
ABSTRACT
Oncolytic adenoviruses (Ads) are cancer selective tumoricidal agents; however their mechanism of Ad-mediated cancer cell lysis, or oncolysis, remains undefined. This report focuses upon the autophagy mediator c-JUN n-terminal kinase (JNK) and its effects upon Ad oncolysis and replication. Previously, E1b-deleted Ads have been used to treat several hundred cancer patients with limited clinical efficacy. We hypothesize that by studying the potential interactions between E1b and JNK, mechanisms to improve oncolytic Ad design and cancer therapeutic efficacy may be elucidated. To test this hypothesis, E1b was selectively deleted from the Ad genome. These studies indicated that Ads encoding E1b induced JNK phosphorylation predominately occurred via E1b-19K. The expression of another crucial Ad gene E1a was then overexpressed by the CMV promoter via the replication competent Ad vector Adhz69; these data indicated that E1A also induced JNK phosphorylation. To assess the effects of host cell JNK expression upon Ad oncolysis and replication, siRNA targeting JNK1 and JNK2 (JNK1/2) were utilized. The oncolysis and replication of the E1b-19K wild-type Ads Ad5 and Adhz63 were significantly attenuated following JNK1/2 siRNA transfection. However the oncolytic effects and replication of the E1b-19K deleted Ad Adhz60 were not altered by JNK1/2 siRNA transfection, further implicating the crucial role of E1b-19K for Ad oncolysis and replication via JNK phosphorylation. This study has demonstrated for the first time that JNK is an intriguing molecular marker associated with enhanced Ad virotherapy efficacy, influencing future Ad vector design.
1. Introduction
Oncolytic Ads represent an emerging class of cancer therapeutics with clear cancer selectivity in vitro.Citation1–Citation3 Specifically, E1b-deleted Ads have been shown to selectively lyse cancer cells very efficiently in vitro; however, these viruses were relatively ineffective during phase II and III clinical trials.Citation4–Citation7 To enhance the efficacy of Ad therapy, a more complete understanding of Ad-mediated cancer cell lysis or “oncolysis” is necessary. Among the programmed cell death pathways (apoptosis, necrosis, and autophagy), autophagy appears to be associated with increased Ad oncolysis and replication which is discussed in greater detail in this report.Citation8,Citation9
Many viruses have been shown to induce autophagy to either suppress or stimulate specific stages of their respective life cycles. For example, autophagy has been shown to inhibit HIV-1,Citation10 HSVCitation11 and Influenza A viruses.Citation12 However, autophagy has been shown to promote Ad oncolysis and replication in glioma,Citation8,Citation13 ovarianCitation14 and lungCitation8,Citation9,Citation15 tumors, indicating the significance of autophagy to enhance oncolytic Ad therapy in these tissues. Furthermore, the autophagy inhibitors 3MA and Baf-A1 were shown to protect against Ad oncolysis and inhibit Ad replication.Citation9,Citation15,Citation16 The deletion of E1b has also been shown to inhibit the conversion of LC3-I to LC3-II and the degradation of p62 which are markers of autophagy.Citation9,Citation17
Rapamycin treatment was observed to enhance E1b-deleted Ad oncolysis and replication. Studies have indicated that mTOR activation supported the S-phase entry of G0-phase primary cells and that this activation was dependent upon the interaction of the adenovirus protein E4-ORF4 and the protein phosphatase 2A (PP2A) binding domain.Citation18
Interestingly, Ads have been shown to induce autophagy independently of the AMPK/ULK1/mTOR cell starvation pathway in cancer cells.Citation13 Previous studies indicated that Ads expressing E1b induced autophagy via JNK phosphorylation.Citation8,Citation13,Citation19 Mechanistically, JNK induces autophagy via the phosphorylation of BCL-2 to overcoming the inhibition of Beclin-1 via un-phosphorylated BCL-2 in the cytosol which freely forms an inhibitory complex with Beclin-1.Citation13,Citation19–Citation24 JNK and the map kinase (MAPK) pathway also play a crucial role to upregulate ATG5 expression.Citation25 JNK1 and JNK2 are also known known as MAPK8 and MAPK9 respectively. The first report, to our knowledge, to demonstrate that Ads induced JNK phosphorylation via E1b was published by See and Shi in 1998; however, this report did not indicate if Ads induced JNK phosphorylation to enhance their own oncolytic and replicative phenotypes respectively.Citation26 Klein et al. reported that JNK host cell expression was essential for Δ24RGD to induce autophagy and cancer oncolysis in vitro and in vivo, however these effects were not determined using E1b-deleted Ads.Citation13 The effects of JNK upon E1b-deleted Ads and their dependence upon JNK phosphorylation for efficient oncolysis and replication remains unknown.
As it pertains to Ad cancer therapy, Ads have been shown to manipulate both apoptosis and autophagy via the expression of E1a, E1b-19K, and E1b-55K to lyse cancer cells in a highly-coordinated manner. JNK phosphorylation has been shown to induce both apoptosis and autophagy due to its role in ER-stress responses.Citation27 E1A is known to stabilize TP53 to induce apoptosis,Citation28 while E1b-19K and E1b-55K proteins counteract these effects by antagonizing BAK/BAX and TP53 respectively. E1b-19K and E1b-55K are also known to stimulate MKK7-mediated JNK phosphorylation to induce autophagy.Citation26,Citation29 Therefore, it appears E1b inhibits apoptosis and drives Ad oncolysis towards an autophagy-mediated mechanism of oncolysis.
