
ABSTRACT
Approximately half of all human cancers contain mutations in the TP53 tumor suppressor. In addition to mutations, there are single nucleotide polymorphisms (SNPs) in TP53 that can dampen p53 function, and can increase cancer risk and decrease the efficacy of cancer therapy. Approximately 6% of Africans and 1% of African-Americans express a p53 allele with a serine instead of proline at position 47 (Pro47Ser, or S47). The S47 variant is associated with increased breast cancer risk in pre-menopausal African Americans, and in a mouse model for the S47 variant, mice are predisposed to spontaneous cancers. We recently showed that the S47 variant is impaired for p53-mediated apoptosis in response to radiation and some genotoxic agents, particularly cisplatin. Here we identify the mechanism for impaired apoptosis of S47 in response to cisplatin. We show that following cisplatin treatment, the S47 variant shows normal stabilization and serine 15 phosphorylation, but reduced ability to bind to the peptidyl prolyl isomerase PIN1, which controls the mitochondrial localization of p53. This is accompanied by impaired mitochondrial localization of S47, along with decreased induction of cleaved caspase-3. Interestingly, we show that this defect occurs only for cisplatin and not for camptothecin. These findings show that normal tissues may respond differently to genotoxic stress depending upon this TP53 genotype. These data suggest that toxicity to cisplatin may be decreased in S47 individuals, and that this compound may be a superior treatment option for these individuals.
KeyWords:
Introduction
In normal cells and tissues, the p53 tumor suppressor is found at nearly undetectable levels. However, the steady state level of p53 is rapidly up-regulated by a variety of biological stresses, including oncogenic and genotoxic stress. In turn, p53 plays a critical role in suppressing tumorigenesis by directly regulating the expression of genes that promote apoptosis, cell cycle arrest, and senescence.Citation1, Citation2 In addition to this transcription-dependent pathway of programmed cell death, p53 also has a direct transcription-independent mitochondrial role in cell death. Specifically, p53 can directly translocate to the mitochondria in response to genotoxic stress.Citation3, Citation4 Once at the mitochondria, p53 can activate BCL-2 family proteins, such as BAX and BAK, to promote mitochondrial membrane permeabilization, in turn allowing for cytochrome c release, caspase activation and induction of apoptosis.Citation5, Citation6 At the mitochondria p53 can also bind and inhibit the function of anti-apoptotic BCL2 family members like Bcl-xl.Citation4
Given its critical role as a suppressor of tumorigenesis, it is not surprising that over fifty percent of human tumors contain inactivating mutations in TP53. In addition to mutation, the TP53 gene also contains a number of functionally-significant single nucleotide polymorphisms (SNPs) that impair p53 pathway function, and contribute to increased cancer risk and decreased efficacy of cancer therapy.Citation7 We recently characterized a non-synonymous SNP at codon 47 in TP53 that is predominantly found in people of African descent. Approximately 6% of Africans and 1% of African-Americans express a p53 allele with a serine residue at codon 47 (Pro47Ser, rs1800371, hereafter S47). In non-transformed cell lines expressing this variant, p53 demonstrates reduced phosphorylation on serine 46, as well as decreased ability to induce cell death.Citation8 Mice expressing the S47 variant show a significant increase in cancer incidence between 12-18 months of age, particularly hepatocellular carcinoma.Citation9 Finally, this variant is over-represented in African American women with pre-menopausal breast cancer.Citation10 We previously reported that both human and mouse cells containing the S47 variant were particularly resistant to cell death by cisplatin.Citation9 However, we did not define the mechanism for cisplatin resistance in these cells. Here-in we report that the S47 variant of p53 has a defect in the translocation to mitochondria following cisplatin treatment. This defect is associated with a reduced ability of this variant to interact with PIN1, which is a peptidyl prolyl isomerase that regulates p53 trafficking to mitochondria. Our data support the premise that impaired PIN1-binding and mitochondrial trafficking underlies the resistance of non-transformed S47 cells to cisplatin. These data have implications for personalized medicine approaches to cancer in S47 individuals.
