
ABSTRACT
In an elegant report, Corbet et alCitation1 recently demonstrated the much needed insight to exploit cancer's metabolic reprogramming for potential therapeutic intervention. In brief, the findings underscore the principle that abrogation of mitochondrial pyruvate metabolism upregulates glycolysis, and sensitizes cancer cells to radiation. Distinctive from the conventional approach of inhibition/ down-regulation of glycolysis, this emerging paradigm of forced-upregulation of glycolysis (i.e., a “hyperglycolytic” phenotype) concomitant with a reduced mitochondrial capacity turns the metabolic plasticity into vulnerability that may have implications in therapeutic targeting. Nevertheless, this commendable reportCitation1 also provokes scientific curiosity and future directions of research on the opportunities and challenges of such forced upregulation of glycolysis in cancer.
Metabolic plasticity in cancer is exemplified by the ability to switch between glycolysis and mitochondrial (mito) oxidative phosphorylation (OxPhos) to metabolize glucose. Such a metabolic flexibility enables cancer cells to maintain uninterrupted proliferation, and overcome challenges posed by therapeutics (e.g., antiglycolytic agents). Conventionally, targeting glucose metabolism essentially focused on the tactics of disruption of glycolysis, with the objective to abolish one of the biochemical signatures of cancer, the “aerobic glycolysis”. Ever since Otto Warburg reported the seminal discoveryCitation2 that cancer cells metabolize bulk of the glucose via glycolytic pathway and produce lactate, the focus on aerobic glycolysis (also referred as tumor glycolysis) gained a ceaseless momentum. The fact that tumor glycolysis occurs despite the availability of sufficient oxygen (i.e., aerobic glycolysis) imply that the evasion of mito-OxPhos, and embracing the non-OxPhos (glycolysis) mechanisms facilitate cancer growth. Although, Warburg originally believed that defective-mitochondria as the underlying cause of the glycolytic phenotype, later studies have established that aerobic glycolysis occurs in cancer despite the presence of functionally active mitochondria.Citation3 Yet, the relevance or necessity of glycolytic phenotype in cancer remained obscure.Citation4 Technological advancements in metabolomics, and constant effort of research groups worldwide, have illuminated the implications of aerobic glycolysis in cancer progression. For example, rapid synthesis of macromolecules, competitive advantage of accelerated glucose uptake, prevention of excessive ROS (reactive oxygen species) accumulation, extrusion of lactate to create a microenvironment (that is protective against anticancer agents or immune cells) are some of the principal advantages of tumor glycolysis.Citation5 With the recognition of the role of aerobic glycolysis in cancer growth, as mentioned earlier, much of the therapeutic strategies focused on the inhibition of glycolysis (). Meanwhile, converging reports from several studies including the inhibition of LDH,Citation6 abrogation of lactate extrusionCitation7 have indicated that glycolytically challenged cancer cells switch to mito-OxPhos and survive through TCA (tricarboxylic acid)-cycle anaplerosis. Furthermore, insights on the metabolic symbiosis between hypoxic and normoxic cancer cells to utilize lactate via TCA cycle,Citation8,9 and also the pivotal role of glutamine-metabolismCitation10 substantiated that cancer cells invoke mitochondria-dependent metabolic processes if necessary. Comprehensibly, cancer cells manifest the metabolic plasticity to switch between glycolysis and mito-OxPhos, with the precondition that sufficient oxygen and functional mitochondria are available. Thus, in principle, an efficient anticancer, antimetabolite perhaps have to downregulate lactate production, block mitochondrial pyruvate metabolism via TCA cycle, and inhibit the glutamine-dependent TCA anaplerosis.
Figure 1. Evolving paradigm of abrogation of mitochondrial pyruvate metabolism and the associated hyperglycolytic phenotype. a Conventional strategy to target glycolysis by inhibition or down-regulation. b MPC-inhibition dependent augmentation of glycolysis disrupts metabolic plasticity. GLUTs, glucose transporters; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; MPC, mitochondrial pyruvate carrier; Gln, glutamine; TCA, tricarboxylic acid cycle.