In this study, the effects of Ads upon JNK phosphorylation via E1a or E1b were assessed using Ads with selective E1a overexpression or E1b-deletions in A549 lung cancer cells. These data indicate that both E1b and E1a are crucial for efficient JNK phosphorylation. The suppression of JNK1/2 by siRNA protected A549 cancer cells against E1b-19K wild-type Ad oncolysis and replication. These studies also demonstrated for the first time that JNK phosphorylation is not only essential for Ad oncolysis, but also for Ad replication, underscoring the potential of JNK as a molecular target to enhance the therapeutic effects of oncolytic Ads.
2. Materials and methods
2.1. Cell lines and culture conditions
HEK293 (ATCC, CRL-1573) human embryonic kidney and A549 (ATCC, CCL-185) human lung carcinoma cells were purchased from the American Type Culture Collection (ATCC). A549 and HEK293 cells were maintained in DMEM supplemented with 10% fetal bovine serum, L-glutamine, and penicillin/streptomycin (100 U/mL). All other cell culture reagents were obtained from VWR. All cells were cultured and maintained in humidified 5% CO2 incubators at 37°C.
2.2. Adenoviral vectors
The Ad dl309 (Ad5) is E1 wild-typeCitation30 and was used as a non-selective Ad control for these studies. Adhz63 is an E1b55K-deleted Ad similar to dl1520 and H101. Therefore, Adhz63 was used as a cancer selective control.Citation28 Adhz69 is an E1b55K-deleted Ad that over-expresses E1A via the CMV promoter.Citation31 Adhz60 is an E1b55K and 19K-deleted vector used as a cancer selective and E1b19K-deleted Ad control for these studies.Citation28 AdUV is an oncolytic Ad mutated by sequential UV-light treatment with an E1b wild-type backbone; its construction and complete DNA sequence were described previously.Citation32,Citation33 AdGFP is E1 deleted (E1a and E1b), with green fluorescent protein (GFP) expression driven by the CMV promoter.Citation2 AdGFP was utilized as a negative control and does not replicate nor induce cytopathic effect (CPE) in infected cells.Citation17,Citation34 All Ads used in this study are based upon Ad5 backbone sequences (GENEID# AC_000008.1).
2.3 Plasmid amplification and transfection
Bacterial transformation was carried out accordingly to manufacturer instructions (Invitrogen). Transformed bacteria were then processed to isolate DNA plasmids using Qiagen Mini, or Maxi-scale kits according to the manufacturer’s instructions. The CMV-E1b19K plasmid was a generous gift from Dr. Eileen White at Rutgers University.Citation35 The pCycE-GFP plasmid was constructed by Dr. Heshan Zhou at the University and Louisville and was used as a negative control; pCycE-GFP was also of similar size to CMV-E1b19K and was therefore used to determine plasmid transfection efficiency.Citation36 2 × 105 A549 cells were seeded onto 6-well plates overnight. The next day, these cells were 60–70% confluent the next day at the time of transfection to ensure maximal transfection efficiency. These cells were then transfected with 2 µg of plasmid DNA using Jetprime according to the manufacturer’s protocol. The media of these transfected cells were then changed after 4 h to maximize transfected cell viability. Using these conditions, a transfection efficiency of at least 85% was observed.
2.4 siRNA transfection
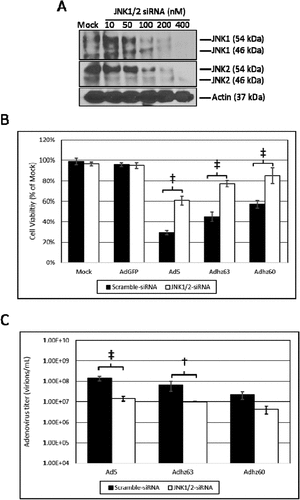
To ascertain the role of JNK expression upon Ad oncolysis and replication, siRNA transfection was utilized. Interestingly, JNK1 and JNK2 are functionally homologous in their autophagy induction phenotypes during Ad infection as shown in Ref. 13, therefore the suppression of both JNK1 and JNK2 was required to observe the effects of JNK upon Ad oncolysis.Citation13 Considering these factors, siRNA species targeting both JNK 1 and 2 (JNK1/2-siRNA) were utilized in this study: JNK1/2 5ʹ- AAAGAAUGUCCUACCUUCU-3ʹ was initially described by Dr. Li et al..Citation37 This siRNA sequence targets a common mRNA sequence found in JNK1 and JNK2 at 377 and 425 nucleotides downstream from their respective start codons.Citation37 As the JNK1/2-siRNA was ordered by email using the sequence from Dr. Li and its duplex complement from Thermo Fisher Scientific.Citation37 The optimal siRNA concentration to suppress JNK1 and JNK2 was assessed by transfecting A549 cells with 10 to 400 nM of JNK1/2-siRNA. These transfected cells were then lysed and studied by Western blot analysis after 24 hours to determine whether JNK1 and JNK2 protein production was suppressed. 120 µg of these protein isolates were electrophoresed on 10% SDS-PAGE gels and JNK1 and JNK2 production was probed using JNK1 (1:100; Santa Cruz, sc-137018) and JNK2 (1:200; Santa Cruz, sc-271133) selective antibodies from Santa Cruz.
2.5 Virus release and titration
A549 cells were seeded onto 12-well plates at a density of 4 × 104 cells per well and allowed to adhere overnight. Cells were then treated as indicated and harvested with a cell scraper. These samples were then subjected to 3 freeze-thaw cycles to release all the Ads into solution. For the determination of total Ad titer by the median tissue culture infective dose (TCID50) method as described previously.Citation3 HEK293 cells were seeded overnight onto 96-well plates at a density of 1 × 103 cells per well and infected with virus samples serially diluted 10 fold. The presence or absence of CPE in HEK293 cells was then recorded after a minimum of 7 days to calculate the viral titers.