Results
The S47 variant of p53 shows a defect in programmed cell death and mitochondrial trafficking in response to cisplatin
We previously reported that human lymphoblastoid cells (LCLs), mouse embryo fibroblasts (MEFs) and murine tissues containing the S47 variant of p53 were resistant to cell death by cisplatin.9 In order to probe the underlying mechanism, we treated early passage cultures of MEFs from mice containing wild type (WT) p53 or the S47 variant with cisplatin and assessed a time course for serine 15 phosphorylation (a marker for p53 activation) and the appearance of two cell death markers, cleaved lamin A and cleaved caspase 3. This experiment indicated that the level of total p53, and serine-15 phosphorylated p53, was nearly identical in WT and S47 MEFs treated with cisplatin, but the level of cell death was markedly different, with over 10-fold increased cleaved lamin A in WT MEFs compared to S47 MEFs after 24 hours of cisplatin (). QPCR of the pro-apoptotic p53 target genes PUMA, NOXA, BAX and others revealed modest or no differences in the upregulation of these targets in these cells (T. Barnoud, unpublished data). Additionally, cisplatin predominantly utilizes the transcription independent mitochondrial pathway of cell death.Citation11 Therefore we chose to investigate the direct mitochondrial pathway of p53-mediated cell death in these cells.
Figure 1. The S47 variant of p53 is impaired for transcription-independent apoptosis and mitochondrial localization of p53 in response to cisplatin.
(A) WT and S47 MEFs were treated with 10 μM CDDP for 0, 6, 24 hours and subjected to analysis by western blot using antibodies for the proteins indicated. HSP90 is included as a loading control. The data depicted are representative of three independent experiments from several independent MEF cultures of each genotype. (B,C) Western blot analysis of whole cell lysate (WCL) (B) and lysate from purified mitochondria (Mito) versus cytosolic fraction (Cyto) (C) isolated from WT and S47 MEFs untreated or treated with 10 μM CDDP for 12 hours. Lysates were probed for the mitochondrial proteins HSPA9 (GRP75) and cytochrome c, and the nuclear/cytosolic protein PCNA as an assessment of purity.
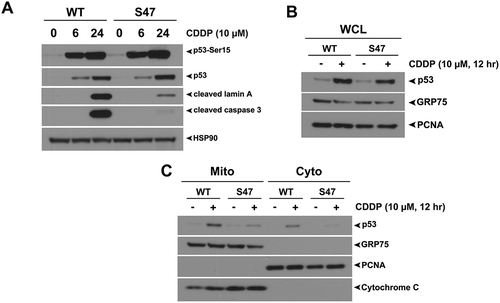
To assess the ability of WT and S47 forms of p53 to traffic to mitochondria in MEFs, we purified mitochondria using mannitol gradients from untreated and cisplatin-treated WT and S47 MEFs and used Western blotting to assess the level of p53 co-localized with this organelle. While lysates from whole cell extracts (WCL) show similar induction of p53 between WT and S47 treated cells (), we found markedly increased amount of the WT protein at the mitochondria of treated cells compared to S47 (). These data indicate there is a correlation between impaired cell death and impaired mitochondrial trafficking of the S47 variant in response to cisplatin. We next sought to determine whether this defect of S47 was true following other forms of genotoxic stress.
Comparable mitochondrial localization and transcription-independent apoptosis in WT and S47 MEFs treated with camptothecin
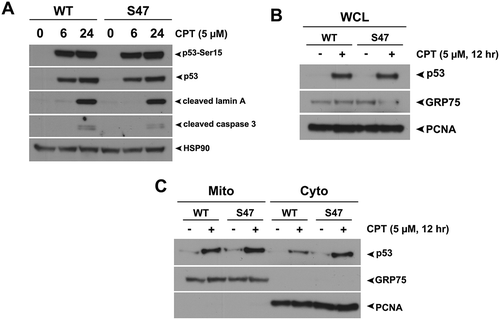
We next treated WT and S47 MEFs with camptothecin (CPT) for 24 hours and analyzed cell death and mitochondrial localization of p53. CPT led to similar induction of total p53 and phosphorylation at Serine-15. Interestingly, unlike cisplatin, we saw no differences in apoptosis (cleaved lamin A or cleaved caspase 3) between WT and S47 treated MEFs treated with CPT (). Additionally, similar levels of WT and S47 p53 were found at the mitochondria of CPT-treated cells, suggesting no difference in transcription-independent cell death between WT and S47 MEFs ( and ). These data suggest that the impaired mitochondrial localization and cell death of S47 is specific to certain genotoxic stresses, such as cisplatin.
