
ABSTRACT
Despite evidence that estrogens and insulin are related to type 1 endometrial carcinoma (EC), their synergistic role has not been analyzed. Here, we investigated how estrogens cooperate with insulin to promote type 1 EC progression. We examined the clinical significance of serum estrogen and insulin levels using type 1 EC patients and control subjects. Univariate and multivariate logistic regression analyses for total, premenopausal, and postmenopausal subjects were performed. Type 1 EC risk was evaluated with respect to estrone, estradiol, and insulin levels based on odds ratios (ORs) using stratified data. Cell growth in vitro and in vivo, effects of insulin and estradiol on apoptosis and cell cycle distribution were measured after estradiol and insulin stimulation. Estrone and insulin concentrations were significantly high in type 1 EC patients and retained positive associations with type 1 EC after adjustment for BMI, WHR, diabetes, and hypertension. The odds ratio was significantly high for type 1 EC patients with higher levels of estrone/estradiol and insulin than for patients with higher levels of either estrone/estradiol or insulin, suggesting that estrogen and insulin play a synergistic role in type 1 EC carcinogenesis and progression. Compared to EC cells and cell-based xenografts treated with estradiol or insulin alone, those treated with estradiol and insulin exhibited stronger stimulation. Estrogen and insulin play synergistic roles in type 1 EC carcinogenesis and progression, extending our understanding of EC risks.
Abbreviations
| CI | = | confidence interval |
| EC | = | endometrial cancer |
| IGF | = | insulin-like growth factor |
| OR | = | odds ratio |
| SHBG | = | sex hormone binding globulin |
Introduction
Endometrial cancer (EC) represents the most common gynecological malignancy in developed countries and its incidence is growing worldwide. In 2016, EC was estimated to be diagnosed in 63,400 patients and to account for more than 21,800 deaths in China,Citation5 exceeding those of the United States.Citation19 Despite these trends, the etiology and pathogenesis of EC remain poorly defined. It is known that an imbalance in estrogen/progesterone is involved in type 1 EC progression,Citation2 as epidemiologic studies have found that estrogens, insulin, and insulin-like growth factors are higher in patients with type 1 EC than in healthy individuals. Indeed, both steroid hormones, such as estrogen, and growth factors, such as insulin-like growth factor (IGF)/insulin, can be major drivers of type 1 EC.
Insulin can regulate the growth and differentiation of cells through its downstream signaling pathways. Notably, it was reported that insulin-binding sites were expressed in the endometrial stroma of women with type 1 EC,Citation18 providing evidence that an excess of insulin signaling could result in endometrial changes with a pro-proliferative change similar to that of unopposed estrogen. Besides, insulin also promotes the development of type 1 EC in less direct ways. Specifically, it has been observed that insulin resistance and compensatory hyperinsulinemia provoke androgen synthesis at the expense of estrogen production.Citation20 Increased free androgens supply more substrate for peripheral estrogen conversion, which is especially dangerous for postmenopausal women. After menopause, the ovaries cease to produce estrogen and progesterone, making peripheral estrogen conversion the main source of estrogen in the circulation.Citation22 Also, Insulin has been reported to inhibit the synthesis of sex hormone binding globulin (SHBG), which tightly binds and regulates the activity of sex hormones.Citation15 Thus, when insulin levels increase due to insulin resistance, this inhibition results in an increase in free sex hormone levels (of both estrogens and androgens) and further stimulates type 1 EC tumorigenesis. As to premenopausal women, increased levels of androgen induce anovulation,Citation6 resulting in insufficient progesterone to counterbalance the proliferation promoting and antiapoptotic effects of estrogen.
On the other hand, ovarian hormones regulate normal human endometrial cell proliferation, regeneration, and function, and are therefore implicated in endometrial carcinogenesis.Citation9 Furthermore, recent evidence suggests that all types of ECs might share common etiological factors, including their response to/stimulation by estrogen and other ovarian steroid hormones.Citation17 Although previous studies have described the individual roles of estrogens,Citation13 progesterone, and insulin in endometrial carcinogenesis, they have largely disregarded the putative synergistic effects of these hormones, especially their crosstalk effects. As sex hormones, estrogens not only regulate normal body growth and maintain female characteristics, but can also function as growth factors that promote endometrial proliferation and malignant transformation.Citation7
Generally, risk factors for type 1 EC include obesity, diabetes, and hypertension, arising from long-term estrogen stimulation without progesterone,Citation1,8,10–12,16,21 and that the common pathophysiological features of these conditions are insulin resistance and hyperinsulinemia.Citation14 Many researchers have reported the coexistence of insulin resistance and hyperinsulinemia in cases of hyper-estrogen status such as infertility, dysfunctional uterine bleeding with anovulation, and late menopause.Citation4,14 These clinical phenomena indicate that estrogen and insulin may play important roles in EC progression.