2.6 Western blot analysis
A549 cells were seeded onto 60 mm dishes overnight at a density of 3 × 105 cells per dish. Cells were collected and centrifuged at 1500 RPM (453 RCF) at 4°C for 5 min using an Eppendorf 15 amp 5810R refrigerated centrifuge equipped with a A-4–62 rotor (Fisher Scientific, 05–413-112). The cell pellets were then washed with PBS prior to lysis with RIPA buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS with PI containing 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), pepstatin A, trans-epoxysuccinyl-l-leucylamido-(4-guanidino)butane (E-64), bestatin, leupeptin, and aprotinin (10 mL/1 × 106 cells; Sigma, P9599). The cell lysates were then incubated on ice in PI containing RIPA buffer for 30 min and homogenized every 10 min using a vortex-genie 2 (Scientific Industries, SKU-0236). These lysates were then centrifuged at 14,500 RPM (196,000 RCF) at 4°C for 10 min using a micromax RF microcentrifuge equipped with an IEC 851 rotor to pellet cell debris formed during lysate preparation (Thermo Fisher Scientific, 05–112-114D). The pellet was discarded and the supernatant was stored at −80°C for further experimentation. Protein concentrations were determined using the Pierce BCA protein assay kit according to the manufacturer’s instructions (Thermo Fisher Scientific, 23225). Equal amounts of cellular protein were resolved by electrophoresis through 8% (E1A), 10% (JNK, p-JNK, JNK1, JNK2, E1b-19K, actin) or 12% (LC3-I and LC3-II) SDS-polyacrylamide gels prior to transfer to methanol activated PVDF membranes (GE healthcare, 10600058) using a semi-dry transfer apparatus (BIO-RAD, 1703940). Membranes were then blocked using 5% nonfat milk prepared in TBST for 1 h at room temperature. To detect protein expression, membranes were incubated with the following primary antibodies: rabbit-anti-human-LC3 (1:3000; Novus Biologicals, NB100-2220), rabbit-anti-human-actin (1:2000; Sigma, A5316), rabbit-anti-human p-JNK which binds the phosphorylated T183/Y185 residues (1:1000; Cell Signaling, 9251), rabbit-anti-human-JNK (1:1000; Cell signaling, 9252), mouse-anti-human-JNK1 (1:100; Santa Cruz, sc-1648), mouse-anti-human JNK2 (1:200; Santa Cruz, sc-271133), mouse-anti-Adenovirus2-E1b19K (1:150; Calbiochem, no longer commercially available) or mouse-anti-adenovirus-E1A (1:1000; BD Pharmagen, 554155) at 4°C overnight on a lab-line thermal rocker (Thermo Fisher Scientific, 4637Q). The mouse-anti-Adenovirus2-E1b19K antibody is cross-reactive with Ad serotype 5 (Ad5). Primary antibody binding was detected by the incubation with the horseradish peroxidase (HRP) linked anti-mouse or anti-rabbit immunoglobulin (Ig) diluted 1:5000 for 1 h at room temperature (Amersham, NA931 & NA934). All antibodies were diluted in TBST. Enhanced chemiluminescence (ECL) reagents were used to detect HRP-linked secondary antibody binding according to the manufacturer’s instructions (Amersham, RPN2106).
2.7 Western blot quantification and densitometry
Western blots were quantified via densitometric analysis using Gel-pro analyzer 4.0 software to determine the respective IOD values for each band. These IOD values were then normalized to their respective actin loading control bands. To determine the fold-changes for a given protein, these normalized experimental values were divided by them the values of the mock treated cells. Using this approach, mock treated cells were standardized as 1-fold change relative to the experimental groups as indicated. For the quantification of JNK and p-JNK, the IOD values of both 46 and 54 kDa bands (p-JNK; T183 and Y185) were amalgamated prior to the calculation of p-JNK fold changes. For LC3 quantification, the top (LC3-I) and bottom (LC3-II) bands were quantified separately and both band intensities were normalized to actin. These normalized LC3-II values were then divided by the LC3-I values to generate the LC3-II/I ratio and these values were then divided again by the mock LC3-II/I ratio (1-fold) to determine the fold-changes in LC3-II/I in these experimental groups respectively.
2.8 Cytotoxicity assay
A549 cells were seeded at a density of 2 × 104 cells per well onto 24-well plates and allowed to adhere overnight. Cytotoxicity was assessed by crystal violet staining at the time-points indicated.Citation38 Suspended cells were aspirated, and the adherent cells were then fixed via incubation with 3.7% formaldehyde for 25 min at room temperature (RT). Excess formaldehyde was washed away using PBS. Cells were then stained using 1% crystal violet for 30 min at RT. Excess crystal violet was washed away with water. These crystal violet stained wells were then scanned using an HP Scanjet 4070 scanner (HP, Palo Alto, CA, USA). The remaining crystal violet was then solubilized with a 2% SDS solution, and the sample ODs were measured at 590 nm using a Synergy HT Multi-Mode Microplate Reader (Bio-Tek, BTS1A). These OD values were then normalized to mock-treated cells, converting each sample OD into the percent (%) cell viability by the formula, cell viability % = (OD of treated cells/OD of mock-treated cells) × 100.
2.9 Statistical analysis
All experiments were repeated at least 3 times. Quantification of results was reported as means of 3 independent experiments plus or minus (±) the SD. Statistical significance was set at p-value less than 0.05. The statistical analysis of the p-JNK fold changes induced by Ad5 treatment across time (, )) was conducted using one-way ANOVA with Dunnett’s post-test. All other statistical tests were conducted using one-way ANOVA with Tukey’s post-test for multiple comparisons. All statistical tests were conducted using GraphPad Prism 6 software (Microsoft).
