
ABSTRACT
In this study, we report here a rare case of polycythemia and cRCC in the same patient, which may be helpful in understanding clinical features and molecular mechanisms underlying VHL–mutation-associated cRCC and polycythemia induced by germline mutation of HIF2A. Firstly, we identified a rare but well studied germline mutation resulting in polycythemia in HIF2A (c.1609G>A, p.Gly537Trp) in the blood of the patient and his daughter. Meanwhile, we identified an inactivating VHL mutation (c.391A>T, p.N131Y), as well as TP53 mutation(c.977A>T, p.E326V) and mTOR mutation(c.7498A>T, p.I2500F) in renal cancer tissue. Moreover, protein levels of VHL, HIF1A, HIF2A, EPO, and VEGF estimated by immunohistochemical staining substantiated hyperactivation of the oxygen-sensing pathway. In addition, we identified 158 somatic SNP/indel mutations, including 90 missense/nonsense/splice/stop-loss mutations by whole-exome sequencing (WES) of the tumor specimen and matched normal DNA.
KEYWORDS:
Introduction
Renal-cell carcinoma (RCC) is the most common kidney cancer, with the overwhelming majority of cases resulting from mutations in the von Hippel-Lindau tumor suppressor (VHL) gene.Citation1 Germline mutations in VHL cause VHL disease, including RCC, which results from biallelic VHL inactivation. Somatic VHL mutations occur in the majority of clear-cell (c)RCC cases, accounting for 50%–70% of cases.Citation2 Mutation-induced loss-of-function VHL protein allows hypoxia inducible factors (HIFs) to act as transcription factors regulating expression of various pro-tumorigenic genes, including that encoding vascular endothelial growth factor (VEGF). Upregulation of VEGF subsequently leads to RCC development and progression by inducing angiogenesis.Citation3 Tumor development tends to occur in patients carrying one mutant VHL allele when loss of heterozygosity occurs in somatic cells. Biallelic inactivation of VHL is an early and critical event in the pathogenesis of cRCC in VHL disease and in many sporadic RCC cases; however, additional genetic and epigenetic events are required for the development of cRCC.Citation4
Polycythemia refers to an increased number of circulating red blood cells, which can be categorized as primary or secondary polycythemia. Primary polycythemia occurs when excess red blood cells are produced as a result of an abnormality of bone marrow due to mutation in the gene encoding erythropoietin receptor (EPOR) or JAK2 exon 14 (V617F)/exon 12 (K539L, N542-E543del, E543-D544del).Citation5 According to Online Mendelian Inheritance in Man (OMIM), familial polycythemia consists of four subtypes, which are associated with mutations in EPOR, VHL, EGLN1, and HIF2A, respectively. It is noteworthy that EGLN1, VHL, and HIF2A are the key regulators in the HIF signaling pathway.Citation6 Recent studies showed that mutations in HIF2A exon 12 are involved in regulation of EPO synthesis, resulting in polycythemia.Citation7,8 Secondary polycythemia is usually caused by excessive production of EPO due to high altitude, hypoxic disease, or neoplasms. Renal cancer is the most frequent cause of paraneoplastic polycythemia linked to deregulation of the pVHL-HIF2A pathway in renal clear carcinoma.
Here, we report a rare case of cRCC and hereditary polycythemia. Genotyping revealed that the patient carried both a germline HIF2A mutation and a somatic VHL mutation. Both mutations result in overactivation of HIF2A and its downstream target genes.
Case report
On November 25, 2015, a 61-year old male was referred to our hospital due to the finding of a well-defined and heterogeneous contrast–medium-enhanced mass in the upper pole of the left kidney () identified by an abdominal computed tomography (CT) scan. T1N0M0 kidney cancer was diagnosed by a subsequent positron emission tomography (PET)/CT scan and the finding of a high–FDG-uptake mass in the left kidney with no metastasis (data not shown). Nephron-sparing enucleation was conducted by retroperitoneal laparoscopic procedure on December 1, 2015. The size of the mass was about 1.0 × 1.0 cm and pathological examination revealed cRCC ().