In the present study, we found that type 1 EC patients exhibited significantly higher serum levels of estradiol and insulin than control subjects. Moreover, the odds ratios for type 1 EC patients with higher levels of estrone/estradiol and insulin were increased significantly, compared to those of patients with higher levels of either estrone/estradiol or insulin alone. We next employed Ishikawa and ECC-1 epithelial adenocarcinoma cell lines to characterize the effects of estradiol and insulin on EC cell proliferation, cell cycle progression, and apoptosis in vitro. Finally, we established EC xenografts into nude mice to analyze the in vivo effects of action of insulin and estrogen in EC.
Materials and methods
Serum sample collection
A total of 510 type 1 EC patients and 510 control subjects were enrolled in this study at Tianjin Medical University General Hospital between 2003 and 2014. Patients were identified among women with first diagnosis of histologically confirmed type 1 EC, none of whom had undergone radiotherapy or chemotherapy prior to surgery. Healthy subjects were selected among women who presented for routine examination in the general physical examination center. None had a history of cancer, and all were age- and nationality-matched to the patients. The study protocol was approved by the Ethics Committee of Tianjin Medical University General Hospital, and informed consent was obtained from each patient and healthy subject enrolled in the study. The clinicopathological features of patients with EC were recorded. Information on obesity, diabetes, hypertension, and family history of cancer was collected from each subject. A single 5-mL blood specimen was collected early in the morning after 6 h of fasting and prior to surgery (for patients), or on the day of regular examination (for healthy subjects). The collected samples were centrifuged for serum separation, and the resulting serum samples were stored at −80°C.
Regents, kits, drugs and antibodies
Human recombinant insulin and dimethyl sulfoxide solutions were purchased from Sigma-Aldrich (St. Louis, MO, USA). Propidium iodide and RNase A were obtained from Dingguo Biotechnology (Beijing, China). The Estrone Radioimmunoassay Kit used to quantify serum levels of estrone was purchased from Diagnostic Systems Laboratories, Inc. (Webster, TX, USA), chemiluminescence reagents used to quantify serum levels of insulin and estradiol were obtained from Siemens Medical Solutions (Malvern, PA, USA), and the In Situ Cell Death Detection Kit used for TUNEL assays was purchased from Roche (Basel, Switzerland). The Enhanced Chemiluminescence Detection Kit was obtained from Pierce Biotechnology (Waltham, MA, USA), and the Annexin V-FITC/PI Apoptosis Detection Kit was purchased from BD Biosciences (San Jose, CA, USA). Estradiol pellets were purchased from Sigma. Antibodies specific to Ki-67 and peroxidase-conjugated goat anti-rabbit IgG secondary antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell lines, and animals
Ishikawa and ECC-1 human EC cell lines were from MD Anderson Cancer Center (TX, USA), and the immunodeficient female BALB/c-nu mice (5–6 weeks old, 12–13 g) used for in vivo experiments [production permit no. SCXK (JING) 2014-0004] were purchased from Beijing HFK Bioscience Co. (Beijing, China). All animal experiments were performed under standard guidelines approved by the State Key Laboratory of Experimental Hematology [license no. SYXK (JIN) 2009-0002], and all procedures were carried out in accordance with the approval of the ethics committee on animal care of Tianjin Medical University. Four mouse groups were utilized in this study with five mice (n = 5) per group.
Serum analysis
Insulin, estrone, and estradiol assays were performed according to the manufacturer's instructions. Experimental results were expressed as the means ± standard deviations (SD) of the results from three independent experiments for each sample.
Cell culture
Cells were grown in Dulbecco's modified Eagle's medium without phenol red (Gibco, Grand Island, NY, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco). Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. Assays were performed as described elsewhere.Citation23,24
BrdU cell proliferation enzyme-linked immunosorbent assay (ELISA) analysis
The effects of insulin and estradiol on cell growth and proliferation were measured using a BrdU Cell Proliferation ELISA, as described previously.Citation23 Briefly, Ishikawa cells and ECC-1 cells were treated with a combination of insulin and estradiol, both at a concentration of 10−8 M, for 24, 48, or 72 h prior to measurement.Citation23
Cell cycle analysis
The cell cycle distribution of Ishikawa cells and ECC-1 cells was evaluated via flow cytometry. Cells (5 × 106) were treated with insulin and/or estradiol for 24, 48, or 72 h, after which they were collected, washed with cold phosphate buffered saline (PBS), and fixed in ice-cold 70% ethanol. The samples were then stored at −20°C for 24 to 48 h. Before analysis, the cells were washed with cold PBS and then suspended in PBS solution containing 0.1% Triton-X and 30 mg/mL DNase-free RNase A (Sigma) for 30 min at room temperature. Propidium iodide was added at a final concentration of 10 μg/mL. The samples were then analyzed using a FACS Calibur instrument (BD Bioscience). Data were processed with BD Bioscience software. Data for at least 10,000 cells were collected for each sample. All experiments were repeated three times.
Annexin V-FITC/PI assay
Apoptosis levels were measured using an Annexin V-FITC/PI Apoptosis Detection Kit, according to the manufacturer's directions and as described previously.Citation23,24 Briefly, Ishikawa cells and ECC-1 cells were treated with a combination of insulin and estradiol, both at a concentration of 10−8 M, for 24, 48, or 72 h. The percentage of apoptotic cells was determined from the number of Annexin V (+)/PI (−) cells relative to the number of Annexin V (−)/PI (−) cells.