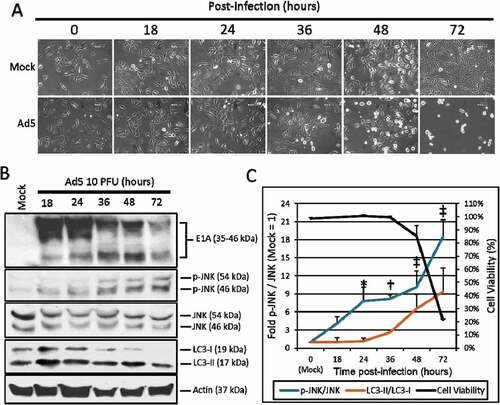
Figure 1. The effect of Ad5 upon JNK phosphorylation. A549 cells were mock treated or infected with Ad5 at 10 PFU/cell and then observed from 0 to 72 h post-infection. (a) A549 cell morphology following mock or Ad5 treatment was photographed at the indicated time-points. Mock treated cells were non-infected, while only Ad5 treated A549 cells displayed CPE. Pictures were taken using an inverted microscope at 200x total magnification. 600 µm scale bar is located in the top right corner. (b) A549 cell lysates were treated and harvested at the time-points indicated and observed for protein production via Western blot analysis. (c) These Western blot data were quantified via densitometric analysis using Gel-pro analyzer 4.0 software and these IOD values were normalized to actin. These values were then converted to fold changes by dividing the values by the values for mock treated cells. Cell viability was determined by crystal violet staining and plotted on the secondary y-axis. Results are expressed as the average of 3 experiments plus or minus the standard deviation. Statistical analysis was performed using one-way ANOVA by Dunnett’s test for multiple comparisons relative to the mock control (1 fold). * indicates p-value < 0.05, † indicates p-value < 0.01, ‡ indicates p-value < 0.001.
3. Results
3.1 Adenovirus serotype 5 induced JNK phophorylation in A549 cells
To observe the effect of Ads upon JNK phosphorylation, A549 lung cancer cells were infected with the wild-type serotype 5 Ad dl309 (Ad5) at 10 plaque forming units per cell (PFU/cell). Infected or non-infected (Mock) cells were then observed for CPE ()) and the phosphorylation of JNK via Western blot analysis from 18 to 72 h post-infection ()). JNK phosphorylation was observed at 18 h (4.1 fold) reaching the greatest level of JNK phosphorylation at 72 h (18.4 fold) post-infection. The highest levels of JNK phosphorylation (48 and 72 h) was associated with changes in A549 cell morphology consistent with Ad CPE (, )). To quantify the effects of Ad5 upon A549 cell viability, A549 cells were stained with crystal violet at the time-points indicated. By plotting these fold changes in JNK phosphorylation, cell viability, and LC3-II production over time, the effects of Ad5 upon the induction of JNK phosphorylation occurred first (18 h), prior to the induction of LC3-II (36 h), and A549 oncolysis, and CPE induction by Ad5 at 48 and 72 h, respectively ()). These data indicate that Ad5 oncolysis and replication were associated with increased JNK phosphorylation in A549 cells.
3.2 The effects of e1b upon JNK phosphorylation
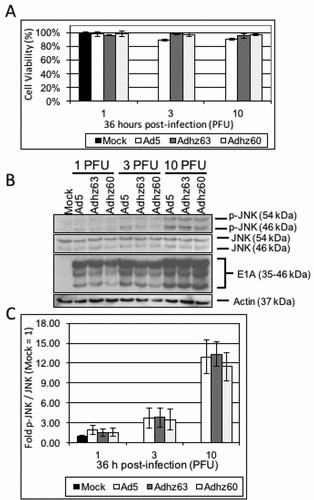
To determine whether E1b-deleted Ads induced JNK phosphorylation, A549 cells were infected with the E1b wild-type Ad5, the E1b55K-deleted Adhz63 and the E1b55K and 19K-deleted Adhz60 at 10 PFU/cell to observe p-JNK production by Western blot analysis. Differences in p-JNK production were first assessed at 36 h post-infection. The 36h time-point was selected as it was prior to A549 cells lysis at 10 PFU/cell ()). However, no significant differences in JNK phosphorylation were observed between E1b wild-type and E1b-deleted Ad at any of the PFU/cell treatments at 36 h (1, 3, and 10 PFU/cell; , )).
Figure 2. The effect of selective E1b-deleted Ads upon JNK phosphorylation at 36 h. A549 cells were mock-treated or infected with Ad5, Adhz63, and Adhz60 at 1, 3 and 10 PFU/cell respectively. (a) Crystal violet staining of A549 cells treated as indicated at 36 h post-infection. (b) A549 cell lysates were prepared at 36 h post-infection and observed for the production of the indicated proteins via Western blot analysis. (c) These Western blot data were quantified via densitometric analysis using Gel-pro analyzer 4.0 software and these IOD values were normalized to actin. These values were then converted to fold changes by dividing the values by the values for mock treated cells. Results were expressed as the average of 3 experiments plus or minus the standard deviation.
The effects of Ad5, Adhz63 and Adhz60 upon JNK phosphorylation were then assessed at 48 and 72 h post-infection when A549 cell death was nearly complete at 10 PFU/cell ()). These data indicated that p-JNK production by Ad5 (32.2 fold), Adhz63 (28.1 fold) and Adhz60 (10.9 fold) were significantly different at 72 h post-infection ()).