Figure 1. Characterization of the patient. (A) Computed tomography (CT) scan. Renal tumor is indicated by red arrow. (B) Hematoxylin and eosin staining (HE) and immunohistochemical staining of HIF1α, HIF2α, EPO, and VEGF in the tumor tissue. Magnification, 400 ×. (C) Sequence of HIF2α exon 12 in DNA purified from blood of patient (upper) and his daughter (lower). The heterozygous G>A substitution is indicated by a red arrow. (D) Patient family pedigree. □/○, normal male/female; ▪/•: male/female carrying the mutation. Patient and daughter are represented by a filled square (arrow) and filled circle, respectively.
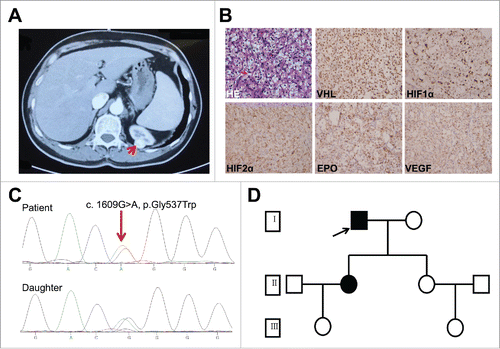
The patient had “red face” from a very young age and, based on elevated hemoglobin, was initially diagnosed in his 30 s as having polycythemia, however, no any treatment was initiated. There was no history of hypertension (blood pressure was 106/64 mmHg at admission) or thromboembolic event. At the time of admission, his hemoglobin was 227 g/L (normal range 115–150 g/L) and red blood cell count was 6.83 × 1012/L (normal range 3.8-5.1 × 1012/L). As expected, neither the hemoglobin (211 g/L) nor red blood cell count (6.82 × 1012/L) returned to normal range after tumor removal, suggesting that the polycythemia was not due to the tumor itself. Since then, intermittent phlebotomies (400 ml about every 6 months) have been performed to alleviate the red face associated with polycythemia. The overall clinical course of the patient is summarized in . No recurrence or metastatic lesions have been detected during follow-up. His second daughter was also found with increased red blood cell count (6.37 × 1012/L) and elevated hemoglobin level (190 g/L), as well as EPO level (28.4 mIU/mL, normal range 2.59-18.5/L). And she gets intermittent phlebotomies once every year.
Table 1. Patient clinical course.
Because of the positive family history, we speculated that polycythemia in our patient is the result of germline mutation of some genes. Based on this hypothesis, we screened for mutations in four genes (EPOR, VHL, EGLN1, and HIF2A) that are strongly related to polycythemia.Citation7,8 A rare but well studied germline mutation in HIF2 (c.1609G>A, p.Gly537Trp) was found in both the patient and his daughter (). No mutations were identified in the other three genes. Subsequently, we conducted whole-exome sequencing (WES) of the tumor specimen and matched normal DNA, and identified 158 somatic single nucleotide polymorphism (SNP)/insertion-deletion (indel) mutations, including 90 missense/nonsense/splice/stop-loss mutations (Supplementary Table S1). Of these, a VHL mutation (c.391A>T, p.N131Y) was reported in renal cancer, and mutations in TP53 (c.977A>T, p.E326V) and mTOR (c.7498A>T, p.I2500F) were assumed to be deleterious, suggesting that any of these mutations alone or a combination may play important roles in the occurrence of cRCC. The clinical significance of other mutations was unclear. We also analyzed WES data for EPOR, VHL, EGLN1, HIF2A, and 152 cancer-susceptibility genes (Supplementary Table S2) for the presence of germline mutations. The HIF2A mutation was confirmed to be the only pathogenic mutation, based on the American College of Medical Genetics and Genomics (ACMG) guidelines (Supplementary Table S3). Furthermore, protein levels of VHL, HIF1A, HIF2A, EPO, and VEGF estimated by immunohistochemical staining () suggested hyperactivation of the oxygen-sensing pathway.
