to RNA binding proteins that regulate stability (Fig. S4). In contrast, although MSH2 variant R534P was expressed at wt levels, this VUS was associated with complete loss of function in regulation of basal or drug treatment-associated cell viability or DDR signaling ( and ).
Unclassified variants present a variety of unique challenges to the genetic counselor and the clinician as well as the patient and their family. Previous studies have advised application of standardized approaches to assess the pathogenicity of the VUS. Rasmussen et. al. suggested a 3-step diagnostic decision process,Citation62 incorporating computational methods to predict effects on mRNA splicing, analysis of evolutionary sequence conservation, and assaying the activity of the VUS protein in vitro. They also suggested studying the biologic, biochemical and biophysical properties of the VUS to further aid in diagnosis. The International Society for Gastrointestinal Hereditary Tumors (InSiGHT) recently published a 5-tiered standardized scheme with methods to deal with MMR VUS.Citation63 They suggested that computational methods (MAPP-MMR and Polyphen-2.1) may exhibit high accuracy in variant prediction. By contrast, in our small test set of 15 rare or novel variants, all of the computational predictors performed rather poorly in assessing function of the genes in regulating viability, apoptosis, and DDR signaling. The best results came from BA-SVM,Citation26 which correctly classified 10 out of the 15 mutants, according to our 3-tiered system.
Gene-specific predictors are an alternative to generic predictors. However, for the 15 VUS in this study, the results were not promising. Using a 3-tiered classification system (Functional, Moderate/Intermediate, and Defective), MAPP-MMR correctly classified 2 out of 8 variants, but was not capable of making predictions on the 5 PMS2 variants and 2 of the MHS2/MLH1 variants. PON-MMR2 correctly classified 3 out of 15 variants, based on empirically determined functionality (summarized in ). Additionally, these gene-specific methods have been trained on rather small variant sets (< 200), with few or no PMS2 variants. Moreover, the experimental data upon which these methods are trained may contain some errors, as a variant may affect a protein function that was not assayed. For example, a pathogenic MLH1 c.2041G>A (p. Ala681Thr) mutation exhibited MSI-high tumors but was functional in various in vitro assays of expression, MMR complementation and interaction with PMS2.Citation63 More refinement and training on larger data sets would be beneficial.
Both generic and gene-specific predictors are based on evolutionary conservation of protein sequences and amino acid properties. Most of them do not consider important biologically relevant details; such as RNA splice sites, codon usage bias, biologic assembly interfaces, post-translational modification sites, etc. Incorporating such data could improve results, as evidenced by the superior performance of the biologic assembly SVM predictor. Computational methods that predict protein dysfunction most often rely on amino acid substitution in relation to evolutionary conservation across species. In only a very few cases do these feature structural assessment of the protein.Citation62 There is not only a need for protein specific computational predictors, but also programs that are trained on a large balanced set of (pathogenic and neutral) mutations per protein for machine learning and dysfunction prediction.Citation26,64 In case of PMS2 and MSH6 gene variants, limited variants are known as well as limited information is available on the disease spectrum.Citation65
Another possible confounder for structural prediction methods is that a given mutation may affect a specific function that is not standardly assayed. Most VUS studies focus on biologic assays in yeast and cell-free MMR assays which do not reflect the cellular environment.Citation39,62 Our alignment of VUS with structural features of MSH2, MLH1, and PMS2 () indicated multiple domains were targeted, leading to suggestion of a wide range of potential mechanisms of action (summarized in and ). The approaches used here were specifically focused on defining in vivo effects of VUS, rather than providing a biochemical assessment of their mismatch repair capacity. While there are strengths to this approach, in simulating in vivo consequences, there are also potential caveats for surrogate assays based on viability and apoptosis. For example, it has been shown that MRE11, a component of the ATM-regulated double strand break signaling, is inactivated in several MMR-deficient cell lines, including the HCT116 and LoVo lines.Citation66,67 Hence, behavior of VUS may vary based on the genetic background of the models in the experimental study. While it is not possible to develop clinical tests for all possible activities, our data suggest that that MMR repair should not be the sole criterion upon which variant function should be judged. Rather, apoptotic signaling and therapeutic-dependent viability should be evaluated when considering how a novel MMR mutation will affect a patient.