Figure 3. The effect of E1b-19K upon JNK phosphorylation. A549 cells were mock treated or infected with AdGFP, Ad5, Adhz63 and Adhz60 at 10 PFU/cell. (a) CPE induction in mock, AdGFP, Ad5, Adhz63 and Adhz60 treated cells. Pictures were taken using an inverted microscope at 200x total magnification at 48 and 72 h post-infection. 600 µm scale bar is located in the top right corner. (b) A549 cell lysates were observed for the production of the indicated proteins via Western blot analysis. (c) These Western blot data were quantified via densitometric analysis using Gel-pro analyzer 4.0 software and these IOD values were normalized to actin. These values were then converted to fold changes by dividing the values by the values for mock treated cells; mock = 1-fold. One of three representative experiments is shown. (d) Cell viability was determined by crystal violet staining and expressed as the percent cell viability relative to non-treated cells. One of three representative experiments is shown. (e) A549 cells were mock transfected, or were transfected with GFP or CMV-E1b19K plasmids as indicated. These cells were then lysed and observed for the production of the indicated proteins. * indicates p-value < 0.05, † indicates p-values < 0.01.
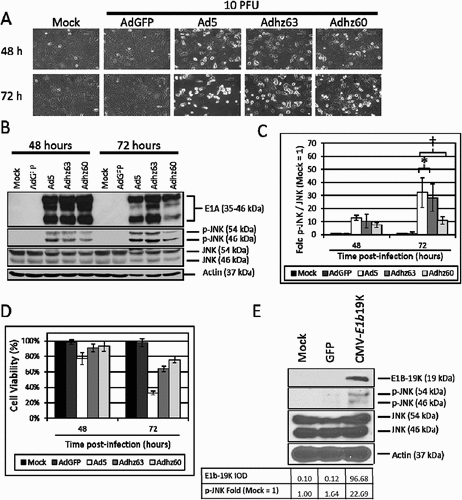
Interestingly, Adhz63 was shown to induce similar JNK phosphorylation as Ad5 at 72 h (28.1 fold vs. 32.2 fold respectively), but the E1b-19K deleted Adhz60 only induced 10 fold JNK phosphorylation (, )). These data indicate that E1b-19K deleted in Adhz60 may be critical for inducing JNK phosphorylation. Therefore, a plasmid that expresses E1b-19K under control of the CMV promoter (CMV-E1b19K) was utilized to selectively assess the role of E1b19K as a JNK phosphorylation inducer ()). These data indicated that E1b-19K expression was sufficient to induce JNK phosphorylation (22.7 fold) in A549 cells alone ()), demonstrating the potentially critical role of E1b-19K to activate JNK signaling during Ad infection.
3.3 The effect of e1a upon JNK phosphorylation
The total E1b-deleted Ad Adhz60 was also shown to induce JNK phosphorylation (10.9 fold), indicating that other non-E1b Ad genes such as E1a, may also play a role to induce JNK phosphorylation ()). Furthermore, E1b-deleted Ads have been shown to depend upon efficient E1a expression to lyse cancer cells,Citation39 implicating the role of E1A as a potential inducer of JNK phosphorylation during Ad oncolysis. E1a has been shown to induce JNK phosphorylation in murine 3T3 fibroblast culturesCitation40 however, the effect of oncolytic Ads expressing E1a upon JNK phosphorylation in cancer cells is unknown at this time.
To assess the effects of E1A upon JNK phosphorylation, A549 cells were treated with Ad5, the E1a wild-type E1b55K-deleted Adhz63, and the CMV-E1a expressing E1b55K-deleted Adhz69. Therefore, by comparing between the Adhz63 and Adhz69 treatment groups, the effects of E1a overexpression upon JNK phosphorylation was determined. E1a over-expression by Adhz69 was shown to increased E1A production and JNK phosphorylation (39.9 fold) relative to Ad5 (12.4 fold) and Adhz63 (3.8 fold; )).
Figure 4. The effect of E1a expression upon JNK phosphorylation. A549 cells were mock-treated or infected with AdGFP, Ad5, Adhz63, and Adhz69 at a PFU of 10. (a) Cell viability was determined by crystal violet staining and expressed as the percent cell viability relative to non-treated cells at 48 h post-infection. (b) A549 cell lysates were treated as indicated at 48 h post-infection and observed for the protein production of the indicated proteins via Western blot analysis. (c) A549 cells were mock treated or infected with AdGFP, Ad5, Adhz63, and Adhz69 at a PFU of 10 as in Figure 4B; Adhz60 was included as a negative control for E1b-19K. E1b-19K protein production was determined via Western blot analysis. These Western blot data were quantified via densitometric analysis using Gel-pro analyzer 4.0 software and these IOD values were normalized to actin. These values were then converted to fold changes by dividing the values by the values for mock treated cells.
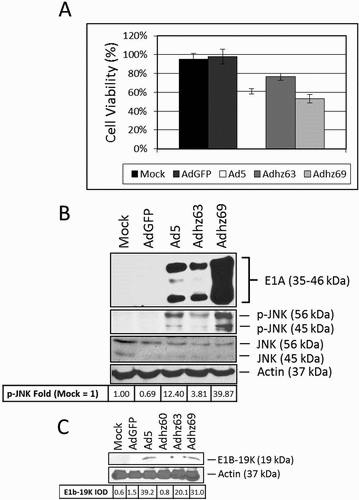
E1A is a well-known transactivator of many Ad and host cell genes.Citation41 One of the transcriptional targets of E1A is E1b-19K.Citation42 Therefore, the effect of E1A over-expression upon E1b-19K was observed to determine if Adhz69 induced JNK phosphorylation was due, at least in part, to enhanced E1b-19K protein levels ()). Ad5 and Adhz60 were used as E1b-19K positive and negative controls respectively. E1A overexpression by Adhz69 did not significantly alter E1b-19K protein production compared to Adhz63 (20.1 to 31.0 E1b-19K IOD), but less than as Ad5 (39.2 IOD, )). Therefore, Ads may induce JNK phosphorylation via an E1b-19K independent mechanism of action when E1a is expressed above endogenous levels.