Figure 2. Equivalent transcription-independent apoptosis and mitochondrial localization of WT p53 and S47 in response to camptothecin.
(A) WT and S47 MEFs were treated with 5 μM camptothecin (CPT) for 0, 6, 24 hours and subjected to analysis by western blot using antibodies for the proteins indicated. HSP90 is included as a loading control. The data depicted are representative of three independent experiments from several independent MEF cultures of each genotype. (B,C) Western blot analysis of whole cell lysate (WCL) (B) and lysate from purified mitochondria (Mito) versus cytosolic fraction (Cyto) (C) isolated from WT and S47 MEFs untreated or treated with 5 μM CPT for 12 hours. Lysates were probed for the mitochondrial proteins HSPA9 (GRP75) and cytochrome c, and the nuclear/cytosolic protein PCNA as an assessment of purity.
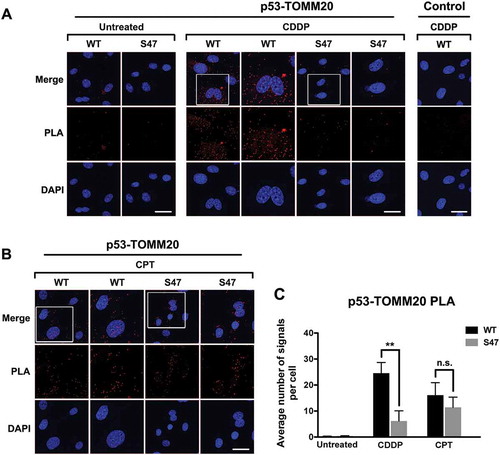
To confirm and extend our findings on mitochondrial trafficking of p53, we performed Proximity Ligation Assays (PLA) on WT and S47 MEFs following cisplatin treatment and monitored p53 localization at the mitochondria by assessing its interaction with the mitochondrial protein TOMM20, which is known to interact with mitochondrial p53.Citation12 In untreated cells there was little evidence for complexes between p53 and TOMM20. However, in cisplatin-treated MEFs there was a significant increase in p53-TOMM20 signal by PLA, but the S47 variant was markedly decreased (p<0.01 between WT and S47 treated MEFs) ( and ). As a control we performed immunostaining for p53 and TOMM20 in WT and S47 MEFs, and noted no differences in abundance or localization (Supp. Fig. 1). We next performed similar experiments using CPT. This analysis revealed no difference in the association between WT p53 and S47 protein with TOMM20 following CPT treatment ( and ).
Figure 3. Proximity ligation assays (PLA) reveal a cisplatin-specific defect for S47 in mitochondrial localization.
(A, B) An in-situ proximity ligation assay (PLA) was performed in WT and S47 MEF cells treated with either 10 μM cisplatin (CDDP) (A) or 5 μM camptothecin (CPT) (B) for 12 hours. Each red dot represents an interaction between endogenous p53 and TOMM20 proteins (scale bar, 25 μm). Cells stained with p53 antibody alone were used as a negative control. DAPI nuclear staining is shown in blue. The white boxed image is magnified in the panel to the right. S47 cells treated with CDDP show a significantly lower number of PLA-positive dots compared to WT cells, indicating a defect in S47 trafficking to the mitochondria.(C) Quantification of (A and B), measured as the average number of PLA signals per nuclei. Data were quantitated by counting the number of cells in five random fields of view per experimental group.