Finally, our data imply that incorporating RNA and biologic assembly interface features could aid in the development of future predictive programs. Other studies have also emphasized the desirability of combining predictive analyses at the DNA, RNA (splicing and secondary structures) and protein level as well as incorporating additional features such as codon usage bias,Citation68,69 with one study suggesting that many VUS would be predicted to influence RNA splicing, stability or translation of encoded variant proteins.Citation62 Although the incorporation of computational RNA analyses requires empirical confirmation, they provide workable hypotheses to explain experimental results, and potentially should be incorporated in next-generation prediction models.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
S.A. designed, coordinated, performed most studies and writing, P.J.H.: writing, structural modeling and discussion; R.S., M.S., SD, A.B., R.L.: mutagenesis, expression, and in vitro analyses, E.N.: RNA analysis, sequencing, M.J.H., R.D. & E.A.G.: discussion, designing, planning studies and writing.
Supplementary_Materials.pdf
Download PDF (1.8 MB)Acknowledgments
The Fox Chase Cancer Center (FCCC) Cell Culture, Genomics, DNA Sequencing, Molecular Modeling, Biostatistics and High Throughput Facilities provided resources and expertise. The authors, facilities, and this work were supported by NCI Core Grant P30 CA06927 (CURE supplement, FCCC), Temple Genomic Research Funding, R01 DK108195 (to E.A.G.), T32 CA09035 (to S.A. and P.J.H.), R01 GM084453 (to R.L.D), and FCCC Summer Undergraduate Research Fellowships (to M.S. and R.L.). We thank Dr. Mark D. Andrake (FCCC, Molecular Modeling Facility) for assistance with modeling, and Dr. Sandy Markowitz (Case Western Reserve University) for providing Vaco481 CRC cells. All authors declare that there is no conflict of interest regarding this manuscript.
References
- Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993; 75:1027-38; PMID:8252616; https://doi.org/10.1016/0092-8674(93)90546-3
- Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomäki P, Sistonen P, Aaltonen LA, Nyström-Lahti M, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993; 75:1215-25; PMID:8261515; https://doi.org/10.1016/0092-8674(93)90330-S
- Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer 2015; 15:181-94; PMID:25673086; https://doi.org/10.1038/nrc3878
- Andersen SD, Liberti SE, Lutzen A, Drost M, Bernstein I, Nilbert M, Dominguez M, Nyström M, Hansen TV, Christoffersen JW, et al. Functional characterization of MLH1 missense variants identified in Lynch syndrome patients. Hum Mutat 2012; 33:1647-55; PMID:22753075; https://doi.org/10.1002/humu.22153
- Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell 2006; 126:297-308; PMID:16873062; https://doi.org/10.1016/j.cell.2006.05.039
- Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem 2006; 281:30305-9; PMID:16905530; https://doi.org/10.1074/jbc.R600022200
- Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 2006; 7:335-46; PMID:16612326; https://doi.org/10.1038/nrm1907
- Provenzale D, Gupta S, Ahnen DJ, Bray T, Cannon JA, Cooper G, David DS, Early DS, Erwin D, Ford JM, et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2016; 14:1010-30; PMID:27496117; https://doi.org/10.6004/jnccn.2016.0108
- Sijmons RH, Greenblatt MS, Genuardi M. Gene variants of unknown clinical significance in Lynch syndrome. An introduction for clinicians. Fam Cancer 2013; 12:181-7; PMID:23525798; https://doi.org/10.1007/s10689-013-9629-8
- Eshleman JR, Markowitz SD. Mismatch repair defects in human carcinogenesis. Hum Mol Genet 1996; 5(Spec No):1489-94; PMID:8875255; https://doi.org/10.1093/hmg/5.Supplement_1.1489
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39:1-10; PMID:23890059; https://doi.org/10.1016/j.immuni.2013.07.012
- Thompson BA, Greenblatt MS, Vallee MP, Herkert JC, Tessereau C, Young EL, Adzhubey IA, Li B, Bell R, Feng B, et al. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat 2013; 34:255-65; PMID:22949387; https://doi.org/10.1002/humu.22214
