
ABSTRACT
Undifferentiated pleomorphic sarcomas (UPSs) are aggressive mesenchymal malignancies with no definitive cell of origin or specific recurrent genetic hallmarks. These tumors are largely chemoresistant; thus, identification of potential therapeutic targets is necessary to improve patient outcome. Previous studies demonstrated that high expression of activated protein kinase B (AKT) in patients with UPS corresponds to poor disease-specific survival. Here, we demonstrate that inhibiting phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/mTOR) signaling using a small molecule inhibitor reduced UPS cell proliferation and motility and xenograft growth; however, increased phosphorylation of insulin-like growth factor 1 receptor (IGF1R) indicated the potential for adaptive resistance following treatment through compensatory receptor activation. Co-treatment with a dual PI3K/mTOR inhibitor and an anti-IGF1R kinase inhibitor reduced in vivo tumor growth rates despite a lack of antiproliferative effects in vitro. Moreover, this combination treatment significantly decreased UPS cell migration and invasion, which is linked to changes in p27 subcellular localization. Our results demonstrate that targeted inhibition of multiple components of the IGF1R/PI3K/mTOR pathway was more efficacious than single-agent therapy and suggest that co-targeting this pathway could be a beneficial therapeutic strategy for patients with UPS.
Introduction
Soft tissue sarcoma (STS) comprise a heterogeneous group of over 80 histologically distinct subtypes of rare mesenchymal malignancies that are characterized according to their tissue of origin.Citation1 Approximately 5%–15% of soft tissue sarcomas cannot be differentiated by current molecular technologies or immunohistochemical criteria and are therefore classified as undifferentiated pleomorphic sarcomas (UPSs).Citation2,3 Although the development of most UPSs is sporadic, approximately 3% of UPSs develop in areas of the body that received radiation therapy to treat an unrelated disease after a median latency of 10 y.Citation4,5 Local recurrence develops in 19%–31% of patients with UPS; metastatic disease occurs in 26%–35% of patients with UPS, and nearly 5% of patients present with metastases at the time of initial diagnosis, reflecting the aggressive nature of the disease.Citation6,7 Furthermore, the current 5-year overall survival rate for patients with UPS is only 65%–70%, highlighting the need for more efficacious treatment options.Citation6
Currently, the standard of care for patients with UPS is surgical resection; for patients with unresectable or metastatic disease, neoadjuvant or adjuvant chemotherapy and/or radiotherapy may be considered.Citation8 Cytotoxic chemotherapies generally elicit low to moderate response rates in patients with UPS.Citation8-10 Furthermore, although UPSs are karyotypically complex malignancies and exhibit a wide array of chromosomal alterations, specific recurring genetic aberrations are rare.Citation1,11 Therefore, it is essential to identify specific molecular dysregulations in UPS that could be therapeutic targets.
Activation of phosphatidylinositol-3-kinase (PI3K) by receptor tyrosine kinases elicits a series of phosphorylation events leading to activation of mammalian target of rapamycin (mTOR) complex 1 and 2 (mTORC1 and mTORC2) and the promotion of cell growth, proliferation, motility, and survival.Citation12,13 The PI3K/mTOR pathway is frequently dysregulated in cancer through increased expression and/or activation of receptor tyrosine kinases, mutations in pathway components, or loss of negative pathway regulators, such as phosphatase and tensin homolog (PTEN).Citation14 Consequently, PI3K/mTOR pathway members have been intensely scrutinized as potential therapeutic targets in cancer, promoting the development of inhibitors against this pathway's components.Citation12,14 Small molecule inhibitors originally designed to block mTOR activity also inhibit PI3K catalytic activity owing to the high structural homology between the adenosine triphosphate–binding domains of these two kinases and thus prevent the feedback activation of protein kinase B (AKT) observed with single-agent inhibition of mTORC1.Citation12,15,16 However, other adaptive resistance mechanisms can occur after the pharmacologic blockade of PI3K and mTOR.Citation14,17-19 Previous studies have demonstrated an increase in insulin-like growth factor 1 receptor (IGF1R) expression and activation in response to targeted inhibition of the PI3K/mTOR pathway; furthermore, increased IGF1R activity promotes resistance to inhibitor-mediated blockade of PI3K and mTOR.Citation20,21 Therefore, the combination of a PI3K/mTOR dual inhibitor with a targeted agent against IGF1R could enhance antitumor effects and limit the possibility of resistance.
PI3K/mTOR pathway activation is associated with an adverse prognosis in patients with soft tissue sarcoma.Citation22-24 Our research group found that elevated phosphorylated AKT at serine 473 (pAKT; detected in approximately 20% of patients with UPS) was significantly correlated with poor disease-specific survival, indicating that PI3K/mTOR signaling may contribute to the aggressive nature of the disease and that targeted pathway inhibition may be beneficial for these patients.Citation25 However, the role of the PI3K/mTOR pathway in UPS has not been explored. In this study, we assessed the antitumor effects of BGT226, a dual PI3K/mTOR inhibitor, as a single agent and in combination with AEW541, a small molecule inhibitor against IGF1R, in patient-derived models of UPS in vitro and in vivo.
Results
In vitro assessment of PI3K/mTOR signaling in UPS cells
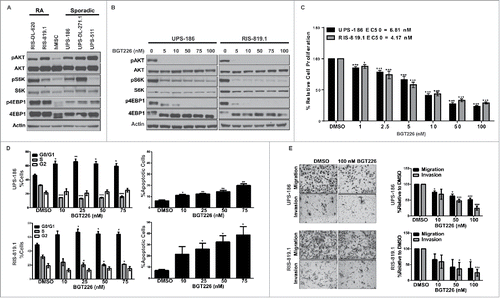
Previously, we found that a subset of UPS tumor samples expressed high levels of pAKT (S473), which corresponded to poor overall survival for those patients.Citation25 To expand upon this finding, we investigated the necessity of intact PI3K/mTOR signaling for UPS progression. Bone marrow-derived human mesenchymal stem cells (hMSCs) were used as a normal cell control, as previous studies have demonstrated that UPS-like tumors can develop following transformation of these cells.Citation26 AKT and S6K (Ribosomal Protein S6 Kinase I) phosphorylation status are commonly used as markers of PI3K and mTOR kinase activity so these markers were examined by western-blot.Citation27-30 The majority of radiation-associated (RA-UPS) and sporadic UPS cell strains used in this study had elevated levels of activated AKT and phosphorylated S6K, and 4EBP1, when compared to the human mesenchymal stem cell control (hMSC), demonstrating an increase of PI3K/mTOR signaling in UPS cells ().