Impaired binding of S47 to the prolyl-isomerase PIN1 following cisplatin treatment
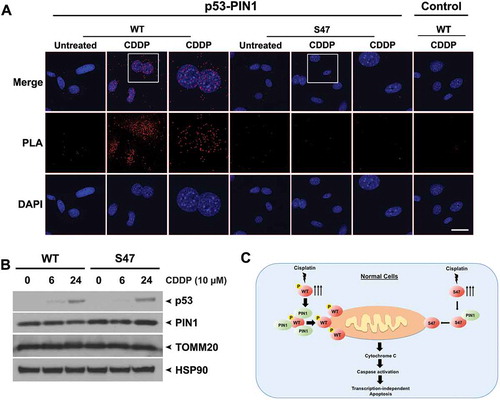
PIN1 is a peptidyl prolyl isomerase that interacts directly with p53 and is required for efficient localization of p53 to mitochondria.Citation13 Interaction with PIN1 requires phosphorylation of serine and threonine residues that are followed by a proline, and some reports indicate that serine 46 phosphorylation, which is defective in S47 cells, is a major docking site on p53 for PIN1.Citation14 To test the hypothesis that the defect of S47 localization to mitochondria is due to impaired PIN1 binding, we performed PLA in cisplatin-treated cells to assess the interaction of WT and S47 p53 with PIN1. PLA analyses showed that the association between S47 and PIN1 was markedly decreased, compared to WT p53, following cisplatin treatment (). As a control, western blot analysis shows equivalent levels of p53 and PIN1 in WT and S47 cells (). These data support the premise that interaction with PIN1 may regulate mitochondrial trafficking of p53, and that the decreased interaction of PIN1 with S47 may underlie its defect in mitochondrial trafficking and cisplatin-mediated cell death (see model ).
Figure 4. Decreased binding of S47 to PIN1 following cisplatin treatment.
(A) An in-situ proximity ligation assay (PLA) was performed in WT and S47 MEFs untreated or treated with 10 μM cisplatin (CDDP) for 12 hours. Each red dot represents an interaction between endogenous p53 and PIN1 proteins (scale bar, 25 μm). Cells stained with p53 antibody alone were used as a negative control. DAPI nuclear staining is shown in blue. The white boxed image is magnified in the panel to the right.(B) Western blot analysis of PIN1 and TOMM20 protein levels in WT and S47 MEFs treated with 10 μM CDDP for 0, 6, 24 hours. HSP90 is included as a loading control.(C) Proposed model highlighting the defect in mitochondrial trafficking and cell death in S47 cells treated with cisplatin.
Discussion
In addition to somatic mutations, coding region SNPs in the TP53 gene, and in p53 pathway genes like MDM2, can have a marked impact on p53 function.Citation7 For example, the SNP309 polymorphism in the MDM2 promoter is associated with increased cancer risk in a mouse model and in humans.Citation15-Citation17 At codon 72 of p53, the Arg72Pro polymorphism is associated with increased body mass index and risk for type 2 diabetes.Citation18-Citation20 We and others have identified a coding region polymorphism in TP53 (Pro47Ser) that appears to be predominantly found in people of African descent. We previously showed that the S47 variant of p53 demonstrates reduced phosphorylation on serine 46, as well as decreased ability to induce cell death by radiation and genotoxic stress.Citation8 We also generated a mouse model and found that S47 mice have an increased incidence of hepatocellular carcinoma and other cancers.Citation9 Finally, we showed that this variant conferred considerable resistance to cisplatin in non-transformed cells and in cells from the kidney of S47 mice.Citation9 However, the mechanism underlying this cisplatin resistance was not clear.