Tumor growth assay
Approximately 5 × 106 Ishikawa cells were injected into the dorsal scapular regions of mice in each group to generate EC tumor xenografts. Resultant tumors were measured with a caliper each week (7 days), and tumor volume was calculated using the following formula: volume = [length × (width)2]}/2. After 56 days of observation, the mice were sacrificed and the tumors were dissected and weighed. Other detailed characteristics were detected by immunohistochemistry and TUNEL assay.
Immunohistochemistry
Immunohistochemistry was performed as described elsewhere.Citation21,24 After anti-Ki-67 antibody binding, peroxidase-conjugated goat anti-rabbit antibody IgG was used as a secondary antibody. Tissue sections were then stained with diaminobenzidine (ZSGB-BIO, ZLI-9031), counterstained with hematoxylin (ZSGB-BIO, ZLI-9643), and examined under a light microscope (CKX41; Olympus Corp., Tokyo, Japan).
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining
The xenograft sections were digested by 0.1% trypsin (Sigma) for 3 min. After rinsing twice with PBS for 5 mins each, the sections were incubated with 0.1% TritonX-100 for 2 min followed by incubation with the TUNEL reaction mixture (In Situ Cell Death Detection Kit). The following steps were exactly according to the instruction manual.
Statistical analysis
Comparisons between serum concentrations of estradiol, estrone, and insulin in EC patients and healthy subjects were performed using non-parametric tests. Conditional logistic regression was used to analyze the risk factors for EC. Experimental results are expressed as the means ± SD of the results from at least three independent experiments. Multiple comparisons were analyzed using Wilcoxon's rank test or one-way analysis of variance (ANOVA). Subsequently, a least significant difference test was adopted if variances were equal, and Tamhane's T2 test was used if the variances were unequal. Statistical significance was assumed at P < 0.05. All calculations were performed using SPSS 13.0 software (SPSS Statistics, Inc., Chicago, IL, USA).
Results
Basic characteristics of type 1 EC and control subjects
Here, only the type 1 endometrial cancer was under investigation. The basic characteristics of the study subjects (EC cases and healthy controls), including marker concentrations in serum, are displayed in . Compared with the control subjects, the weight, waist circumference, abdominal circumference, hip circumference, body mass index (BMI), and waist-hip ratio (WHR) were significantly higher in patients with EC (P < 0.01). Additionally, diabetes, hypertension, and family history of cancer were more prevalent among patients with EC than the control subjects (P < 0.001). Conversely, there were no significant differences in age, height, and self-reported nulliparous history between the two groups (P > 0.05). Compared with the control subjects, patients with EC had significantly higher serum concentrations of insulin and estrone (P < 0.001); however, no significant differences in serum estradiol were identified (P > 0.05). Additionally, the clinicopathological characteristics, including histologic subtype, histopathology grade, myometrial invasion, and International Federation of Gynecology and Obstetrics (FIGO) staging, of EC patients are summarized in .
Table 1. Basic characteristics of patients with endometrial cancer and healthy subjects
Serum estrone and insulin concentrations are independent risk factors for EC
Using a univariate logistic model, we detected positive associations between EC risk and serum estrone and insulin levels, BMI, WHR, diabetes, and hypertension (P < 0.001, ). In contrast, there was no correlation between estradiol levels and EC risk (P = 1, ). Per multivariate logistic modeling, estrone [odds ratio (OR), 2.131; 95% confidence interval (CI), 1.604−2.832; P = 0.000] and insulin concentrations (OR, 1.578; 95% CI, 1.171−2.128; P = 0.003) both retained a positive association with EC after adjustment for BMI and serum estradiol, suggesting that estrone and insulin levels represented independent risk factors for EC. Furthermore, multivariate logistic regression analyses revealed that WHR, diabetes, and hypertension remained positively correlated with EC.
Table 2. Odds ratios (OR) for endometrial cancer risk using logistic regression.
Relationship between estradiol, estrone, and insulin levels with EC risk after stratification for BMI, WHR, diabetes, and hypertension
After EC risk factors including BMI, WHR, diabetes, and hypertension were stratified, the relationships between estradiol, estrone, and insulin concentrations and EC risk were analyzed in total, premenopausal and postmenopausal women, respectively (). In postmenopausal subjects, estrone was associated with increased EC risk upon stratification for BMI over 24 kg/m2 (OR, 2.401; 95% CI, 1.640–3.516; P = 0.000), WHR over 0.85 (OR, 2.426; 95% CI, 1.705–3.452; P = 0.000), and for women without diabetes (OR, 2.282; 95% CI, 1.632–3.193; P = 0.000), whereas there was no significant association in subjects with BMI less than 24 kg/m2, WHR less than 0.85, and in postmenopausal women with diabetes (P > 0.05). Insulin was found to increase EC risk in all of the risk factor stratifications (P < 0.05) except in postmenopausal women with diabetes (P > 0.05). Conversely, estradiol was not significantly correlated with EC risk in any of the stratified risk factors. Similar comparisons for estrone and insulin were significant in total group, but not in premenopausal subjects ().