Figure 1. Blockade of the phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/mTOR) pathway inhibits tumor-supportive processes in vitro. A, Endogenous PI3K/mTOR pathway activity was assessed by western blot analysis for total and activated (phosphorylated) protein kinase B (AKT), S6K, and 4EPB1 in whole cell lysates obtained from a panel of radiation-associated undifferentiated pleomorphic sarcoma (RA-UPS) and sporadic UPS cell strains and cell lines, with human mesenchymal stem cells (hMSCs) serving as the normal control. B, Representative western blots of whole cell lysates from a sporadic UPS cell line (UPS-186) and a RA-UPS cell line (RIS-819.1) after incubation with increasing concentrations of BGT226 for 2 hours. C, MTS assay results show diminished UPS-186 and RIS-819.1 cell proliferation relative to controls treated with dimethyl sulfoxide (DMSO) after 96 hours of BGT226 treatment, and the individual median effective concentrations (EC50) were determined. D Cell-cycle distribution (left) and apoptosis levels (right) were evaluated in the two UPS cell lines by fluorescence-activated cell sorting analysis after 48 or 96 hours of BGT226 treatment, respectively. DMSO, control. E, The effects of BGT226 on UPS-186 and RIS-819.1 cell migration and invasion were assessed by modified Boyden chamber assays. Representative images of cells treated with DMSO or BGT226 (100 nM) are displayed (magnification, 200 ×). The graphs depict the means of triplicate experiments; error bars are the standard errors of the mean. *p < 0.05; **p < 0.01; ***p < 0.001.
In vitro efficacy of BGT226, a dual PI3K/mTOR inhibitor
To evaluate the role of PI3K/mTOR signaling in UPS growth, we assessed the effects of BGT226, a dual PI3K/mTOR inhibitor, in UPS-186 (a sporadic cell line) and RIS-819.1 (a RA-UPS cell line). BGT226-mediated inhibition of PI3K and mTOR in UPS-186 and RIS-819.1 cells suppressed phosphorylation of AKT and downstream kinases S6K and 4EBP1 after 2 hours; dephosphorylation was maintained after 96 hours of treatment (, Supplementary Fig. S1). Substantial antiproliferative effects were detected after 96 hours of treatment with low nanomolar concentrations of BGT226, with calculated median effective concentration (EC50) values of 6.81 nM for UPS-186 and 4.17 nM for RIS-819.1 (). Diminished cell proliferation was linked to the induction of apoptosis measured by Annexin V staining (). Interestingly, the RA-UPS cell line RIS-819.1 was more sensitive to BGT226 than the sporadic UPS-186 cell line. A greater percentage of RIS-819.1 cells were positive for Annexin V after treatment when compared to UPS-186.
UPS cell migration is suppressed in vitro after treatment
PI3K/mTOR pathway activation facilitates tumor growth and metastasis through the regulation of cell migration and invasion.Citation13,31 To examine the effects of BGT226 on UPS cell migration and invasion, we cultured UPS-186 and RIS-819.1 cells in low-serum medium (1% fetal bovine serum [FBS] in Dulbecco modified Eagle medium F-12 [DMEM/F12]) to suppress proliferation and to promote migration toward a chemoattractant (5% FBS in DMEM/F12). Cell migration and invasion were reduced in a dose-dependent manner in both cell lines; however, these processes were not fully inhibited even at the highest dose of BGT226 (100 nM; ).
PI3K/mTOR inhibition, slows tumor growth and promotes IGF1R activation in vivo
On the basis of the substantial effects of BGT226 treatment seen in vitro, we then assessed the efficacy of BGT226 treatment in the RIS-819.1 xenograft mouse model. After initially treating all groups daily (7 d/week) for 1 week, we reduced treatment to 5 d/week in the 15-mg/kg BGT226 group to remedy the observed weight loss in these mice; treatment continued for an additional 17 d. Oral administration of BGT226 once daily was sufficient to reduce RIS-819.1 xenografts by 47.4% and 63.6% in the 10-mg/kg and 15-mg/kg groups, respectively, compared with the control group; additionally, the endpoint tumor volumes in the 15-mg/kg BGT226 group were significantly smaller than those in the control group (p = 0.05) (). A trend of decreased tumor weight in the BGT226-treated groups compared with the control group was noted (Supplementary Fig. S2A). Immunohistochemical analysis of downstream effectors of PI3K/mTOR signaling revealed that pAKT, pS6K, and p4EBP1 were downregulated in the treated xenografts compared with the control xenografts, indicating that target inhibition was achieved; however, no differences in Ki67 or cleaved caspase 3 immunostaining were noted ().
Figure 2. Daily administration of BGT226 reduces tumor volume and blocks PI3K/mTOR signaling and activates insulin-like growth factor 1 receptor (IGF1R) in vivo. A, Hairless severe combined immunodeficient mice harboring RIS-819.1 xenografts were treated with either vehicle (n = 6) or BGT226 (10 mg/kg or 15 mg/kg, n = 7 for both groups) daily via oral gavage; tumor volumes were measured three times per week. The red arrow indicates the change in the treatment schedule for the 15-mg/kg BGT226 group (from 7 days/week to 5 days/week). Values displayed are the mean volumes ± the standard error of the mean (*p < 0.05). B, Representative photographs (magnification, 200 ×) of immunohistochemical analysis performed on RIS-819.1 xenografts from vehicle- and BGT226-treated mice for markers of proliferation (Ki67), of apoptosis (cleaved caspase 3 [CC3]), and of PI3K/mTOR activity (pAKT, pS6RP, and p4EBP1). H&E: hematoxylin and eosin stain. C, Detection of IGF1R activation (phosphorylated IGF1R [pIGF1R]) in vehicle and BGT226-treated xenografts via immunohistochemical analysis (upper panel; magnification, 200 ×) and western blot analysis (lower panel).
![Figure 2. Daily administration of BGT226 reduces tumor volume and blocks PI3K/mTOR signaling and activates insulin-like growth factor 1 receptor (IGF1R) in vivo. A, Hairless severe combined immunodeficient mice harboring RIS-819.1 xenografts were treated with either vehicle (n = 6) or BGT226 (10 mg/kg or 15 mg/kg, n = 7 for both groups) daily via oral gavage; tumor volumes were measured three times per week. The red arrow indicates the change in the treatment schedule for the 15-mg/kg BGT226 group (from 7 days/week to 5 days/week). Values displayed are the mean volumes ± the standard error of the mean (*p < 0.05). B, Representative photographs (magnification, 200 ×) of immunohistochemical analysis performed on RIS-819.1 xenografts from vehicle- and BGT226-treated mice for markers of proliferation (Ki67), of apoptosis (cleaved caspase 3 [CC3]), and of PI3K/mTOR activity (pAKT, pS6RP, and p4EBP1). H&E: hematoxylin and eosin stain. C, Detection of IGF1R activation (phosphorylated IGF1R [pIGF1R]) in vehicle and BGT226-treated xenografts via immunohistochemical analysis (upper panel; magnification, 200 ×) and western blot analysis (lower panel).](/cms/asset/05d419da-3e25-42e1-b3da-b26f62bdd1af/kcbt_a_1373230_f0002_oc.gif)
Previous studies have demonstrated that IGF1R is upregulated in response to targeted inhibition of the PI3K/mTOR pathway in other cancer types.Citation20,21 We detected increased levels of pIGF1R in BGT226-treated xenografts via both immunohistochemical analysis and immunoblotting (). The same phenomenon was seen in vitro via western blot analysis of whole cell lysates harvested from UPS-186 and RIS-819.1 cells treated with BGT226 for 2 or 96 hours (, Supplementary Fig. S1). This finding is specific for pIGF1R and not for phosphorylated insulin receptor as there was no increase in the phosphorylated insulin receptor signal according to western blot analysis of whole cell lysates harvested from UPS-186 and RIS-819.1 cells treated with BGT226 for 2 or 96 hours (Supp Fig 1).