Discussion
In this study we present a rare case with polythemia and a left cRCC. The patient was shown to carry both a point germline mutation of HIF2A (c.1609G>A, p.Gly537Arg) and a somatic mutation of VHL (c.391A>T, p.N131Y). To the best of our knowledge, this is the first report of the coexistence of germline HIF2A–mutation-induced polycythemia and somatic VHL–mutation-associated cRCC.
Polycythemia can be a paraneoplastic manifestation, presumably due to inappropriate production of erythropoietin (EPO) by the tumor cells, which often occurs in advanced stages of renal cancer and is associated with poor prognosis.Citation9 Because the patient in our study has a kidney tumor, one may argue that the polycythemia may be secondary to dysregulation of EPO production. Usually this form of polycythemia is resolved upon removal of the tumor, which did not occur in this patient. On the other hand, the polycythemia persisted after surgical removal of the kidney tumor, with no evidence of recurrent or new tumors. Previous studies also suggested that polycythemia can be acquired as a paradoxical drug effect in patients receiving an anti-VEGF receptor therapy.Citation10 This possibility was excluded here because the patient did not receive any drug in this family. Given the fact that both the patient and one of his two daughters carry the same HIF2A germline mutation and both showed not only high red blood cell count but also elevated hemoglobin level, it is reasonable to conclude that the polycythemia is congenital and the result of the germline HIF2A mutation.
Since the first case of HIF2A germline mutation (Gly537Trp)-associated polycythemia was reported by Percy MJ. et al. in 2008,Citation7 multiple cases have been reported. Further screening of HIF2A mutation in a cohort of 75 erythrocytosis patients identified two novel heterozygous HIF2A mutations, Met535Val and Gly537Arg.Citation8 Patients with germline mutation of HIF2A rarely develop paraganglioma-pheochromocytoma (PGL/PCC) syndrome. To date, only one patient with a germline mutation in HIF2A exon 9 (c.1121T>A, p.F374Y) was reported to have polycythemia and PCC/PGL simultaneously,Citation11 but somatic mutations of HIF2A are frequent genetic events in PCC/PGL and polycythemia. Karel Pacak et al.Citation12 first reported two cases of congenital polycythemia and multiple and recurrent PGL or PCC or somatostatinoma induced by somatic mutations in HIF2A exon 12. Since then, about 14 cases have been reported by several groups,Citation13 and a new syndrome, Pacak-Zhuang syndrome, has been confirmed. Although the molecular mechanisms underlying the different phenotypes associated with germline and somatic HIF2A mutation are unknown, it appears that gain-of-function mutation of HIF2A is insufficient, and loss-of-function of one or more tumor suppressors may be needed, for PCC/PGL to develop. HIF2A, an important component of the VHL-HIF oxygen-sensing pathway, plays a pivotal role in VHL–mutation-induced cRCC, and inhibition of HIF2A is sufficient to suppress pVHL-defective tumor growth in vivo.Citation14 However, until now there has been no evidence to suggest that HIF2A germline mutation itself is associated with cRCC. Polycythemia and cRCC in this particular case appear to be independent events, with polycythemia resulting from HIF2A germline mutation and cRCC likely caused by the combination of somatic mutations in VHL, TP53, and mTOR.
It has also been reported that loss of pVHL alone give rise to only premalignant renal cysts, and additional mutations at non-VHL loci are required for the development of frank RCC.Citation4 This patient meets the criteria of the 2-hit hypothesis: in addition to the mutations in the VHL-HIF pathway, two other known or predicted disease-causing mutations (TP53: c.977A>T, p.E326V; mTOR: c.7498A>T, p.I2500F) exist. A previous studyCitation15 demonstrated that in cRCC the mutation frequencies of mTOR and TP53 are 6% and 3.1%, respectively. It is noteworthy that the mTOR mutation (c.7498A>T, p.I2500F), which results in hyperactivation of the PI3K-AKT-mTOR signaling axis, has been reported in breast cancer.Citation16 The TP53 mutation (c.977A>T, p.E326V) has not been previously reported, but was predicted to be deleterious by SIFT and PolyPhen. In addition, based on the 2-hit hypothesis, Friedrich CA et al.Citation9 suggested that inherited VHL mutation favors the occurrence of secondary and acquired mutations. Not germline HIF2A gain-of-function mutation but somatic VHL loss-of-function mutation is driver mutation for cRCC. Whether the somatic mutations of VHL, TP53, and mTOR have anything to do with HIF2A germline mutation needs further investigation.