Table 3. Relationship between estradiol, estrone, insulin, and endometrial cancer risk after stratification for BMI, WHR, diabetes, or hypertension using a logistic regression model in total, premenopausal and postmenopausal women, respectively.
Higher serum estrogen and insulin concentrations exhibit a synergistic risk for EC
To investigate whether insulin and estrogen synergistically enhance patient risk for EC, we analyzed the odds ratios of estrone, estradiol, and insulin for EC using a logistic regression model, according to stratified data. First, the estrone, estradiol, and insulin concentrations were stratified into median values according to their distribution among EC patients and controls. The cut-off points for serum estradiol, estrone, and insulin concentrations were 21.01, 50.18, and 18.36 pg/mL, 42.70, 58.53, and 37.51 pg/mL, and 8.82, 8.74, and 8.90 mIU/mL in the entire EC population, in premenopausal, and in postmenopausal women, respectively.
Compared with the lower stratification groups, ORs for the higher stratifications in total, premenopausal, and postmenopausal women for estrone were 2.041 (95% CI, 1.590–2.619; P = 0.000), 1.025 (95% CI, 0.664–1.580; P = 0.912), and 2.192 (95% CI, 1.618–2.970; P = 0.000), respectively. However, similar comparisons for estradiol were not significant (P > 0.05; ). Additionally, increased insulin levels were positively correlated with EC risk in the whole population (OR, 2.934; 95% CI, 2.275–3.783; P = 0.000), as well as in premenopausal (OR, 4.280, 95% CI, 2.695–6.796; P = 0.000), and postmenopausal women (OR, 2.414; 95% CI, 1.779–3.277; P = 0.000), respectively ().
Table 4. Higher serum estrogen and insulin concentrations synergistically influence the risk for endometrial cancer, as determined by logistic regression.
We next evaluated whether estrone and insulin synergistically influence EC risk. Compared with the lower estrone and insulin stratifications, ORs for the higher insulin with lower estrone stratification and for both higher insulin and estrone stratifications were increased in the whole population, and in premenopausal and postmenopausal women (). Likewise, compared with lower estradiol and insulin stratification, ORs for higher insulin with lower estradiol stratification and for both higher insulin and estradiol stratifications were increased in all three groups (). These results indicate that higher serum estrogen and insulin concentrations synergistically enhance EC risk.
Serum estrogen concentration was positively correlated with insulin for EC
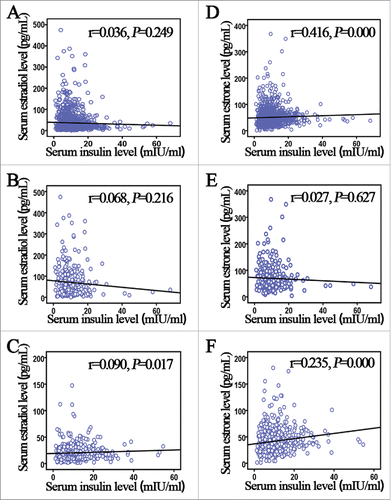
Furthermore, we investigated the correlation between estrogens and insulin in EC. Among the total EC population, insulin was positive correlated with estrone (r = 0.416, P = 0.000; ) but not estradiol (r = 0.036, P = 0.249; ). Meanwhile, in premenopausal women, there was no significantly correlation between estrogens (estradiol and estrone) and insulin (P > 0.05, , ). Conversely, insulin was positively correlated with estradiol (r = 0.090, P = 0. 017; ) and estrone (r = 0. 235, P = 0.000; ) in postmenopausal women.
Figure 1. Correlation between serum estrogen and insulin concentration in endometrial cancer (EC) patients. (A, B) Correlation between serum estradiol and insulin levels in EC cases among the total study population. Insulin was not positively correlated with (A) estradiol (P = 0.249), but was positively correlated with (B) estrone (P = 0.000). (C, D) Correlation between serum estradiol and insulin levels in EC cases in premenopausal women. Insulin was not positively correlated with (C) estradiol or (D) estrone (both P > 0.05). (E, F) Correlation between serum estradiol and insulin levels in postmenopausal women. Insulin was positively correlated with both (E) estradiol (P = 0. 017) and (F) estrone (P = 0.000).
Estradiol induces EC cell proliferation, promotes cell cycle progression, and inhibits apoptosis
We employed estradiol to stimulate Ishikawa and ECC-1 lines. Except for the 10−10 M stimulation, estradiol-induced proliferation was significantly increased in a dose- and time-dependent manner in Ishikawa cells, compared to controls (P < 0.05; ). Likewise, the S phase rate within the cell cycle distribution of Ishikawa cells stimulated with estradiol was significantly increased in a dose-dependent manner compared to that in control cells (P < 0.05, , ). We next examined the anti-apoptotic effect of estradiol in Ishikawa cells at 24, 48, and 72 h. As shown in , the apoptosis rate of Ishikawa cells stimulated with estradiol was significantly decreased compared to that of the control cells, and this decrease appeared to be dose-dependent (P < 0.05, ). Similar results were observed upon estradiol stimulation of ECC-1 cells ().