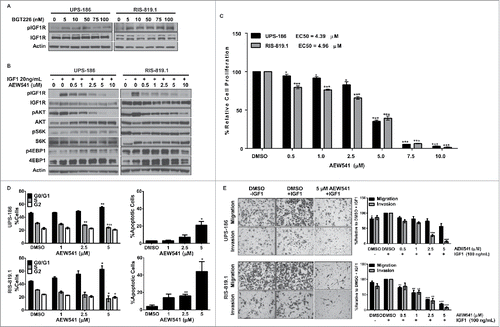
Figure 3. IGF1R activation in response to BGT226-mediated PI3K/mTOR blockade is attenuated by AEW541. A, Detection of phosphorylated IGF1R (pIGF1R) and total IGF1R in undifferentiated pleomorphic sarcoma (UPS) cell lines incubated with BGT226 for 2 hours. B, UPS-186 and RIS-819.1 cells pre-treated with increasing concentrations of AEW541 and then stimulated with IGF1 (20 ng/mL) were subjected to western blot analysis. C, UPS cell proliferation decreased in the presence of AEW541 + IGF1 (20 ng/mL) relative to controls treated with dimethyl sulfoxide (DMSO). D, AEW541 treatment caused cells to accumulate in the G1 phase of the cell cycle and induced apoptosis. DMSO, control in the presence of 20ng/mL IGF1. E, Incubation with AEW541 for 16 hours reduced UPS cell migration and invasion. Representative images of cells treated with DMSO, DMSO + IGF1 (100 ng/mL), or AEW541 (5 µM) + IGF1 (100 ng/mL; magnification: 200 ×). The graphical representations of the data are the means of triplicate experiments; error bars are the standard errors of the mean. *p < 0.05; **p < 0.01; ***p < 0.001.
IGF1R inhibitor AEW541 blocks UPS tumor-supportive processes
Small molecule inhibitors against IGF1R also may target the highly homologous insulin receptor. Therefore, we selected the small molecule inhibitor AEW541 to block IGF1R activation following BGT226 treatment because AEW541 displays a higher affinity for IGF1R than insulin receptor in cellular assays.Citation32 To evaluate the ability of AEW541 to block IGF1R activation, we incubated UPS-186 and RIS-819.1 cells with increasing micromolar concentrations of AEW541 and subjected the cells to acute exposure to recombinant IGF1. Western blot analysis results demonstrated that AEW541 reduced IGF1R activation at all concentrations, with the highest level of inhibition achieved at concentrations greater than 1 µM (). Although AEW541 downregulated pAKT in both cell lines, it did not strongly affect the activation status of S6K or 4EBP1, suggesting that IGF1R is not solely responsible for PI3K/mTOR pathway activation in UPS.
IGF1R can regulate cell growth and proliferation through PI3K/mTOR and/or mitogen-activated protein kinase pathway activationCitation33; accordingly, inhibition of the receptor via AEW541 elicited strong antiproliferative effects in the UPS cell lines (EC50 values: UPS-186, 4.39 µM; RIS-819.1, 4.96 µM) resulting from a significant increase of cells in G1 phase, a concomitant decrease of cells in S phase, and the induction of apoptosis in both cell lines ( and ). Recombinant IGF1 was used as an additional chemoattractant in these assays to assess the ability of AEW541 to prevent IGF1R activation and downstream signaling in the presence of ligand. As with BGT226 treatment, a larger proportion of apoptotic cells was detected in RIS-819.1 samples than in UPS-186 samples.
Because activation of IGF1R has been linked to tumor invasion and metastasis, we investigated the inhibitory effects of AEW541 on IGF1-mediated cell migration and invasion using modified Boyden chamber assays with both UPS-186 and RIS-819.1 cells.Citation34 The sensitivity of RIS-819.1 cells to AEW541 was further reflected by the significant dose-dependent decline in both migration and invasion after treatment (). In contrast, the only significant effects observed in UPS-186 cells were decreases in invasion with AEW541 concentrations greater than 2.5 µM. Taken together, these results demonstrate that AEW541 successfully attenuated IGF1R activation even in the presence of exogenous ligand and exerted antitumor effects in vitro in both cell lines. Although RIS-819.1 cells were more sensitive to drug treatment than UPS-186 cells, our data suggest that AEW541 could be used to prevent IGF1R activation following BGT226 treatment in both cell lines.
Co-treatment and antitumor effects in vitro
To investigate potential synergistic drug interactions, we treated UPS-186 and RIS-819.1 cells with fixed concentrations of BGT226 (range, 0–10 nM) and/or AEW541 (range, 0–2.5 µM) for 96 hours and calculated the combination index values (Fig. S4). Low doses of AEW541 and BGT226 were used (1/2 of EC50) to determine the synergy using combination therapy.Citation35-37 The combinations were determined to be antagonistic from the Chou-Talalay method.Citation38 They may yield the greatest drug effects, but according to the calculations, it is categorized as an antagonistic interaction. We chose drug combinations that were, according to the calculations performed, synergistic by Chou-Talalay cutoffs. According to the combination index values, synergistic interactions occurred in 80% of the drug combinations in UPS-186 cells and in 60% of drug combinations in RIS-819.1 cells; however, the corresponding drug effects for most of the synergistic drug combinations were not substantial, with less than 50% cell death after treatment with AEW541 and BGT226 concentrations less than their EC50 values. Treatment with synergistic concentrations of AEW541 and BGT226 minimally affected cell-cycle distribution and apoptosis levels in UPS-186 and RIS-819.1 samples, further confirming the lack of antiproliferative effects (Supplementary Fig. S3A).
Figure 4. BGT226 and AEW541 act synergistically to block PI3K/mTOR signaling and IGF1R activation in vitro. A, Combination index (CI) values (top) were calculated using CompuSyn software, and drug effect percentages ([1 – relative cell proliferation] × 100%; bottom) were obtained for undifferentiated pleomorphic sarcoma (UPS) cell lines UPS-186 and RIS-819.1 using MTS results in triplicate. B, Effects on IGF1R and effector molecules of PI3K/mTOR signaling in the same cells were determined by western blot analysis following co-treatment with AEW541 and BGT226 and subsequent stimulation with recombinant IGF1.
![Figure 4. BGT226 and AEW541 act synergistically to block PI3K/mTOR signaling and IGF1R activation in vitro. A, Combination index (CI) values (top) were calculated using CompuSyn software, and drug effect percentages ([1 – relative cell proliferation] × 100%; bottom) were obtained for undifferentiated pleomorphic sarcoma (UPS) cell lines UPS-186 and RIS-819.1 using MTS results in triplicate. B, Effects on IGF1R and effector molecules of PI3K/mTOR signaling in the same cells were determined by western blot analysis following co-treatment with AEW541 and BGT226 and subsequent stimulation with recombinant IGF1.](/cms/asset/cff614e5-144a-4267-9450-612d68ac57b1/kcbt_a_1373230_f0004_oc.gif)
Despite the lack of antiproliferative effects seen in vitro, synergistic drug combinations prevented IGF1-mediated PI3K/mTOR signaling cascade activation downstream of IGF1R more effectively than either drug alone (). Furthermore, combination treatment did not alter the phosphorylation status of mitogen-activated protein kinase pathway components MAP2K1/2 and EPHB2, suggesting that this signaling cascade is not activated as a compensatory response to PI3K/mTOR and IGF1R blockade (Supplementary Fig. S3B).