3.4 The effects of JNK upon A549 cell oncolysis and ad replication
Ad5 infection induces the phosphorylation of both JNK1 and JNK2,Citation13,Citation26 therefore, the suppression of both JNK1 and JNK2 using siRNA was necessary to elucidate effects of JNK upon oncolysis and Ad replication. Using this approach, JNK1/2-siRNA was utilized to selectively knock-down the mRNA of both genes simultaneously ()). JNK1/2-siRNA transfection protected A549 cells against oncolysis relative to scramble-siRNA transfected cells ()). This protective effect was observed for Ad5, Adhz63, and Adhz60. The greatest differences were observed in Ad5-infected cells (cell viability 29% vs. 61% for scramble-siRNA vs. JNK1/2-siRNA) and Adhz63-infected cells (cell viability 45% vs. 77% for scramble-siRNA vs. JNK1/2-siRNA, )). Using the same siRNA transfection approach, JNK1/2 suppression was also shown to inhibit the replication of all 3 Ads studied ()). These differences were only statistically significant for the E1b-19K wild-type Ads Ad5 and Adhz63 between the siRNA transfected groups. We observed that the titers for Ad5 and Adhz63 were respectively repressed from 1.45 × 108 and 6.6 × 107 when transfected with scramble-siRNA to 1.43 × 107 and 1 × 107 when transfected with the JNK1/2-siRNA ()). These data indicated for the first time that higher levels of host cell JNK protein supports greater oncolysis and Ad replication in A549 cancer cells.
Figure 5. The effect of JNK expression upon A549 cell oncolysis and oncolytic Ad replication. (a) The concentration of JNK1/2-siRNA that efficiently repressed JNK1 and JNK2 expression was determined at 48 h by Western blot analysis using JNK1 and JNK2 selective antibodies. (b) The effect of JNK1/2-siRNA upon cell viability was assessed using crystal violet staining. Statistical analysis was performed by one-way ANOVA using Tukey’s post-test for multiple comparisons. (c) The effect of JNK1/2-siRNA upon Ad replication was assessed via the TCID50 method. Statistical analysis was performed by one-way ANOVA using Tukey’s post-test for multiple comparisons. † indicates p-value < 0.01, ‡ indicates p-value < 0.001.
4. Discussion
The goal of these studies was to ascertain the role of JNK in Ad oncolysis. Specifically, we seeked to understand why E1b-deleted Ads produced disappointing results during phase II and III clinical trials and to observe if JNK could play a role to enhance the efficacy of Ad therapy. To our knowledge, these studies have shown for the first time that JNK supports Ad oncolysis and replication in cancer cells.
During Ad infection and oncolysis, many cell death pathways are stimulated. However, several reports have indicated that autophagy may be the predominant mechanism of oncolytic Ad cancer cell known as oncolysis.Citation2,Citation9,Citation15,Citation16,Citation23,Citation43 In this report, expression of the autophagy marker LC3-II was markedly induced following wild-type Ad5 infection beginning at 36 h, continuing to increase until total A549 cell death was observed at 72 h post-infection ()). JNK phosphorylation was markedly attenuated by the E1b-19K deleted Ad Adhz60, indicating the importance of E1b-19K as a JNK phosphorylation inducer ()). This finding was consistent with the only other known report, which indicated that E1b-19K was shown to be the primary inducer of JNK phosphorylation.Citation26
Interestingly, E1b-deletions alone were not sufficient to completely ablate the JNK phosphorylation phenotype of oncolytic Ads, implicating the role of other genes such as E1a in this phenomenon. Infection with Adhz69, which overexpresses E1A,Citation31 led to a marked increase in JNK phosphorylation in A549 cells independently of E1b-19K (). E1A, like E1b-19K, does not have endogenous kinase activity. Therefore, it is likely that E1A induces JNK phosphorylation by the stimulation of the MAPK cascade or another family of kinases during Ad infection. E1a may also activate JNK by binding retinoblastoma protein (pRb), displacing the transcription factor E2F-1 from pRb, transactivating the expression of many autophagy related genes, including LC3 and Beclin-1.Citation44–Citation46
To selectively elucidate the effects of host cell JNK expression upon Ad oncolysis and replication, A549 cells were transfected with siRNA targeting JNK1 and JNK2 mRNA sequences.Citation37,Citation47 A549 cells transfected with JNK1/2 siRNA support less efficient replication of and lysis from treatment with E1b-19K expressing Ads ().
In addition to inducing autophagic cancer cell death, the phosphorylation of JNK has also been reported to promote cancer growth. For example, A549 cells treated with the JNK1/2/3 phosphorylation inhibitor, sp600125, were shown to form significantly smaller tumors in nude mice.Citation48 A549 cells are also known to express k-RAS which indirectly phosphorylates JNK via MKK7 of the MAPK cascade.Citation49 Unfortunately, there are very limited molecular tools to observe the selective effects of JNK phosphorylation upon autophagy without disrupting other cellular processes which also significantly influence Ad replication. For example, Ads have been shown to induce the cyclin E/CDK2 axis to induce an S-phase-like state in G0 normal cells during infection, increasing DNA synthesis in order to more efficiently replicate Ad DNA.Citation3,Citation36 It is possible therefore that the inhibition of oncolytic Ad efficacy following JNK1/2-siRNA transfection could be the result of cell cycle repression in A549 cells and not a direct effect of JNK upon Ad oncolysis. Another intriguing molecular target is the JNK phosphatase M3/6. M3/6 selectively de-phosphorylates JNK; therefore, the inhibition of M3/6 would likely significantly enhance JNK phosphorylation selectively and will be the topic of further research.