Figure 2. Estradiol induces cell proliferation, promotes cell cycle progression, and protects against apoptosis in Ishikawa cells. (A) BrdU enzyme-linked immunosorbent assay (ELISA) analysis showed that estradiol-induced proliferation was significantly increased at 24, 48, and 72 h compared to control cells. (B) The number of cells in S phase of the cell cycle after stimulation with estradiol was significantly higher than that in the control group. Data are presented as the means ± standard deviations (SD) of the results from three independent experiments. (C) Cells stimulated with estradiol exhibited significantly lower rates of apoptosis than the control cells. Data are presented as the means ± SD of the results from three independent experiments. (D) Representative flow cytometry results for the cell cycle assay analysis of estradiol-treated and control cells at 24, 48, and 72 h. (E) Representative flow cytometry results for apoptosis assay analysis of estradiol-treated and control cells at 24, 48, and 72 h. *P < 0.05 for all experiments.
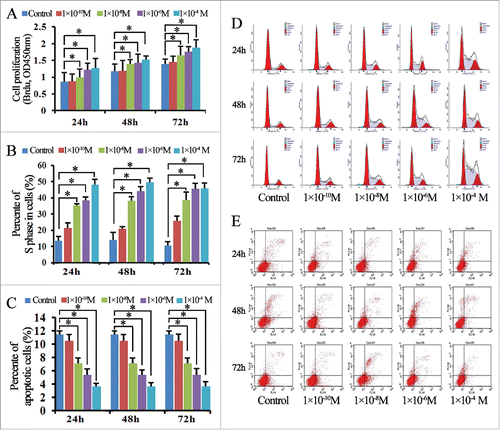
Figure 3. Estradiol induces cell proliferation, promotes cell cycle progression, and protects against apoptosis in ECC-1 cells. (A) BrdU enzyme-linked immunosorbent assay (ELISA) analysis of the levels of estradiol-induced proliferation at 24, 48, and 72 h, compared to control cells. (B) The number of cells in S phase of the cell cycle after stimulation with estradiol, compared with the control group. Data are presented as means ± standard deviations (SD) of the results from three independent experiments. (C) Analysis of the apoptosis rate in cells stimulated with estradiol, compared with the control group. Data are presented as means ± SD of the results from three independent experiments. (D) Representative flow cytometry results for cell cycle assay analysis of estradiol-treated and control cells at 24, 48, and 72 h. (E) Representative flow cytometry results for apoptosis assay analysis of estradiol-treated and control cells at 24, 48, and 72 h. *P < 0.05 for all experiments.
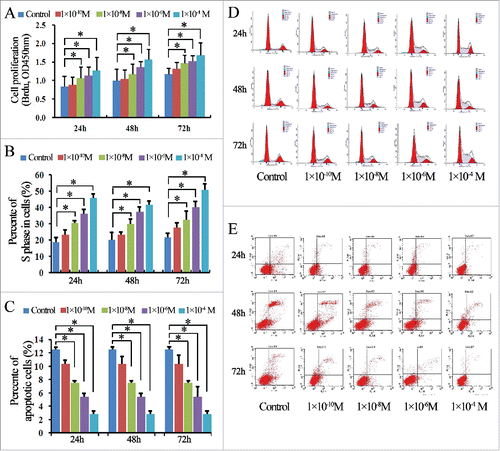
Notably, an estradiol concentration of 10−8 M, which is not substantively higher than physiological concentrations, was sufficient to stimulate EC cell proliferation and S phase progression, and to inhibit apoptosis. Consequently, we used 10−8 M as the reference value for all subsequent experiments.
Insulin and estradiol synergistically induce EC cell proliferation, promote cell cycle progression, and protect against apoptosis in vitro and in vivo
In a previous study, we demonstrated that insulin treatment significantly increased Ishikawa cell growth in a dose- and time-dependent manner and protected these cells from apoptosis in a dose-dependent manner.Citation23 Additionally, the current study revealed that increased serum levels of both estradiol and insulin imposed a higher risk of EC than increased serum levels of estradiol or insulin alone, suggesting that estradiol and insulin synergistically promote EC progression.
To evaluate the synergistic effects of insulin and estrogen in Ishikawa and ECC-1 cells, we treated EC Ishikawa and ECC-1 cells in vitro with a combination of insulin and estrogen (estradiol; both at 10−8 M) and assayed the resultant changes in cell proliferation, cell cycle distribution, and apoptosis. Proliferation was significantly higher in Ishikawa and ECC-1 cells treated with both insulin and estradiol, compared with the untreated control cells and those treated with insulin or estradiol alone (P < 0.05; and ). Furthermore, the combination of insulin and estradiol appreciably increased the S-phase distribution and reduced the G0/G1-phase distribution in both cell types, compared with insulin or estradiol alone or with no treatment (P < 0.05; and ). We also used flow cytometry to quantify the percentage of apoptotic cells after combination treatment; these cells can be seen in the lower right quadrants of and . In both Ishikawa and ECC-1 cells, stimulation with insulin and estradiol for 24 h resulted in reduced levels of apoptosis, compared to treatment with insulin or estradiol alone or to no treatment (P < 0.05; and ).