Combination therapy inhibits the rate of tumor growth in vivo
Given the negative effect on PI3K/mTOR signal transduction from combination treatment in vitro, we evaluated the effects of combined inhibition of PI3K/mTOR and IGF1R in RIS-819.1 xenografts. Mice treated daily with either BGT226 or AEW541 had tumor volumes comparable to those of the control group (). Strikingly, the combination group had significantly lower endpoint xenograft volumes (nearly 85% lower) than the control group (p = 0.0202) and 80% than the single-agent groups (AEW541: p = 0.00689; BGT226: p = 0.00541); tumor weight also was significantly lower in the combination group than in the control and single-agent groups (Supplementary Fig. S2B). One mouse in the combination group had complete regression of its tumor within the first 2 weeks of treatment, while stable disease was achieved in the 5 remaining animals within the combination treatment group. Immunohistochemical analysis revealed that AEW541 and BGT226 co-treatment moderately reduced cell proliferation as demonstrated by decreased Ki67 staining compared with vehicle or single-agent therapy but had little effect on apoptosis as determined by cleaved caspase 3 staining (). To rule out the induction of senescence with combination therapy, we performed β-galactosidase staining. Xenografts from mice in the combination group showed no induction of senescence (Supplementary Fig. S4). PI3K/mTOR signaling was more strongly inhibited by the combination treatment than by other treatments, as shown by the downregulation of pAKT, pS6RP, and p4EBP1; furthermore, AEW541 prevented BGT226-associated IGF1R activation in the combination group.
Figure 5. Combined inhibition of PI3K/mTOR and IGF1R signaling results in stable disease in vivo. A, Tumor volumes of RIS-819.1 xenografts treated with vehicle (n = 7), BGT226 (10 mg/kg, n = 7), AEW541 (40 mg/kg, n = 6), or a combination of BGT226 and AEW541 (n = 6) daily (5 days/week for approximately 3 weeks) are presented as mean ± the standard error of the mean. B, Representative images show control and inhibitor-treated xenografts that were subjected to immunohistochemical analysis for protein markers of cell proliferation (Ki67), apoptotic cell death (cleaved caspase 3 [CC3]), and target inhibition (pAKT, pS6RP, p4EBP1, phosphorylated insulin-like growth factor 1 receptor [pIGF1R], p27, and CD31; magnification, 200 ×). H&E: hematoxylin and eosin stain. *p < 0.05; **p < 0.01.
![Figure 5. Combined inhibition of PI3K/mTOR and IGF1R signaling results in stable disease in vivo. A, Tumor volumes of RIS-819.1 xenografts treated with vehicle (n = 7), BGT226 (10 mg/kg, n = 7), AEW541 (40 mg/kg, n = 6), or a combination of BGT226 and AEW541 (n = 6) daily (5 days/week for approximately 3 weeks) are presented as mean ± the standard error of the mean. B, Representative images show control and inhibitor-treated xenografts that were subjected to immunohistochemical analysis for protein markers of cell proliferation (Ki67), apoptotic cell death (cleaved caspase 3 [CC3]), and target inhibition (pAKT, pS6RP, p4EBP1, phosphorylated insulin-like growth factor 1 receptor [pIGF1R], p27, and CD31; magnification, 200 ×). H&E: hematoxylin and eosin stain. *p < 0.05; **p < 0.01.](/cms/asset/79c1100d-ace4-4322-a239-cedd66058fc0/kcbt_a_1373230_f0005_oc.gif)
Synergistic effects of BGT226 and AEW541 on cell motility
Combination treatment did not significantly alter cell proliferation; however, the substantial antitumor effects of co-targeting PI3K, mTOR, and IGF1R in vivo suggested that combination treatment may affect other tumor-supporting processes such as cell migration and invasion. The addition of IGF1 as a chemoattractant enhanced the migratory and invasive capacities of both UPS-186 and RIS-819.1 cells in modified Boyden chambers (). IGF1 is used as a chemoattractant to draw tumor cells from the upper chamber to the lower chamber. The number of cells able to migrate or invade increased when IGF1 was used as a chemoattractant and allowed us to observe the effects of inhibitor treatment in a larger window and allowed for us to examine the effects of IGF1-mediated cell migration. It is surprising that BGT226-treated cells migrated and invaded as previous experiments demonstrated that inhibition of the PI3K/mTOR pathway sufficiently hindered UPS cell migration and invasion. However, IGF1R activation following BGT226 treatment may positively influence cell migration and invasion through alternate downstream pathways. AEW541 treatment alone decreased migration and invasion by nearly 20% in both cell lines. Surprisingly, BGT226-treated cells migrated and invaded at levels similar to or exceeding those of the controls, suggesting that IGF1R activation may override the inhibitory effects of BGT226. In UPS-186 samples, co-treatment with AEW541 and BGT226 reduced cell migration and invasion by approximately 50% and 75%, respectively, compared with the control samples treated with DMSO + IGF1. Similarly, we observed a significant decrease in both migration and invasion (approximately 60% and 80%, compared with the respective controls) in combination-treated RIS-819.1 samples.
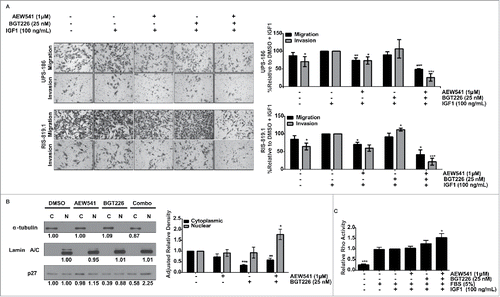
Figure 6. Reduction of migration and invasion after co-treatment is associated with localization of p27 to the nucleus and increased Ras homolog family member A (RhoA) activity. A, A combination of AEW541 and BGT226 reduced undifferentiated pleomorphic sarcoma (UPS) cell migration and invasion according to modified Boyden chamber assay results. B, p27 localization was determined by nuclear (N) and cytoplasmic (C) fractionation of cell lysates (right panel) cells were subjected to immunoblot analysis (left panel), and relative p27 protein levels were calculated using densitometry using α-tublin as the cytoplasmic loading control and lamin A/C as the nuclear loading control. Fraction purity was assessed by α-tubulin (cytoplasmic) and lamin A/C (nuclear). C, The effect of the combination treatment on RhoA activity was assayed. RhoA activity relative to the fetal bovine serum/insulin-like growth factor 1 receptor (FBS/IGF1)-stimulated control is displayed. *p < 0.05; **p < 0.01; ***p < 0.001.