It is well established that p53 can regulate apoptosis by both transcription-dependent and independent mechanisms.Citation21 We find that S47 MEFs, which are more resistant to cisplatin, show decreased trafficking of p53 to the mitochondria, suggesting that S47 cells show decreased apoptosis by virtue of a defective direct mitochondrial cell death program. We also show that PIN1, which is required for mitochondrial localization of p53,Citation13 shows markedly decreased binding to the S47 variant of p53, thus providing a mechanistic explanation for our findings (). The correlation between impaired mitochondrial localization and impaired cell death are consistent with the findings of others, that unlike many other genotoxic stresses, cisplatin preferentially induces cell death by the direct mitochondrial pathway of p53-mediated cell death.Citation11 Our data that PIN1 binding correlates with trafficking of p53 to the mitochondria likewise fits the findings of others that this protein is a key regulator of p53 trafficking to this organelle, and cell death. However, one question remaining is why PIN1 fails to bind to the S47 variant. Because the S47 variant is defective at serine 46 phosphorylation, one answer might be the differences in serine 46 phosphorylation account for the differences in mitochondrial trafficking between WT and S47 protein. However, we find that there are no differences in serine 46 phosphorylation between WT p53 cells treated with camptothecin and cisplatin (Supp. Fig. 2), suggesting that other levels of regulation are clearly involved. Along these lines, some reports have shown that PIN1 binding to p53 can occurs on multiple phosphorylation sites in this protein.Citation22,Citation23 Additionally, other post-translational modifications, such as acetylation and C-terminal phosphorylation of p53, have been implicated in the mitochondrial trafficking of this protein.Citation24,Citation25 Therefore, the reason that S47 is impaired for mitochondrial trafficking only in response to cisplatin remains to be resolved.
Our findings have implications for personalized medicine approaches to treating cancer in African-descent individuals. Specifically, we find that normal non-transformed S47 cells show resistance to cisplatin, shown here due to decreased mitochondrial cell death. Conversely, we find that transformed S47 cells actually show increased sensitivity to cisplatin.Citation26 Therefore, cisplatin would appear an ideal choice for cancer therapy in S47 individuals: normal cells show reduced toxicity, but transformed cells show increased cytotoxicity. In the case of cisplatin, where cytotoxicity in normal cells is not insignificant, these findings augur well for the use of this compound in S47 individuals. We posit that genetic screening of African-descent individuals for tumors where cisplatin is a therapeutic option, such as bladder, lung and ovary, should be performed in order to best inform therapy in these individuals.
Materials and methods
Cell Culture and Reagents
Wild type (WT) and S47 primary MEFs were generated as previously described.Citation27 These were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Corning Cellgro™) supplemented with 10% FBS (HyClone™, GE Healthcare Life Sciences) and 100 units of Penicillin/Streptomycin (Corning Cellgro™). Cells were grown in a 5% CO2 humidified incubator at 37°C. For treatments, cisplatin (Acros Organics, Cat. No. 193760010) and camptothecin (Cayman Chemicals) were used at the indicated concentrations.
Mitochondria Isolation and Immunoblotting
Mitochondria were purified using our previously published protocols.5 Western blotting was performed on 15 µg of whole cell and mitochondrial lysates. The following primary antibodies were used: p53 (2524S), phospho-p53 (Ser15) (9284S), phospho-p53 (Ser46) (2521), Cleaved Caspase-3 (9661S), Cleaved Lamin A (2035S), HSP90 (4877S), GAPDH (2118S) (all from Cell Signaling Technologies), Cytochrome C (556433) (BD Biosciences), PCNA (sc-56), HSPA9/GRP75 (C19), PIN1 (sc-15340), and TOMM20 (sc-11415) (Santa Cruz Biotechnology). Secondary antibodies conjugated to horseradish peroxidase were used at a dilution of 1:10,000 (Jackson Immunochemicals). ECL was purchased from GE Healthcare (RPN2106).
Immunofluorescence and Proximity Ligation Assays
For immunofluorescence analysis of endogenous proteins, primary MEFs were grown overnight on Lab-Tek II 4-well chamber slides (Thermo Fisher Scientific). Following treatment with cisplatin (CDDP) or camptothecin (CPT) for 12 hours, cells were fixed in 4% paraformaldehyde for 10 minutes, permeabilized in 0.5% Triton X-100 for 5 minutes, stained with antibodies to p53 (2524S) and TOMM20 (sc-11415) and then incubated with secondary antibodies at 37°C for 1 hour. Cells were then stained with DAPI prior to microscopy using a Leica TCS SP5 confocal microscope. PLA was performed as described previously.Citation28 Briefly, wild type (WT) and S47 primary MEFs were grown on Lab-Tek II 8-well chamber slides and fixed for endogenous protein detection as described above, using the Duolink kit (Sigma Aldrich). As a negative control, one of the primary antibodies was omitted. Brightness and contrast were adjusted to allow for the clearest resolution of images. PLA signals were quantified using Image J software.