These studies indicated for the first time that JNK not only supports the oncolysis, but also the replication of Ads. Prior studies indicated that E1b induced JNK or that Ads induced JNK phosphorylation led to cancer oncolysis, but this is the first study to correlate the JNK phosphorylation phenotype to E1b-19K and to relate these effects to Ad oncolysis and replication. This study also indicated for the first time that E1a-overexpression was sufficient to induce JNK phosphorylation. In this report, JNK phosphorylation was associated with increased Ad oncolysis while the repression of JNK via JNK1/2-siRNA increased cancer cell survival and inhibited Ad titers. Furthermore, a recent review has also described the importance of JNK to cancer therapeutic effects of oncolytic Ads.50 Taken together, these studies indicate that JNK is an interesting molecular target for oncolytic Ads and that autophagy inducers acting upon the JNK/Beclin-1 axis would likely enhance the cancer therapeutic effects of E1b-deleted oncolytic Ads.
Author contributions
SLW, KMM, and HSZ designed these studies. SLW preformed the experiments with the technical assistance and advice from XMR and JGG. SLW also statistically analyzed these data. All authors edited the manuscript prior to submission.
Acknowledgments
This manuscript represents my final publication from the University of Louisville (UofL). It is dedicated to my daughter Eleanor Elizabeth Wechman who will become 3 years old around the time of this manuscript is published. I am, and always will, be proud of you.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Bischoff, J. R., Kirn D.H, Williams A., Heise C., Horn S., Muna M., Ng L., Nye J. A., Sampson-Johannes A., Fattaey A., McCormick F. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996; 274(5286):373–376.
- Cheng PH, Lian S, Zhao R, Rao XM, McMasters KM, Zhou HS. 2013. Combination of autophagy inducer rapamycin and oncolytic adenovirus improves antitumor effect in cancer cells. Virol J. 10:293. doi:10.1186/1743-422X-10-293.
- Cheng PH, Rao XM, McMasters KM, Zhou HS. Molecular basis for viral selective replication in cancer cells: activation of CDK2 by adenovirus-induced cyclin E. PloS One. 2013;8(2):e57340. doi:10.1371/journal.pone.0057340.
- Khuri, F. R., Nemunaitis J., Ganly I., Arseneau J., Tannock I. F., Romel. L., Gore M., Ironside J., MacDougall R. H., Heise C., Randlev B., Gillenwater A. M., Bruso P., Kaye S. B., Hong W. K., Kirn D. H. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6(8):879–885. doi:10.1038/78638.
- Nemunaitis J., Cunningham C., Buchanan A., Blackburn A., Edelman G., Maples P., Netto G., Tong A., Randlev B., Olson S., Kirn D. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Ther. 2001;8(10):746–759. doi:10.1038/sj.gt.3301424.
- Xia Z. J., Chang J. H., Zhang L., Jiang W. Q., Guan Z. Z., Liu J. W., Zhang Y., Hu X. H., Wu G. H., Wang H. Q., Chen Z. C., Chen J. C., Zhou Q. H., Lu J. W., Fan Q. X., Huang J. J., Zheng X. Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai Zheng = Aizheng = Chinese Journal of Cancer. 2004; 23(12):1666–1670.
- Kimball, K. J., Preuss M. A., Barnes M. N., Wang M., Siegal G. P., Wan W., Kuo H., Saddekni S., Stockard C. R., Grizzle W. E., Harris R. D., Aurigemma R., Curiel D. T., Alvarez R. D. A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research. 2010;16(21):5277–5287. doi:10.1158/1078-0432.CCR-10-0791.
- Ito, H., Aoki H., Kühnel F., Kondo Y., Kubicka S., Wirth T., Iwado E., Iwamaru A., Fujiwara K., Hess K. R., Lang F. F., Sawaya R., Kondo S. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J Natl Cancer Inst. 2006;98(9):625–636. doi:10.1093/jnci/djj161.
- Rodriguez-Rocha H, Gomez-Gutierrez JG, Garcia-Garcia A, Rao XM, Chen L, McMasters KM, Zhou HS. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology. 2011;416(1–2):9–15. doi:10.1016/j.virol.2011.04.017.
- Campbell GR, Bruckman RS, Chu YL, Spector SA. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J Biol Chem. 2015;290(8):5028–5040. doi:10.1074/jbc.M114.605428.
- Yakoub AM, Shukla D. 2015. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci Rep. 5:9730. doi:10.1038/srep09730.
- Zhang R, Chi X, Wang S, Qi B, Yu X, Chen JL. The regulation of autophagy by influenza A virus. Biomed Res Int. 2014;2014:498083.
- Klein S, R, Piya S., Lu Z., Xia Y., Alonso M. M., White E. J., Wei J., Gomez-Manzano C., Jiang H., Fueyo J. C-Jun N-terminal kinases are required for oncolytic adenovirus-mediated autophagy. Oncogene. 2015. doi:10.1038/onc.2014.452.
- Baird SK, Aerts JL, Eddaoudi A, Lockley M, Lemoine NR, McNeish IA. Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene. 2008;27(22):3081–3090. doi:10.1038/sj.onc.1210977.
- Gomez-Gutierrez JG, Nitz J, Sharma R, Wechman SL, Riedinger E, Martinez-Jaramillo E, Sam Zhou H, McMasters KM. 2016. Combined therapy of oncolytic adenovirus and temozolomide enhances lung cancer virotherapy in vitro and in vivo. Virology. 487:249–259. doi:10.1016/j.virol.2015.10.019.