Combination treatment alters p27 subcellular localization
Nuclear p27Kip1 (p27) is a key regulator of the G1/S phase transition in the cell cycle; however, cytoplasmic localization of p27 can promote cell motility through the interaction with and inhibition of the small GTPase RhoA.Citation39 Aberrant PI3K/mTOR signaling can facilitate the cytoplasmic sequestration of p27 through AKT-mediated phosphorylation at T157 and T198, which blocks the nuclear import of p27.Citation40-42 We subjected RIS-819.1 cells to nuclear and cytoplasmic fractionation and to western blot analysis after single-agent or combination treatment to determine the effects of combination treatment on the subcellular localization of p27. p27 was equally distributed in the cytoplasm and the nucleus in DMSO-treated cells (). BGT226 decreased cytoplasmic p27 levels, but nuclear p27 levels were not altered; AEW541 did not significantly affect cytoplasmic or nuclear p27 levels compared with control cells. Combination treatment with AEW541 and BGT226, however, effectively reduced cytoplasmic p27 levels and simultaneously increased nuclear p27 levels. To confirm the results of the fractionation experiment, immunofluorescence for p27 was performed in UPS-186 and RIS819.1 cells that were subjected to single agent or combination treatment. In RIS-819.1 cells we also observed a reduction of the cytoplasmic levels of p27 after combination therapy (Supplementary Fig. S5). Subsequently, we confirmed these results by IHC in the combination animal experiment (). We determined that in the combination therapy, there is decreased signal for p27 when compared to either single agent or vehicle control. To rule out the possibility that the combination effect was a consequence of the anti-angiogenic effects of combination IGF1R/TOR inhibition, we examined the angiogenesis marker CD31 by IHC in these tumors (). In the combination IGF1R/mTOR inhibition we observed similar levels of CD31 in vehicle or single agent therapy; however when the combination therapy is examined there is an observed decrease in the size of the vascularization with no change in the amount of CD31 signal that is observed. To determine the potential downstream effect on RhoA, we used a colorimetric assay to evaluate RhoA activity in RIS-819.1 cells treated with BGT226 and AEW541 alone and in combination. Cells cultured in low-serum conditions exhibited low endogenous RhoA activity, which increased after stimulation with FBS with or without IGF1 (). Combination treatment resulted in a significant 50% increase in RhoA activity, but treatment with either single agent had no significant effect. These data suggest that PI3K/mTOR signaling contributes to UPS cell migration and invasion through the promotion of p27 cytoplasmic localization, further studies will need to be performed to determine if this increased RhoA activity is responsible for the reduced migration. Co-targeting PI3K, mTOR, and IGF1R reduced cytoplasmic p27 while simultaneously increasing nuclear p27 levels.
Discussion
In this study, we investigated the PI3K/mTOR signaling cascade as a target for anti-UPS therapy through pharmacologic pathway blockade using the dual PI3K/mTOR inhibitor BGT226 in vitro and in vivo. In both cell lines tested, BGT226 exhibited strong antiproliferative effects at low nanomolar concentrations because of cell-cycle arrest in G1 phase and the induction of apoptosis; these findings echo the effects of BGT226 in other model systems.Citation43-48 We also found that BGT226 resulted in smaller xenografts in vivo; however, cell proliferation and apoptosis (measured by Ki67 and cleaved caspase 3 staining, respectively) were not altered in treated tumors. Because control and treated xenografts were subjected to immunohistochemical analysis after the mice were euthanized, it is possible that treatment may merely delay tumor growth until compensatory tumor-supportive protumorigenic pathways are activated, which may explain why no negative effects on cell viability were observed.
Although the combined inhibition of PI3K and mTOR catalytic activity by BGT226 suppressed pathway activity and prevented the compensatory increase in pAKT observed with single-agent mTOR inhibitors,Citation16 IGF1R was activated in response to BGT226 treatment in both UPS cell lines and xenografts. Indeed, upregulation of IGF1R protein and activity has been reported as a mechanism of adaptive resistance to PI3K/mTOR inhibition in matrix-attached ovarian cancer cells.Citation21 Chandarlapaty et al reported an increase in the expression and activation of several receptor tyrosine kinases following AKT inhibition, partially owing to the de-repression of FOXO transcriptional activity.Citation20 Although we did not quantify the level of IGF1R transcription, we did notice a slight increase in total IGF1R protein after 96 hours of BGT226 treatment; however, IGF1R activation measured by pIGR1R increased after 2 hours of treatment, indicating that this response may be independent of increased total protein expression in UPS.
Synergistic drug interaction was found at several sub-EC50 concentrations of BGT226 and AEW541; however, these combinations did not elicit substantial antiproliferative effects in vitro despite improved suppression of PI3K/mTOR signaling. Similarly, Wander and colleagues found that inhibition of the PI3K/mTOR pathway using a small molecule inhibitor did not greatly affect the proliferation of metastatic breast cancer cells but nonetheless strongly downregulated migration and invasion through the reduction of cytoplasmic p27 levels.Citation49 Another report demonstrated that following phosphorylation by RSK1, a serine/threonine kinase downstream of PI3K/mTOR, cytoplasmic p27 promoted cell motility by inhibiting RhoA.Citation50 Inhibition of RhoA blocks RhoA-dependent ROCK1 activation and prevents downstream actin remodeling through the ROCK1/LIMK/cofilin pathway, thus reducing cell motility.Citation39,51-53 In our study, combination treatment strongly decreased cell migration and invasion, corresponding with a decrease in cytoplasmic p27 levels and increased RhoA activity. We concluded that this reduction was due in part to a decrease in cytoplasmic p27 and therefore an increase in RhoA activity. Cytoplasmic p27 has also been linked to the disruption of actin fiber stability, possibly also leading to cell motility. Although cytoplasmic p27 levels were significantly reduced after BGT226 treatment, cell motility was not inhibited (). This was most likely due to the fact that cells subjected to fractionation were not stimulated with IGF1 (), possibly increasing the levels of cytoplasmic p27 in single agent–treated cells. Furthermore, combination treatment resulted in an influx of nuclear p27, which may potentially prohibit G1/S checkpoint transition and thus be responsible for the observed reduction in tumor cell proliferation. In Beeson et al, the reduced RhoA activity leads to reduced cell motility; however, in our system we observe increased RhoA activity, but decreased cell motility. UPS cells may have increased motility potentially because of high cytoplasmic levels of p27. It has been observed in fibroblasts that there is high RhoA activity and decreased motility.Citation54 The increased RhoA activity in our system needs further investigation.
Despite the lack of antiproliferative effects in vitro, the combination of BGT226 and AEW541 drastically reduced tumor volume and weight compared with the control or single-agent treatment owing in part to superior target inhibition. Collection of xenograft samples at earlier time points could determine whether the effects of initial therapeutic interventions on cell proliferation or apoptosis are attenuated, as resistance to therapy emerges over time. Combined inhibition of IGF1R and PI3K/mTOR may attenuate invasion and angiogenesis, slowing tumor growth and preventing disease progression. Although one combination-treated mouse displayed complete tumor regression within 2 weeks of treatment, a slight increase in growth was recorded for the remaining xenografts in this group. Additional in vivo survival studies could determine whether long-term co-treatment of UPS xenografts with BGT226 and AEW541 would improve survival times owing to disease stabilization and/or tumor regression or whether other compensatory mechanisms would arise.
Despite the substantial preclinical data supporting IGF1R as a potential therapeutic target in multiple malignancies and early clinical trial results showing response in some patients, large phase II/III trials have shown a lack of efficacy of IGF1R single-agent therapy in unselected patient populations.Citation55 Targeted blockade of IGF1R can induce adaptive responses to therapy, such as increased insulin receptor activation or dysregulated endocrine signaling, which could enhance oncogenic signaling and prove detrimental to patient outcome.Citation56 Combinations of IGF1R monoclonal antibodies and small molecule inhibitors against mTOR have demonstrated some clinical activity in cohorts of patients with sarcoma, including those diagnosed with UPS.Citation57,58 Targeted PI3K inhibition, both as an additional single agent or as a dual inhibitor therapy, could enhance pathway suppression and reduce other oncogenic signaling pathways owing to extensive pathway crosstalk.