Figure 4. Insulin and estrogen have synergistic effects in Ishikawa and ECC-1 cells. (A) The effects of insulin and estradiol on Ishikawa cell proliferation were evaluated using a BrdU enzyme-linked immunosorbent assay (ELISA). (B) Flow cytometry analysis of cell cycle distribution after insulin and estradiol stimulation of Ishikawa cells. (C) The percentage of apoptotic Ishikawa cells after insulin and estradiol stimulation, as detected via flow cytometry analysis using annexin V-FITC antibodies. (D) Analysis of the effects of insulin and estradiol on ECC-1 cell proliferation via BrdU ELISA analysis. (E) Flow cytometry analysis of cell cycle distribution after insulin and estradiol stimulation in ECC-1 cells. (F) The percentage of apoptotic ECC-1 cells after insulin and estradiol stimulation, as detected via flow cytometry analysis using annexin V-FITC antibodies. (G) Analysis of the effects of insulin and estradiol on xenografts of Ishikawa cells with estrogen receptor (ER)-α (+) and insulin receptor (InsR)-β (+) cells. (H) Levels of Ki-67 expression and apoptosis in xenograft tissues.
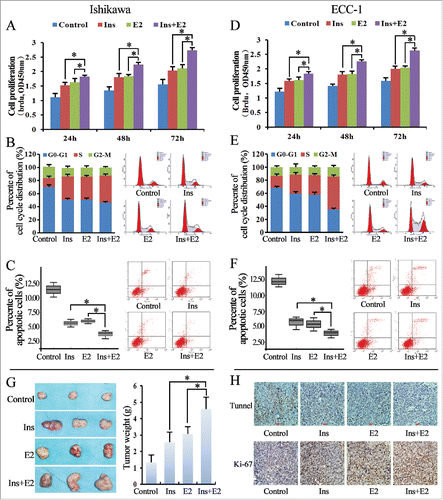
To examine the in vivo effects of insulin and estradiol, we next injected approximately 5 × 106 Ishikawa cells into both flanks of subject mice. Three weeks after transplantation, co-treatment with insulin and estradiol had a stronger effect on normal Ishikawa xenograft growth than treatment with either insulin or estradiol alone or no treatment (; P < 0.05). Xenografts treated with a combination of insulin and estradiol exhibited more Ki67+ nuclei than those treated with insulin or estrogen alone or those receiving no treatment (). Apoptosis of the xenografts was also analyzed by TUNEL assay. Our data showed that treatment with both insulin and estradiol produced fewer TUNEL+ cells than treatment with insulin and estradiol alone or no treatment ()
Discussion
In the present study, we found that increased insulin and estrone levels represented independent risk factors for type 1 EC. In addition, higher levels of insulin and estrone were associated with significantly increased EC risk compared to lower levels of these agents. While our findings concur with many of previous studies, those reports failed to evaluate the synergistic activities of estrogen and insulin, and their signal transduction in EC.
The primary advantages of the current study were the large size and wide age-range of the patient cohort, which enabled us to study associations among insulin, estrogens, and type 1 EC incidence. In the current study, the odds ratio for patients with EC that exhibit higher levels of estrone/estradiol and insulin increased significantly compared to those with higher levels of either estrone/estradiol or insulin alone. This observation indicated that insulin and estrogen may have a combined or synergistic effect in EC; however, the specific effects and underlying mechanism(s) of this synergy remain unclear. Notably, although there was a positive correlation between serum insulin levels and those of estradiol and estrone in postmenopausal women, there were no significant correlations between estradiol and insulin levels in the total female population or in premenopausal women. This discrepancy might be due to the cyclic variation in estrogen levels observed in premenopausal women and in certain subjects among the total population, which likely contributed non-determinacy to the correlation between estrogens and insulin. Consequently, while studying the synergistic effects of estrogens and insulin on EC with serum samples, we placed more importance on postmenopausal subjects.
Generally, serum analysis demonstrated that insulin and estrogens could exert combined or synergistic effects on EC progression (). To confirm this finding, we further investigated the effects of insulin and estradiol on EC cells. We found that estradiol alone promoted cell proliferation, increased S phase distribution, and inhibited cell apoptosis in a dose- and time- dependent manner ( and ). Insulin, as a growth factor and a member of the insulin/IGF system, also play a role in EC pathogenesis. In particular, our previous study showed that insulin promotes EC cell proliferation and inhibits cell apoptosis in a dose- and time-dependent manner.Citation23,24 Further, it was previously reported that estrogens and IGF-I, when combined, synergistically contribute to the development and progression of breast cancer.Citation3,25 We have similar evidences here to support the synergy between the insulin and estrogen effects on EC tumorigenesis () though the signaling pathways were not discussed here.