In summary, we report here a rare case of polycythemia and cRCC in the same patient, with polycythemia resulted from a germline HIF2A mutation and cRCC likely due to multiple (VHL, P53 and mTOR) somatic mutations.
Conflicts of interest
Conflict of interest relevant to this article was not reported.
Disclosure
The Institutional Review Board of Daping Hospital of Third Military Medical University waived the IRB approval for the study; however, the written informed consent was obtained from the patients for the use of medical records and related images, before the publication of the study. The authors have nothing to disclose.
New_folder__4_.zip
Download Zip (47 KB)References
- Diaz JI, Mora LB, Hakam A. The Mainz Classification of Renal Cell Tumors. Cancer control: journal of the Moffitt Cancer Center. 1999;6(6):571–9. doi:10.1177/107327489900600603.
- Richards FM. Molecular pathology of von HippelLindau disease and the VHL tumour suppressor gene. Expert reviews in molecular medicine. 2001;2001:1–27.
- Wiesener MS, Munchenhagen PM, Berger I, Morgan NV, Roigas J, Schwiertz A, Jurgensen JS, Gruber G, Maxwell PH, Loning SA, et al. Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1alpha in clear cell renal carcinomas. Cancer Res. 2001;61(13):5215–22.
- Zhuang Z, Bertheau P, Emmert-Buck MR, Liotta LA, Gnarra J, Linehan WM, Lubensky IA. A microdissection technique for archival DNA analysis of specific cell populations in lesions < 1 mm in size. Am J Pathol. 1995;146(3):620–5.
- Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, Ferrari M, Gisslinger H, Kralovics R, Cremonesi L, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111(3):1686–9.
- Eltzschig HK, Eckle T, Grenz A. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2009, 360(13):1361–2; author reply 1362.
- Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, Lee FS. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358(2):162–8. doi:10.1056/NEJMoa073123.
- Percy MJ, Beer PA, Campbell G, Dekker AW, Green AR, Oscier D, Rainey MG, van Wijk R, Wood M, Lappin TR, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood. 2008;111(11):5400–2. doi:10.1182/blood-2008-02-137703.
- Friedrich CA. Genotype-phenotype correlation in von Hippel-Lindau syndrome. Human molecular genetics. 2001;10(7):763–7. doi:10.1093/hmg/10.7.763.
- Richard S, Croisille L, Yvart J, Casadeval N, Eschwege P, Aghakhani N, David P, Gaudric A, Scigalla P, Hermine O. Paradoxical secondary polycythemia in von Hippel-Lindau patients treated with anti-vascular endothelial growth factor receptor therapy. Blood. 2002;99(10):3851–3. doi:10.1182/blood.V99.10.3851.
- Lorenzo FR, Yang C, Ng Tang Fui M, Vankayalapati H, Zhuang Z, Huynh T, Grossmann M, Pacak K, Prchal JT. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med. 2013;91(4):507–12. doi:10.1007/s00109-012-0967-z.
- Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, Prchal JT, Tischler AS, Lechan RM, Zhuang Z. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31(13):1690–8. doi:10.1200/JCO.2012.47.1912.
- Liu Q, Wang Y, Tong D, Liu G, Yuan W, Zhang J, Ye J, Zhang Y, Yuan G, Feng Q, Zhang D, Jiang J. A Somatic HIF2α Mutation-induced Multiple and Recurrent Pheochromocytoma/Paraganglioma with Polycythemia: Clinical Study with Literature Review. Endocr Pathol. 2017;28(1):75–82. doi:10.1007/s12022-017-9469-4.
- Kondo K, Kim WY, Lechpammer M, Kaelin WG, Jr.. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biology. 2003, 1(3):E83. doi:10.1371/journal.pbio.0000083.
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–9.
- Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014;4(5):554–63. doi:10.1158/2159-8290.CD-13-0929.