Identifying predictive biomarkers is key to selecting patient populations that could benefit from targeted therapies. We found that single-agent PI3K/mTOR or IGF1R inhibition induced higher rates of apoptosis in the RA-UPS cell line RIS-819.1 than in the sporadic cell line UPS-186. RA-UPSs are far more aggressive and result in dramatically shorter patient survival than sporadic UPSs.Citation59 This difference in biological behavior has yet to be linked to molecular variation; however, unique expression of some markers, such as IGF1R and/or effectors of the PI3K/mTOR pathway, could be exploited as predictive biomarkers to identify patient populations that could benefit from specific targeted agents. Insulin-like growth factor 2 (IGF2) may be an excellent candidate biomarker, as Rüping et al recently demonstrated that high IGF2 expression levels were detected in most UPS samples and corresponded to tumor progression.Citation60 It is possible that, with proper patient selection, combination therapy could enhance the antitumor activity of targeted PI3K/mTOR and IGF1R inhibition, while minimizing or eliminating compensatory signaling.
Overall, our data suggest that combined targeted inhibition of the PI3K/mTOR pathway and IGF1R is a promising therapeutic strategy for patients with UPS. Further studies are necessary to investigate the effect of combination treatment on long-term survival and metastatic disease; members of the IGF1R/PI3K/mTOR signaling axis also should be investigated as predictive biomarkers for selecting patients with UPS and for predicting survival outcomes.
Materials and methods
UPS cell isolation and tissue culture
We isolated UPS cells from patient tumor samples after obtaining approval from the Institutional Review Board at The University of Texas MD Anderson Cancer Center and patients' written informed consent. Surgical specimens were obtained from five patients who were treated at MD Anderson between July 2010 and November 2011. RIS-DL-620, UPS-186, and UPS-DL-511 cells were isolated as previously described,Citation61 and RIS-819.1 and UPS-DL-271.1 cells were isolated from patient surgical specimens that were serially passaged in hairless severe combined immunodeficient (SCID) mice. Two of the tumors were associated with previous radiation (RIS-DL-620 and RIS-819.1) and the other three were sporadic. UPS cell strains and cell lines were authenticated by short tandem repeat (STR) DNA fingerprinting using the Promega PowerPlex 16 High Sensitivity STR Kit (cat# DC2100). Additional information, including cell source details, STR results, and culture conditions, is described in the Supplementary Information and Table S1. Bone-derived human mesenchymal stem cells (hMSCs) were purchased from PromoCell (cat# C-12974) and maintained in Mesenchymal Stem Cell Growth Medium (PromoCell, cat# C-28010). After initial results, the RIS-819.1 and UPS-186 cells were chosen as representative of radiation-associated UPS (RA-UPS) and sporadic UPS, respectively, for further studies.
Drugs and vehicles
Dual PI3K/mTOR inhibitor BGT226 (NVP-BGT226, cat# S2749)Citation62 and small molecule IGF1R inhibitor AEW541 (NVP-AEW541, cat# S1034)Citation63 were purchased from Selleck Chemicals. Both drugs were suspended in dimethyl sulfoxide (DMSO) for in vitro applications. For in vivo use, BGT226 was suspended in N-methyl-2-pyrrolidone (NMP). Immediately before administration, the NMP/BGT226 stock solution was diluted in PEG300 (10% NMP/BGT226 plus 90% PEG300). AEW541 was suspended in L(+)-tartaric acid (25 mM) for use in mice.
Cellular assays
Detailed descriptions of in vitro cellular assays are available in the Supplementary Information. Briefly, cell proliferation was assessed by MTS assays, as previously described.Citation64 Drug synergy was determined for drug combinations by the Chou-Talalay method using CompuSyn software.Citation38 Standard protocols for western blot analyses were followed.Citation64 Cell-cycle distribution and apoptosis were assessed by fluorescence-activated cell sorting (FACS) analysis. Migration and invasion were evaluated using modified Boyden chamber assays, as previously described.Citation61 Nuclear and cytoplasmic fractionation was performed per the manufacturer's instructions (Pierce NE-PER™ Nuclear and Cytoplasmic Extraction Reagents Cat# 78835). Ras homolog family member A (RhoA) activity was assessed using a colorimetric kit according the manufacturer's instructions (Cytoskeleton, Inc. G-LISA RhoA Activation Assay Cat# BK124)
In vivo xenograft studies
All animal procedures were approved by the MD Anderson Institutional Animal Care and Use Committee (IACUC) at UTMDACC. Six-week old female hairless SCID mice (Charles River Laboratory, Strain Code 488) were implanted with pieces of RIS-819.1 tumor (approximately 5 mm in diameter) in the flank. UPS-186 was not tested in vivo as its growth in SCID mice is not reproducible. Therapeutic interventions began when xenografts reached an average volume of 100 mm3 in each group. Vehicle, BGT226, and/or AEW541 were administered daily (5 d/week or 7 d/week; indicated in the figure legends) via oral gavage. When tumors in the control group reached a maximum volume of 1500 mm3, all animals were euthanized following IACUC guidelines. Immunohistochemical analysis of xenografts is detailed in the Supplementary Information.
Statistical analysis
Data are presented as means ± the standard error of the mean. Each experiment was repeated in triplicate unless otherwise indicated in the figure legends. The Student's t-test was used to evaluate the statistical significance of differences between groups. P values < 0.05 were considered statistically significant.
Disclosure of potential conflicts of interest
The authors do not have any conflicts of interest to disclose.
Financial support
We gratefully acknowledge the support of NIH/NCI K08CA160443 (to KET), the Sally M. Kingsbury Sarcoma Research Foundation (supporting CDM), the Jay Vernon Jackson Fund (supporting SML and XM), Michael and Mary Kay Poulos (supporting KLW), Friends of Sarcoma Fund (to KET), the Johnnie M Lowe Sarcoma Research Endowment (to KET) and NIH/NCI K08CA160443 (to KET). Short tandem repeat DNA fingerprinting was done by the Characterized Cell Line Core (funded by NCI P30CA016672). None of the funding organizations had a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
2017CBT10318R-file002.docx
Download MS Word (22.1 KB)Acknowledgments
We gratefully acknowledge the support of the National Institutes of Health/National Cancer Institute (NIH/NCI Cancer Center Support Grant P30CA016672) and use of the Flow Cytometry and Cellular Imaging Facility. Short tandem repeat DNA fingerprinting was done by the Characterized Cell Line Core (also funded by NCI P30CA016672). We greatly appreciate the support of the Amschwand Sarcoma Cancer Foundation (supporting CDM) and the Sally M. Kingsbury Sarcoma Research Foundation. This research was supported in part by an NIH grant (NIH/NCI K08CA160443 to KET).
References
- Romeo S, Bovee JVMG, Jundt G. Undifferentiated high grade pleomorphic sarcoma. In Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. (eds). WHO Classification of Tumours of Soft Tissue and Bone. Fourth Edition. Lyon: IARC Press; 364-365.