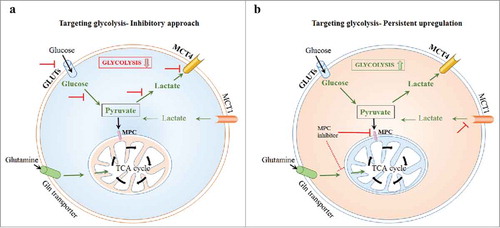
With this background, the mitochondrial pyruvate carrier (MPC), which is pivotal for the transport of pyruvate into mitochondria for subsequent oxidation by TCA cycle, is emerging as an attractive target. Transcriptional repression as well as pharmacological inhibition of MPC abrogated pyruvate metabolism by TCA cycle with a concomitant upregulation of glycolysis.Citation11-13 Intriguingly, differences between MPC knock-down and chemical inhibition of MPC have also been reported, particularly on the lack of sensitivity to docetaxel and radiation therapy, and in the upregulation of TCA cycle-related intermediary metabolism.Citation12 Nevertheless, MPC inhibitorsCitation1,Citation13 have been consistent in affecting mitochondrial metabolism, and have been found to enforce a persistent, constitutive glycolytic phenotype in cancer cells. In other words, cancer cells' capacity to shift between glycolysis and mito-OxPhos has been contorted by MPC inhibition, coercing them (cancer cells) to rely on glycolysis with an impaired mitochondrial pyruvate metabolism. Consequently, such unalterable, glycolytic phenotype presents vulnerability of cancer metabolism to therapeutic targeting (). However, based on the vast literature on the versatility of cancer's metabolic machinery it is imperative to adopt a fervent and cautious approach on the future directions of research on MPC-inhibition related hyperglycolytic strategy. Particularly, with the emerging knowledge on the link between tumor glycolysis and immune evasion,Citation14 and therapeutic resistance which is frequently encountered in targeting glycolytic cancer cells, it concerns that any upregulation of aerobic glycolysis may resist potential chemo-, immune-interventions.
Unlike the pharmacological inhibition of MPC, data from MPC-knockdown studies show the maintenance of TCA-related anaplerosis via glutamine oxidation. Similarly, while the pharmacological inhibitors (e.g., 7ACC2, lonidamine) increase cancer's sensitivity to radiation, MPC-null cells exhibit reduced sensitivity.Citation12 Thus, the pharmacological agents though primarily target MPC, the outcome of increased sensitivity to therapy may be attributed to other inhibitory effects on lactate influx and the glutamine/glutamate metabolism, besides MPC inhibition. Accordingly, effective metabolic sensitization of cancer cells to therapy may require, besides MPC inhibition, the blockade of other molecular targets (e.g., MCT4) as well other molecular targets (e.g., MCT4) may be necessary. Similarly, it remains to be known if there are alternative isoforms of MPC in cancer cells, as in lower organisms such as yeastsCitation15 that may compensate for specific MPC inhibition. Next, as MPC-knockdown has been shown to increase the stemness, invasive or migratory potentialCitation12 it may be relevant to verify that the pharmacological inhibition of MPC does not facilitate a metastatic or stem-cell like phenotype. As opined by Vacanti et al.,Citation11 blockade of pyruvate transport may in turn favor the utilization of fatty acids and amino acids as metabolic fuels. Taken together, further research on the possibility of emergence of a resistant phenotype and understanding the prevalence of alternative rescue mechanisms will have a significant impact on the translational potential of MPC-inhibition strategy in targeting cancer metabolism.
Disclosure of potential conflicts of interest
The author declares that there is no conflict of interest.
References
- Corbet C, Bastien E, Draoui N, Doix B, Mignion L, Jordan BF, Marchand A, Vanherck JC, Chaltin P, Schakman O, et al. Interruption of lactate uptake by inhibiting mitochondrial pyruvate transport unravels direct antitumor and radiosensitizing effects. Nat Commun. 2018;9:1208-018-03525-0. doi:10.1038/s41467-018-03525-0.
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. PMID:13351639.
- DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi:10.1126/sciadv.1600200. PMID:27386546.
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi:10.1038/nrc1478. PMID:15516961.
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi:10.1126/science.1160809. PMID:19460998.
- Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi:10.1016/j.ccr.2006.04.023. PMID:16766262.
- Parks SK, Pouyssegur J. Targeting pH regulating proteins for cancer therapy-progress and limitations. Semin Cancer Biol. 2017;43:66–73. doi:10.1016/j.semcancer.2017.01.007. PMID:28137473.
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. PMID:19033663.
- Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab. 2016;24:311–323. doi:10.1016/j.cmet.2016.07.003. PMID:27508876.
- Altman BJ, Stine ZE, Dang CV. From krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–634. doi:10.1038/nrc.2016.71. PMID:27492215.
- Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, Murphy AN, Metallo CM. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol Cell. 2014;56:425–435. doi:10.1016/j.molcel.2014.09.024. PMID:25458843.
- Li X, Han G, Li X, Kan Q, Fan Z, Li Y, Ji Y, Zhao J, Zhang M, Grigalavicius M, et al. Mitochondrial pyruvate carrier function determines cell stemness and metabolic reprogramming in cancer cells. Oncotarget. 2017;8:46363–46380. PMID:28624784.
- Nath K, Guo L, Nancolas B, Nelson DS, Shestov AA, Lee SC, Roman J, Zhou R, Leeper DB, Halestrap AP, et al. Mechanism of antineoplastic activity of lonidamine. Biochim Biophys Acta. 2016;1866:151–162. PMID:27497601.
- Ganapathy-Kanniappan S. Linking tumor glycolysis and immune evasion in cancer: Emerging concepts and therapeutic opportunities. Biochim Biophys Acta. 2017;1868:212–220. PMID:28400131.
- Bender T, Pena G, Martinou JC. Regulation of mitochondrial pyruvate uptake by alternative pyruvate carrier complexes. EMBO J. 2015;34:911–924. doi:10.15252/embj.201490197. PMID:25672363.