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31:3812-4; PMID:12824425; https://doi.org/10.1093/nar/gkg509
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248-9; PMID:20354512; https://doi.org/10.1038/nmeth0410-248
- Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011; 39:e118; PMID:21727090; https://doi.org/10.1093/nar/gkr407
- Niroula A, Vihinen M. Variation Interpretation Predictors: Principles, Types, Performance, and Choice. Hum Mutat 2016; 37:579-97; PMID:26987456; https://doi.org/10.1002/humu.22987
- Grimm DG, Azencott CA, Aicheler F, Gieraths U, MacArthur DG, Samocha KE, Cooper DN, Stenson PD, Daly MJ, Smoller JW, et al. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum Mutat 2015; 36:513-23; PMID:25684150; https://doi.org/10.1002/humu.22768
- Chao EC, Velasquez JL, Witherspoon MS, Rozek LS, Peel D, Ng P, Gruber SB, Watson P, Rennert G, Anton-Culver H, et al. Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR). Hum Mutat 2008; 29:852-60; PMID:18383312; https://doi.org/10.1002/humu.20735
- Terui H, Akagi K, Kawame H, Yura K. CoDP: predicting the impact of unclassified genetic variants in MSH6 by the combination of different properties of the protein. J Biomed Sci 2013; 20:25; PMID:23621914; https://doi.org/10.1186/1423-0127-20-25
- Niroula A, Vihinen M. Classification of Amino Acid Substitutions in Mismatch Repair Proteins Using PON-MMR2. Hum Mutat 2015; 36:1128-34; PMID:26333163; https://doi.org/10.1002/humu.22900
- Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res 2004; 64:517-22; PMID:14744764; https://doi.org/10.1158/0008-5472.CAN-03-2957
- Drotschmann K, Topping RP, Clodfelter JE, Salsbury FR. Mutations in the nucleotide-binding domain of MutS homologs uncouple cell death from cell survival. DNA Repair (Amst) 2004; 3:729-42; PMID:15177182; https://doi.org/10.1016/j.dnarep.2004.02.011
- Arora S, Yan H, Cho I, Fan HY, Luo B, Gai X, Bodian DL, Vockley JG, Zhou Y, Handorf EA, et al. Genetic Variants That Predispose to DNA Double-Strand Breaks in Lymphocytes From a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology 2015; 149:1872-83 e9; PMID:26344056; https://doi.org/10.1053/j.gastro.2015.08.052
- Nicolas E, Arora S, Zhou Y, Serebriiskii IG, Andrake MD, Handorf ED, Bodian DL, Vockley JG, Dunbrack RL, Ross EA, et al. Systematic evaluation of underlying defects in DNA repair as an approach to case-only assessment of familial prostate cancer. Oncotarget 2015; 6:39614-33; PMID:26485759; https://doi.org/10.18632/oncotarget.5554
- Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015; 31:2745-7; PMID:25851949; https://doi.org/10.1093/bioinformatics/btv195
- Wei Q, Xu Q, Dunbrack RL, Jr. Prediction of phenotypes of missense mutations in human proteins from biological assemblies. Proteins 2013; 81:199-213; PMID:22965855; https://doi.org/10.1002/prot.24176
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res 2005; 33:W382-8; PMID:15980494; https://doi.org/10.1093/nar/gki387
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37:e67; PMID:19339519; https://doi.org/10.1093/nar/gkp215
- Halvorsen M, Martin JS, Broadaway S, Laederach A. Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet 2010; 6:e1001074; PMID:20808897; https://doi.org/10.1371/journal.pgen.1001074
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406-15; PMID:12824337; https://doi.org/10.1093/nar/gkg595
- Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell 2007; 26:579-92; PMID:17531815; https://doi.org/10.1016/j.molcel.2007.04.018
- Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA–a self-parameterizing force field. Proteins 2002; 47:393-402; PMID:11948792; https://doi.org/10.1002/prot.10104
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 2005; 33:W244-8; PMID:15980461; https://doi.org/10.1093/nar/gki408