- Fletcher CD. The evolving classification of soft tissue tumours: an update based on the new WHO classification. Histopathology. 2006;48(1):3-12. doi:10.1111/j.1365-2559.2005.02284.x
- Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Annals Surg. 2014;260(3):416-21; discussion 21–2. doi:10.1097/SLA.0000000000000869
- Gladdy RA, Qin LX, Moraco N, Edgar MA, Antonescu CR, Alektiar KM, Brennan MF, Singer S. Do radiation-associated soft tissue sarcomas have the same prognosis as sporadic soft tissue sarcomas? J Clin Oncol. 2010;28(12):2064-9. doi:10.1200/JCO.2009.25.1728
- Riad S, Biau D, Holt GE, Werier J, Turcotte RE, Ferguson PC, Griffin AM, Dickie CI, Chung PW, Catton C, et al. The clinical and functional outcome for patients with radiation-induced soft tissue sarcoma. Cancer. 2012;118(10):2682-92. doi:10.1002/cncr.26543
- Goldblum JR. An approach to pleomorphic sarcomas: can we subclassify, and does it matter? Mod Pathol. 2014;27(Suppl 1):S39-46. doi:10.1038/modpathol.2013.174
- Fletcher CDM, Unni KK, Mertens F. Pathology and Genetics of Soft Tissue and Bone: World Health Organization Classification of Tumors. Fletcher CDM, editor. Lyon, France: IARC Press; 2002.
- Donato Di Paola E, Nielsen OS, Tissue ES, Bone Sarcoma G. The EORTC soft tissue and bone sarcoma group. Eur Organisation for Res Treatment Cancer. 2002;38 Suppl 4:S138-41.
- Nascimento AF, Raut CP. Diagnosis and management of pleomorphic sarcomas (so-called “MFH”) in adults. J Surg Oncol. 2008;97(4):330-9. doi:10.1002/jso.20972
- Maki RG, Wathen JK, Patel SR, Priebat DA, Okuno SH, Samuels B, Fanucchi M, Harmon DC, Schuetze SM, Reinke D, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25(19):2755-63. doi:10.1200/JCO.2006.10.4117
- Wardelmann E, Schildhaus HU, Merkelbach-Bruse S, Hartmann W, Reichardt P, Hohenberger P, Büttner R. Soft tissue sarcoma: from molecular diagnosis to selection of treatment. Pathological diagnosis of soft tissue sarcoma amid molecular biology and targeted therapies. Annals Oncol. 2010;21 Suppl 7:vii265-9. doi:10.1093/annonc/mdq381
- Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143-53. doi:10.1038/nrclinonc.2013.10
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science 2003;302(5651):1704-9. doi:10.1126/science.1092053
- Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2014;15(1):7-24. doi:10.1038/nrc3860
- Garcia-Echeverria C. Allosteric and ATP-competitive kinase inhibitors of mTOR for cancer treatment. Bioorganic Med Chem Lett. 2010;20(15):4308-12. doi:10.1016/j.bmcl.2010.05.099
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500-8. doi:10.1158/0008-5472.CAN-05-2925
- Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30(22):2547-57. doi:10.1038/onc.2010.626
- Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig-Mueller N, Colinge J, Duernberger G, Nijman SM. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7(11):787-93. doi:10.1038/nchembio.695
- Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22(6):796-811. doi:10.1016/j.ccr.2012.10.023
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58-71. doi:10.1016/j.ccr.2010.10.031
- Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, Brugge JS. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21(2):227-39. doi:10.1016/j.ccr.2011.12.024
- Tomita Y, Morooka T, Hoshida Y, Zhang B, Qiu Y, Nakamichi I, Hamada K, Ueda T, Naka N, Kudawara I, et al. Prognostic significance of activated AKT expression in soft-tissue sarcoma. Clin Cancer Res. 2006;12(10):3070-7. doi:10.1158/1078-0432.CCR-05-1732
- Valkov A, Kilvaer TK, Sorbye SW, Donnem T, Smeland E, Bremnes RM, Busund LT. The prognostic impact of Akt isoforms, PI3K and PTEN related to female steroid hormone receptors in soft tissue sarcomas. J Translational Med. 2011;9:200. doi:10.1186/1479-5876-9-200
- Dobashi Y, Suzuki S, Sato E, Hamada Y, Yanagawa T, Ooi A. EGFR-dependent and independent activation of Akt/mTOR cascade in bone and soft tissue tumors. Mod Pathol. 2009;22(10):1328-40. doi:10.1038/modpathol.2009.104
- Lahat G, Zhang P, Zhu QS, Torres K, Ghadimi M, Smith KD, Wang WL, Lazar AJ, Lev D. The expression of c-Met pathway components in unclassified pleomorphic sarcoma/malignant fibrous histiocytoma (UPS/MFH): a tissue microarray study. Histopathology. 2011;59(3):556-61. doi:10.1111/j.1365-2559.2011.03946.x
- Matushansky I, Hernando E, Socci ND, Mills JE, Matos TA, Edgar MA, Singer S, Maki RG, Cordon-Cardo C. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J Clin Invest. 2007;117(11):3248-57. doi:10.1172/JCI31377
- Butler DE, Marlein C, Walker HF, Frame FM, Mann VM, Simms MS, et al. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget. 2017;8(34):56698-56713. doi:10.18632/oncotarget.18082
- Ning C, Liang M, Liu S, Wang G, Edwards H, Xia Y, Polin L, Dyson G, Taub JW, Mohammad RM, et al. Targeting ERK enhances the cytotoxic effect of the novel PI3K and mTOR dual inhibitor VS-5584 in preclinical models of pancreatic cancer. Oncotarget. 2017;8(27):44295-44311. doi:10.18632/oncotarget.17869
- Rewcastle GW, Kolekar S, Buchanan CM, Gamage SA, Giddens AC, Tsang KY, Kendall JD, Singh R, Lee WJ, Smith GC, et al. Biological characterization of SN32976, a selective inhibitor of PI3K and mTOR with preferential activity to PI3Kalpha, in comparison to established pan PI3K inhibitors. Oncotarget. 2017;8(29):47725-47740. doi:10.18632/oncotarget.17730
- Riedl A, Schlederer M, Pudelko K, Stadler M, Walter S, Unterleuthner D, Unger C, Kramer N, Hengstschläger M, Kenner L, et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J Cell Sci. 2017;130(1):203-18. doi:10.1242/jcs.188102
- Kim EK, Yun SJ, Ha JM, Kim YW, Jin IH, Yun J, Shin HK, Song SH, Kim JH, Lee JS, et al. Selective activation of Akt1 by mammalian target of rapamycin complex 2 regulates cancer cell migration, invasion, and metastasis. Oncogene. 2011;30(26):2954-63. doi:10.1038/onc.2011.22
- Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro HG, Cozens R, et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5(3):231-9.
- Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12(3):159-69. doi:10.1038/nrc3215
- Sachdev D, Zhang X, Matise I, Gaillard-Kelly M, Yee D. The type I insulin-like growth factor receptor regulates cancer metastasis independently of primary tumor growth by promoting invasion and survival. Oncogene. 2010;29(2):251-62. doi:10.1038/onc.2009.316
- Ning J, Wakimoto H, Peters C, Martuza RL, Rabkin SD. Rad51 degradation: Role in Oncolytic Virus-Poly(ADP-Ribose) Polymerase Inhibitor Combination Therapy in Glioblastoma. J Natl Cancer Inst. 2017;109(3):1-13. doi:10.1093/jnci/djw229
- Chaib I, Karachaliou N, Pilotto S, Codony Servat J, Cai X, Li X, Drozdowskyj A, Servat CC, Yang J, Hu C, et al. Co-activation of STAT3 and YES-Associated Protein 1 (YAP1) pathway in EGFR-Mutant NSCLC. J Natl Cancer Inst. 2017;109(9):1-12. doi:10.1093/jnci/djx014
- Xie J, Liu JH, Liu H, Liao XZ, Chen Y, Lin MG, Gu YY, Liu TL, Wang DM, Ge H, et al. Tanshinone IIA combined with adriamycin inhibited malignant biological behaviors of NSCLC A549 cell line in a synergistic way. BMC Cancer. 2016;16(1):899. doi:10.1186/s12885-016-2921-x
- Chou T-C, Talalay P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enz. Regul. 1984;22:27-55.