Briefly, clarification of the mechanism of this synergistic effect would be expected to identify two primary phenomena: i) that estrogen signaling could act on insulin receptors and activate downstream pathways and ii) that insulin signaling could simultaneously activate estrogen receptors. We have been further focusing on the abovementioned objects.
Conclusion
Taken together, our results present epidemiological evidences of the synergy between estrogen and insulin in endometrial carcinogenesis and reveal synergistic role of estrogen and insulin in type 1 EC cells both in vitro and in vivo.
Conflicts of Interest
All authors approved the manuscript and have agreed to submit it to your esteemed journal. There is no conflict of interest to declare.
Declarations
Authors' contributions
W. Tian conducted all experiments. Y. Wang and F. Xue wrote the manuscript, directed this study, designed the research, and gave key advice. F. Teng, J. Zhao, J. Gao, C. Gao, D. Sun, G. Liu, Y. Zhang, S. Yu, and W. Zhang provided essential assistance during the work.
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee of Tianjin Medical University General Hospital, and informed consent was obtained from each patient and healthy subject enrolled in the study.
All animal experiments were performed under standard guidelines approved by the State Key Laboratory of Experimental Hematology (license no. SYXK (JIN) 2009-0002), and all procedures were carried out in accordance with the approval of the ethics committee on animal care of Tianjin Medical University.
Compliance with Ethical Standards
This manuscript has not been published or presented elsewhere in part or in entirety, and is not under consideration by another journal. All study participants provided informed consent, and the study design was approved by the appropriate ethics review boards.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 30772316 to F. Xue and 81572568 to Y. Wang) and Natural Science Foundation of Tianjin City (No. 12JCQNJC06700 to Y. Wang).
Reference
- Allen NE, Key TJ, Dossus L, Rinaldi S, Cust A, Lukanova A, Peeters PH, Onland-Moret NC, Lahmann PH, Berrino F, Panico S, Larranaga N, Pera G, Tormo MJ, Sanchez MJ, Ramon Quiros J, Ardanaz E, Tjonneland A, Olsen A, Chang-Claude J, Linseisen J, Schulz M, Boeing H, Lundin E, Palli D, Overvad K, Clavel-Chapelon F, Boutron-Ruault MC, Bingham S, Khaw KT, Bueno-de-Mesquita HB, Trichopoulou A, Trichopoulos D, Naska A, Tumino R, Riboli E, Kaaks R. ‘Endogenous sex hormones and endometrial cancer risk in women in the european prospective investigation into cancer and nutrition (Epic)’. Endocr Relat Cancer. 2008;15:485–97. doi:10.1677/ERC-07-0064.
- Bokhman JV. ‘Two pathogenetic types of endometrial Carcinoma’. Gynecol Oncol. 1983;15:10–7. doi:10.1016/0090-8258(83)90111-7.
- Bradley LM, Gierthy JF, Pentecost BT. ‘Role of the insulin-like growth factor system on an estrogen-dependent cancer phenotype in the Mcf-7 human breast cancer cell line’. J Steroid Biochem Mol Biol. 2008;109:185–96. doi:10.1016/j.jsbmb.2007.10.006.
- Brown SB, Hankinson SE. ‘Endogenous estrogens and the risk of Breast, Endometrial, and Ovarian Cancers’. Steroids. 2015;99:8–10. doi:10.1016/j.steroids.2014.12.013.
- Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J, ‘Cancer Statistics in China, 2015’. CA Cancer J Clin. 2016;66:115–32. doi:10.3322/caac.21338.
- Diamanti-Kandarakis E, Papailiou J, Palimeri S, ‘Hyperandrogenemia: Pathophysiology and its role in ovulatory dysfunction in Pcos’. Pediatr Endocrinol Rev. 2006;3 Suppl 1:198–204.
- Hapangama DK, Kamal AM, Bulmer JN. ‘Estrogen receptor beta: The guardian of the Endometrium’. Hum Reprod Update. 2015;21:174–93. doi:10.1093/humupd/dmu053.
- Joung KH, Jeong JW, Ku BJ. ‘The association between Type 2 diabetes mellitus and Women Cancer: The epidemiological evidences and putative mechanisms’. Biomed Res Int. 2015;2015:920618.
- Kamal A, Tempest N, Parkes C, Alnafakh R, Makrydima S, Adishesh M, Hapangama DK. ‘Hormones and endometrial Carcinogenesis’. Horm Mol Biol Clin Investig. 2016;25:129–48.
- Kurita T, Wang YZ, Donjacour AA, Zhao C, Lydon JP, O'Malley BW, Isaacs JT, Dahiya R, Cunha GR. ‘Paracrine regulation of apoptosis by steroid hormones in the Male and Female reproductive system’. Cell Death Differ. 2001;8:192–200. doi:10.1038/sj.cdd.4400797.
- Li Q, Kannan A, DeMayo FJ, Lydon JP, Cooke PS, Yamagishi H, Srivastava D, Bagchi MK, Bagchi IC. ‘The Antiproliferative action of progesterone in uterine epithelium is mediated by Hand2’. Science. 2011;331:912–6. doi:10.1126/science.1197454.