- Perevoztchikova SA, Romanova EA, Oretskaya TS, Friedhoff P, Kubareva EA. Modern aspects of the structural and functional organization of the DNA mismatch repair system. Acta Naturae 2013; 5:17-34; PMID:24303200
- Krivov GG, Shapovalov MV, Dunbrack RL, Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009; 77:778-95; PMID:19603484; https://doi.org/10.1002/prot.22488
- Guarne A, Junop MS, Yang W. Structure and function of the N-terminal 40 kDa fragment of human PMS2: a monomeric GHL ATPase. EMBO J 2001; 20:5521-31; PMID:11574484; https://doi.org/10.1093/emboj/20.19.5521
- Wu H, Zeng H, Lam R, Tempel W, Kerr ID, Min J. Structure of the human MLH1 N-terminus: implications for predisposition to Lynch syndrome. Acta Crystallogr F Struct Biol Commun 2015; 71:981-5; PMID:26249686; https://doi.org/10.1107/S2053230X15010183
- Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 1977; 36:59-74; PMID:886304; https://doi.org/10.1099/0022-1317-36-1-59
- Heinen CD, Juel Rasmussen L. Determining the functional significance of mismatch repair gene missense variants using biochemical and cellular assays. Hered Cancer Clin Pract 2012; 10:9; PMID:22824075; https://doi.org/10.1186/1897-4287-10-9
- Cyr JL, Heinen CD. Hereditary cancer-associated missense mutations in hMSH6 uncouple ATP hydrolysis from DNA mismatch binding. J Biol Chem 2008; 283:31641-8; PMID:18790734; https://doi.org/10.1074/jbc.M806018200
- Cyr JL, Brown GD, Stroop J, Heinen CD. The predicted truncation from a cancer-associated variant of the MSH2 initiation codon alters activity of the MSH2-MSH6 mismatch repair complex. Mol Carcinog 2012; 51:647-58; PMID:21837758; https://doi.org/10.1002/mc.20838
- Trojan J, Zeuzem S, Randolph A, Hemmerle C, Brieger A, Raedle J, Plotz G, Jiricny J, Marra G. Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology 2002; 122:211-9; PMID:11781295; https://doi.org/10.1053/gast.2002.30296
- Brieger A, Trojan J, Raedle J, Plotz G, Zeuzem S. Transient mismatch repair gene transfection for functional analysis of genetic hMLH1 and hMSH2 variants. Gut 2002; 51:677-84; PMID:12377806; https://doi.org/10.1136/gut.51.5.677
- Perera S, Bapat B. The MLH1 variants p.Arg265Cys and p.Lys618Ala affect protein stability while p.Leu749Gln affects heterodimer formation. Hum Mutat 2008; 29:332; PMID:18205192; https://doi.org/10.1002/humu.9523
- Mastrocola AS, Heinen CD. Lynch syndrome-associated mutations in MSH2 alter DNA repair and checkpoint response functions in vivo. Hum Mutat 2010; 31:E1699-708; PMID:20672385; https://doi.org/10.1002/humu.21333
- Blasi MF, Ventura I, Aquilina G, Degan P, Bertario L, Bassi C, Radice P, Bignami M. A human cell-based assay to evaluate the effects of alterations in the MLH1 mismatch repair gene. Cancer Res 2006; 66:9036-44; PMID:16982745; https://doi.org/10.1158/0008-5472.CAN-06-1896
- Watanabe Y, Haugen-Strano A, Umar A, Yamada K, Hemmi H, Kikuchi Y, Takano S, Shibata Y, Barrett JC, Kunkel TA, et al. Complementation of an hMSH2 defect in human colorectal carcinoma cells by human chromosome 2 transfer. Mol Carcinog 2000; 29:37-49; PMID:11020245; https://doi.org/10.1002/1098-2744(200009)29:1%3c37::AID-MC5%3e3.0.CO;2-210.1002/1098-2744(200009)29:1%3c37::AID-MC5%3e3.3.CO;2-U
- Imesch P, Goerens A, Fink D, Fedier A. MLH1-deficient HCT116 colon tumor cells exhibit resistance to the cytostatic and cytotoxic effect of the poly(A) polymerase inhibitor cordycepin (3′-deoxyadenosine) in vitro. Oncol Lett 2012; 3:441-4; PMID:22740928; https://doi.org/10.3892/ol.2011.504
- Ma AH, Xia L, Littman SJ, Swinler S, Lader G, Polinkovsky A, Olechnowicz J, Kasturi L, Lutterbaugh J, Modrich P, et al. Somatic mutation of hPMS2 as a possible cause of sporadic human colon cancer with microsatellite instability. Oncogene 2000; 19:2249-56; PMID:10822375; https://doi.org/10.1038/sj.onc.1203568
- Glaab WE, Risinger JI, Umar A, Barrett JC, Kunkel TA, Tindall KR. Cellular resistance and hypermutability in mismatch repair-deficient human cancer cell lines following treatment with methyl methanesulfonate. Mutat Res 1998; 398:197-207; PMID:9626980; https://doi.org/10.1016/S0027-5107(98)00004-9