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18(8):862-76. doi:10.1101/gad.1185504
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8(10):1153-60. doi:10.1038/nm761
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8(10):1145-52. doi:10.1038/nm759
- Viglietto G, Motti ML, Bruni P, Melillo RM, D'Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8(10):1136-44. doi:10.1038/nm762
- Baumann P, Schneider L, Mandl-Weber S, Oduncu F, Schmidmaier R. Simultaneous targeting of PI3K and mTOR with NVP-BGT226 is highly effective in multiple myeloma. Anti-Cancer Drugs. 2012;23(1):131-8. doi:10.1097/CAD.0b013e32834c8683
- Chang KY, Tsai SY, Wu CM, Yen CJ, Chuang BF, Chang JY. Novel phosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potent growth-inhibitory activity against human head and neck cancer cells in vitro and in vivo. Clin Cancer Res. 2011;17(22):7116-26. doi:10.1158/1078-0432.CCR-11-0796
- Fokas E, Yoshimura M, Prevo R, Higgins G, Hackl W, Maira SM, Bernhard EJ, McKenna WG, Muschel RJ. NVP-BEZ235 and NVP-BGT226, dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitors, enhance tumor and endothelial cell radiosensitivity. Radiat Oncol. 2012;7:48. doi:10.1186/1748-717X-7-48
- Glienke W, Maute L, Wicht J, Bergmann L. The dual PI3K/mTOR inhibitor NVP-BGT226 induces cell cycle arrest and regulates Survivin gene expression in human pancreatic cancer cell lines. Tumour Biol. 2012;33(3):757-65. doi:10.1007/s13277-011-0290-2
- Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Rasp KH, Illing B, Dohner H, Döhner K, Schittenhelm MM. Cell cycle-dependent activity of the novel dual PI3K-MTORC1/2 inhibitor NVP-BGT226 in acute leukemia. Mol Cancer. 2013;12:46. doi:10.1186/1476-4598-12-46
- Sanchez CG, Ma CX, Crowder RJ, Guintoli T, Phommaly C, Gao F, Lin L, Ellis MJ. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011;13(2):R21. doi:10.1186/bcr2833
- Wander SA, Zhao D, Besser AH, Hong F, Wei J, Ince TA, Milikowski C, Bishopric NH, Minn AJ, Creighton CJ, et al. PI3K/mTOR inhibition can impair tumor invasion and metastasis in vivo despite a lack of antiproliferative action in vitro: implications for targeted therapy. Breast Cancer Res Treatment. 2013;138(2):369-81. doi:10.1007/s10549-012-2389-6
- Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, Smith JA, Slingerland JM. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A. 2009;106(23):9268-73. doi:10.1073/pnas.0805057106
- Lee GE, Yu EY, Cho CH, Lee J, Muller MT, Chung IK. DNA-protein kinase catalytic subunit-interacting protein KIP binds telomerase by interacting with human telomerase reverse transcriptase. J Biol Chem. 2004;279(33):34750-5. doi:10.1074/jbc.M401843200
- Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004;18(8):851-5. doi:10.1101/gad.1205304
- Lee S, Helfman DM. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J Biol Chem. 2004;279(3):1885-91. doi:10.1074/jbc.M306968200
- Zhou H, Kramer RH. Integrin engagement differentially modulates epithelial cell motility by RhoA/ROCK and PAK1. J Biol Chem. 2005;280(11):10624-35. doi:10.1074/jbc.M411900200
- King H, Aleksic T, Haluska P, Macaulay VM. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treatment Rev. 2014;40(9):1096-105. doi:10.1016/j.ctrv.2014.07.004
- Yee D. Insulin-like growth factor receptor inhibitors: baby or the bathwater? J Natl Cancer Inst. 2012;104(13):975-81. doi:10.1093/jnci/djs258
- Chawla SP, Staddon AP, Baker LH, Schuetze SM, Tolcher AW, D'Amato GZ, Blay JY, Mita MM, Sankhala KK, Berk L, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol. 2012;30(1):78-84. doi:10.1200/JCO.2011.35.6329
- Schwartz GK, Tap WD, Qin LX, Livingston MB, Undevia SD, Chmielowski B, Agulnik M, Schuetze SM, Reed DR, Okuno SH, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013;14(4):371-82. doi:10.1016/S1470-2045(13)70049-4
- Dineen SP, Roland CL, Feig R, May C, Zhou S, Demicco E, et al. Radiation-associated undifferentiated Pleomorphic Sarcoma is associated with worse clinical outcomes than Sporadic Lesions. Annals Surgical Oncol. 2015;22(12):3913-20. doi:10.1245/s10434-015-4453-z
- Ruping K, Altendorf-Hofmann A, Chen Y, Kampmann E, Gibis S, Lindner L, Katenkamp D, Petersen I, Knösel T. High IGF2 and FGFR3 are associated with tumour progression in undifferentiated pleomorphic sarcomas, but EGFR and FGFR3 mutations are a rare event. J Cancer Res Clin Oncol. 2014;140(8):1315-22. doi:10.1007/s00432-014-1700-9
- Peng T, Zhang P, Liu J, Nguyen T, Bolshakov S, Belousov R, Young ED, Wang X, Brewer K, López-Terrada DH, et al. An experimental model for the study of well-differentiated and dedifferentiated liposarcoma; deregulation of targetable tyrosine kinase receptors. Lab Invest. 2011;91(3):392-403. doi:10.1038/labinvest.2010.185
- Katanasaka Y, Kodera Y, Yunokawa M, Kitamura Y, Tamura T, Koizumi F. Synergistic anti-tumor effects of a novel phosphatidyl inositol-3 kinase/mammalian target of rapamycin dual inhibitor BGT226 and gefitinib in non-small cell lung cancer cell lines. Cancer Letters. 2014;347(2):196-203. doi:10.1016/j.canlet.2014.02.025
- Stauffer F, Cowan-Jacob SW, Scheufler C, Furet P. Identification of a 5-[3-phenyl-(2-cyclic-ether)-methylether]-4-aminopyrrolo[2,3-d]pyrimidine series of IGF-1R inhibitors. Bioorg Med Chem Lett. 2016;26(8):2065-7. doi:10.1016/j.bmcl.2016.02.074
- Zou CY, Smith KD, Zhu QS, Liu J, McCutcheon IE, Slopis JM, Meric-Bernstam F, Peng Z, Bornmann WG, Mills GB, et al. Dual targeting of AKT and mammalian target of rapamycin: a potential therapeutic approach for malignant peripheral nerve sheath tumor. Mol Cancer Ther. 2009;8(5):1157-68. doi:10.1158/1535-7163.MCT-08-1008