- Mathew D, Drury JA, Valentijn AJ, Vasieva O, Hapangama DK. ‘In Silico, in Vitro and in Vivo analysis identifies a potential role for Steroid hormone regulation of Foxd3 in Endometriosis-Associated genes’. Hum Reprod. 2016;31:345–54.
- Meng YG, Han WD, Zhao YL, Huang K, Si YL, Wu ZQ, Mu YM, ‘Induction of the Lrp16 Gene by estrogen promotes the invasive growth of Ishikawa human endometrial Cancer cells through the downregulation of E-Cadherin’. Cell Res. 2007;17:869–80. doi:10.1038/cr.2007.79.
- Mu N, Zhu Y, Wang Y, Zhang H, Xue F. ‘Insulin resistance: A significant risk factor of Endometrial Cancer’. Gynecol Oncol. 2012;125:751–7. doi:10.1016/j.ygyno.2012.03.032.
- Plymate SR, Matej LA, Jones RE, Friedl KE. ‘Inhibition of Sex hormone-binding globulin production in the Human Hepatoma (Hep G2) Cell line by Insulin and Prolactin’. J Clin Endocrinol Metab. 1988;67:460–4. doi:10.1210/jcem-67-3-460.
- Schindler AE. ‘Progestogen deficiency and endometrial Cancer Risk’. Maturitas. 2009;62:334–7. doi:10.1016/j.maturitas.2008.12.018.
- Setiawan VW, Yang HP, Pike MC, McCann SE, Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, van den Brandt PA, van de Vijver K, Thompson PJ, Group Australian National Endometrial Cancer Study, Strom BL, Spurdle AB, Soslow RA, Shu XO, Schairer C, Sacerdote C, Rohan TE, Robien K, Risch HA, Ricceri F, Rebbeck TR, Rastogi R, Prescott J, Polidoro S, Park Y, Olson SH, Moysich KB, Miller AB, McCullough ML, Matsuno RK, Magliocco AM, Lurie G, Lu L, Lissowska J, Liang X, Lacey JV, Jr., Kolonel LN, Henderson BE, Hankinson SE, Hakansson N, Goodman MT, Gaudet MM, Garcia-Closas M, Friedenreich CM, Freudenheim JL, Doherty J, De Vivo I, Courneya KS, Cook LS, Chen C, Cerhan JR, Cai H, Brinton LA, Bernstein L, Anderson KE, Anton-Culver H, Schouten LJ, Horn-Ross PL. ‘Type I and Ii Endometrial Cancers: Have they different risk factors?’. J Clin Oncol. 2013;31:2607–18. doi:10.1200/JCO.2012.48.2596.
- Shao R, Li X, Feng Y, Lin JF, Billig H. ‘Direct effects of Metformin in the Endometrium: A hypothetical mechanism for the treatment of Women with Pcos and Endometrial Carcinoma’. J Exp Clin Cancer Res. 2014;33:41. doi:10.1186/1756-9966-33-41.
- Siegel RL, Miller KD, Jemal A. ‘Cancer Statistics, 2016’. CA Cancer J Clin. 2016;66:7–30. doi:10.3322/caac.21332.
- Suba Z. ‘Interplay between insulin resistance and Estrogen deficiency as Co- Activators in Carcinogenesis’. Pathology & Oncology Research. 2012;18:123–33. doi:10.1007/s12253-011-9466-8.
- Tian W, Zhu Y, Wang Y, Teng F, Zhang H, Liu G, Ma X, Sun D, Rohan T, Xue F. ‘Visfatin, A potential biomarker and prognostic factor for endometrial Cancer’. Gynecol Oncol. 2013;129:505–12. doi:10.1016/j.ygyno.2013.02.022.
- Vermeulen A, Verdonck L. ‘Sex Hormone concentrations in post-menopausal women’. Clin Endocrinol (Oxf). 1978;9:59–66.
- Wang Y, Hua S, Tian W, Zhang L, Zhao J, Zhang H, Zhang W, Xue F. ‘Mitogenic and anti-apoptotic effects of insulin in endometrial cancer are Phosphatidylinositol 3-Kinase/Akt Dependent’. Gynecol Oncol. 2012;125:734–41. doi:10.1016/j.ygyno.2012.03.012.
- Wang Y, Zhu Y, Zhang L, Tian W, Hua S, Zhao J, Zhang H, Xue F. ‘Insulin promotes proliferation, Survival, and Invasion in Endometrial Carcinoma by Activating the Mek/Erk Pathway’. Cancer Lett. 2012;322:223–31. doi:10.1016/j.canlet.2012.03.026.
- Wu MH, Chou YC, Chou WY, Hsu GC, Chu CH, Yu CP, Yu JC, Sun CA. ‘Relationships between critical period of Estrogen exposure and circulating levels of Insulin-Like Growth Factor-I (Igf-I) in Breast Cancer: Evidence from a case-control study’. Int J Cancer. 2010;126:508–14. doi:10.1002/ijc.24722.