Statistical Analysis of Data
All data are reported as the mean ± standard deviation. The two-tailed unpaired Student t-test was performed using GraphPad Prism 6.0 software. p values are as indicated: * = p<0.05, ** = p<0.01, and n.s. = p > 0.05.
Disclosure of potential conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments and Funding
This work was supported by R01 CA201430 and CA102184 (M.E.M.), and F32 CA220972 (T.B.). Support for the Core Facilities used in this study was provided by Cancer Center Support Grant (CCSG) CA010815 to The Wistar Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to acknowledge the Histotechnology, Laboratory Animal, Molecular Screening and Imaging facilities at The Wistar Institute. The authors would also like to thank Donna George and Julie Leu (University of Pennsylvania) for assistance and thoughtful discussions.
Author contributions
Study conception and design: A.B-K., T.B., M.E.M.Acquisition and analysis of data: A.B-K., T.B., M.E.M.Drafting of manuscript: T.B., M.E.M.Proofing and revision of manuscript: A.B-K., T.B., M.E.M.
Supplemental Material
Download Zip (7.1 MB)Supplemental data
Supplementry data can be accessed here
Additional information
Funding
Related Research Data
References
- Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2010; 2:a000935.
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev 2012; 26:1268–86.
- Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 2004; 24:6728–41.
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577–90.
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004; 6:443–50.
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303:1010–4.
- Basu S, Murphy ME. Genetic Modifiers of the p53 Pathway. Cold Spring Harb Perspect Med 2016; 6:a026302.
- Li X, Dumont P, Della Pietra A, Shetler C, Murphy ME. The codon 47 polymorphism in p53 is functionally significant. J Biol Chem 2005; 280:24245–51.
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 2016; 30:918–30.
- Murphy ME, Liu S, Yao S, Huo D, Liu Q, Dolfi SC, et al. A functionally significant SNP in TP53 and breast cancer risk in African-American women. NPJ Breast Cancer 2017; 3:5.
- Bragado P, Armesilla A, Silva A, Porras A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis 2007; 12:1733–42.
- Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 2003; 33:357–65.
- Sorrentino G, Mioni M, Giorgi C, Ruggeri N, Pinton P, Moll U, et al. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell Death Differ 2013; 20:198–208.
- Grison A, Mantovani F, Comel A, Agostoni E, Gustincich S, Persichetti F, et al. Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc Natl Acad Sci U S A 2011; 108:17979–84.
- Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, et al. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res 2006; 66:5104–10.
- Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004; 119:591–602.
- Post SM, Quintas-Cardama A, Pant V, Iwakuma T, Hamir A, Jackson JG, et al. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell 2010; 18:220–30.
- Kung CP, Leu JI, Basu S, Khaku S, Anokye-Danso F, Liu Q, et al. The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell Rep 2016; 14:2413–25.
- Kung CP, Murphy ME. The role of the p53 tumor suppressor in metabolism and diabetes. J Endocrinol 2016; 231:R61–R75.
- Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung C-P, Murphy ME. The p53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Frontiers in Endocrinology 2018; 9.
- Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 2005; 17:631–6.
- Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, et al. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002; 419:853–7.
- Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, et al. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002; 419:849–53.
- Sykes SM, Stanek TJ, Frank A, Murphy ME, McMahon SB. Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53. J Biol Chem 2009; 284:20197–205.
- Castrogiovanni C, Waterschoot B, De Backer O, Dumont P. Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis. Cell Death Differ 2018; 25:190–203.
- Basu S, Barnoud T, Kung CP, Reiss M, Murphy ME. The African-specific S47 polymorphism of p53 alters chemosensitivity. Cell Cycle 2016; 15:2557–60.
- Frank AK, Leu JI, Zhou Y, Devarajan K, Nedelko T, Klein-Szanto A, et al. The codon 72 polymorphism of p53 regulates interaction with NF-{kappa}B and transactivation of genes involved in immunity and inflammation. Mol Cell Biol 2011; 31:1201–13.
- Basu S, Gnanapradeepan K, Barnoud T, Kung CP, Tavecchio M, Scott J, et al. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1alpha. Genes Dev 2018; 32:230–43.