- Li LS, Morales JC, Veigl M, Sedwick D, Greer S, Meyers M, Wagner M, Fishel R, Boothman DA. DNA mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the potential for new therapeutic targets. Br J Pharmacol 2009; 158:679-92; PMID:19775280; https://doi.org/10.1111/j.1476-5381.2009.00423.x
- Cersosimo RJ. Management of advanced colorectal cancer, Part 1. Am J Health Syst Pharm 2013; 70:395-406; PMID:23413162; https://doi.org/10.2146/ajhp11053210.2146/ajhp110532b
- Pabla N, Ma Z, McIlhatton MA, Fishel R, Dong Z. hMSH2 recruits ATR to DNA damage sites for activation during DNA damage-induced apoptosis. J Biol Chem 2011; 286:10411-8; PMID:21285353; https://doi.org/10.1074/jbc.M110.210989
- Meyers M, Hwang A, Wagner MW, Bruening AJ, Veigl ML, Sedwick WD, Boothman DA. A role for DNA mismatch repair in sensing and responding to fluoropyrimidine damage. Oncogene 2003; 22:7376-88; PMID:14576845; https://doi.org/10.1038/sj.onc.1206941
- Luo Y, Lin FT, Lin WC. ATM-mediated stabilization of hMutL DNA mismatch repair proteins augments p53 activation during DNA damage. Mol Cell Biol 2004; 24:6430-44; PMID:15226443; https://doi.org/10.1128/MCB.24.14.6430-6444.2004
- Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013; 5:a012716; PMID:24003211; https://doi.org/10.1101/cshperspect.a012716
- Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther 2015; 149:124-38; PMID:25512053; https://doi.org/10.1016/j.pharmthera.2014.12.001
- Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev 2004; 18:1331-44; PMID:15175264; https://doi.org/10.1101/gad.294404
- Plys AJ, Rogacheva MV, Greene EC, Alani E. The unstructured linker arms of Mlh1-Pms1 are important for interactions with DNA during mismatch repair. J Mol Biol 2012; 422:192-203; PMID:22659005; https://doi.org/10.1016/j.jmb.2012.05.030
- Hamid FM, Makeyev EV. Emerging functions of alternative splicing coupled with nonsense-mediated decay. Biochem Soc Trans 2014; 42:1168-73; PMID:25110020; https://doi.org/10.1042/BST20140066
- Klaff P, Riesner D, Steger G. RNA structure and the regulation of gene expression. Plant Mol Biol 1996; 32:89-106; PMID:8980476; https://doi.org/10.1007/BF00039379
- Rasmussen LJ, Heinen CD, Royer-Pokora B, Drost M, Tavtigian S, Hofstra RM, de Wind N. Pathological assessment of mismatch repair gene variants in Lynch syndrome: past, present, and future. Hum Mutat 2012; 33:1617-25; PMID:22833534; https://doi.org/10.1002/humu.22168
- Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capellá G, den Dunnen JT, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 2014; 46:107-15; PMID:24362816; https://doi.org/10.1038/ng.2854
- Wei Q, Dunbrack RL, Jr. The role of balanced training and testing data sets for binary classifiers in bioinformatics. PLoS One 2013; 8:e67863; PMID:23874456; https://doi.org/10.1371/journal.pone.0067863
- Al-Sukhni W, Aronson M, Gallinger S. Hereditary colorectal cancer syndromes: familial adenomatous polyposis and lynch syndrome. Surg Clin North Am 2008; 88:819-44, vii; PMID:18672142; https://doi.org/10.1016/j.suc.2008.04.012
- Giannini G, Ristori E, Cerignoli F, Rinaldi C, Zani M, Viel A, Ottini L, Crescenzi M, Martinotti S, Bignami M, et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep 2002; 3:248-54; PMID:11850399; https://doi.org/10.1093/embo-reports/kvf044
- Takemura H, Rao VA, Sordet O, Furuta T, Miao ZH, Meng L, Zhang H, Pommier Y. Defective Mre11-dependent activation of Chk2 by ataxia telangiectasia mutated in colorectal carcinoma cells in response to replication-dependent DNA double strand breaks. J Biol Chem 2006; 281:30814-23; PMID:16905549; https://doi.org/10.1074/jbc.M603747200
- Bali V, Bebok Z. Decoding mechanisms by which silent codon changes influence protein biogenesis and function. Int J Biochem Cell Biol 2015; 64:58-74; PMID:25817479; https://doi.org/10.1016/j.biocel.2015.03.011
- Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet 2014; 30:308-21; PMID:24954581; https://doi.org/10.1016/j.tig.2014.04.006