- Jiang H, White EJ, Rios-Vicil CI, Xu J, Gomez-Manzano C, Fueyo J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J Virol. 2011;85(10):4720–4729. doi:10.1128/JVI.02032-10.
- Cheng PH, Rao XM, Duan X, Li XF, Egger ME, McMasters KM, Zhou HS. Virotherapy targeting cyclin E overexpression in tumors with adenovirus-enhanced cancer-selective promoter. J Mol Med. 2015;93(2):211–223. doi:10.1007/s00109-014-1214-6.
- O’Shea C, Klupsch K, Choi S, Bagus B, Soria C, Shen J, McCormick F, Stokoe D. Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J. 2005;24(6):1211–1221. doi:10.1038/sj.emboj.7600597.
- Cheng X, Liu H, Jiang CC, Fang L, Chen C, Zhang XD, Jiang ZW. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int J Mol Med. 2014;34(3):772–781. doi:10.3892/ijmm.2014.1822.
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi:10.1016/j.cell.2005.07.002.
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993;7(4):546–554. doi:10.1101/gad.7.4.546.
- Piya S, White EJ, Klein SR, Jiang H, McDonnell TJ, Gomez-Manzano C, Fueyo J. The E1B19K oncoprotein complexes with Beclin 1 to regulate autophagy in adenovirus-infected cells. PloS One. 2011;6(12):e29467. doi:10.1371/journal.pone.0029467.
- Sui X., Chen R., Wang Z., Huang Z., Kong N., Zhang M., Han W., Lou F., Yang J., Zhang Q., Wang X., He C., Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. doi:10.1038/cddis.2013.350.
- Zhou YY, Li Y, Jiang WQ, Zhou LF. 2015. MAPK/JNK signalling: a potential autophagy regulation pathway. Bioscience Reports. 35:3. doi:10.1042/BSR20150111.
- See RH, Shi Y. Adenovirus E1B 19,000-molecular-weight protein activates c-Jun N-terminal kinase and c-Jun-mediated transcription. Mol Cell Biol. 1998;18(7):4012–4022.
- Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–3470. doi:10.1016/j.bbamcr.2013.06.028.
- Rao XM, Tseng MT, Zheng X, Dong Y, Jamshidi-Parsian A, Thompson TC, Brenner MK, McMasters KM, Zhou HS. E1A-induced apoptosis does not prevent replication of adenoviruses with deletion of E1b in majority of infected cancer cells. Cancer Gene Ther. 2004;11(9):585–593. doi:10.1038/sj.cgt.7700739.
- Han J, Sabbatini P, Perez D, Rao L, Modha D, White E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996;10(4):461–477. doi:10.1101/gad.10.4.461.
- Jones N, Shenk T. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell. 1979;17(3):683–689.
- Zheng X, Rao XM, Snodgrass C, Wang M, Dong Y, McMasters KM, Zhou HS. Adenoviral E1a expression levels affect virus-selective replication in human cancer cells. Cancer Biol Ther. 2005;4(11):1255–1262. doi:10.4161/cbt.4.11.2137.
- Wechman SL, Rao XM, Cheng PH, Gomez-Gutierrez JG, McMasters KM, Zhou HS. 2016. Development of an oncolytic adenovirus with enhanced spread ability through repeated UV irradiation and cancer selection. Viruses. 8:6. doi:10.3390/v8060167.
- Wechman SL, Rao XM, McMasters KM, Zhou HS. 2016. Adenovirus with DNA packaging gene mutations increased virus release. Viruses. 8:12. doi:10.3390/v8120333.
- Cheng PH, Rao XM, Wechman SL, Li XF, McMasters KM, Zhou HS. 2015. Oncolytic adenovirus targeting cyclin E overexpression repressed tumor growth in syngeneic immunocompetent mice. BMC Cancer. 15:716. doi:10.1186/s12885-015-1584-3.
- White E, Cipriani R. Specific disruption of intermediate filaments and the nuclear lamina by the 19-kDa product of the adenovirus E1B oncogene. Proc Natl Acad Sci U S A. 1989;86(24):9886–9890.
- Zheng X, Rao XM, Gomez-Gutierrez JG, Hao H, McMasters KM, Zhou HS. Adenovirus E1B55K region is required to enhance cyclin E expression for efficient viral DNA replication. J Virol. 2008;82(7):3415–3427. doi:10.1128/JVI.01708-07.
- Li G, Xiang Y, Sabapathy K, Silverman RH. An apoptotic signaling pathway in the interferon antiviral response mediated by RNase L and c-Jun NH2-terminal kinase. J Biol Chem. 2004;279(2):1123–1131. doi:10.1074/jbc.M305893200.
- Ishiyama M, Tominaga H, Shiga M, Sasamoto K, Ohkura Y, Ueno K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol Pharm Bull. 1996;19(11):1518–1520. doi:10.1248/bpb.19.1518.
- Zheng X, Rao XM, Snodgrass CL, McMasters KM, Zhou HS. Selective replication of E1B55K-deleted adenoviruses depends on enhanced E1A expression in cancer cells. Cancer Gene Ther. 2006;13(6):572–583. doi:10.1038/sj.cgt.7700923.
- Romanov VS, Brichkina AI, Morrison H, Pospelova TV, Pospelov VA, Herrlich P. Novel mechanism of JNK pathway activation by adenoviral E1A. Oncotarget. 2014;5(8):2176–2186. doi:10.18632/oncotarget.1860.
- Berk AJ. 1986. Adenovirus promoters and E1A transactivation. Annu Rev Genet. 20:45–79. doi:10.1146/annurev.ge.20.120186.000401.
- Herrmann CH, Dery CV, Mathews MB. Transactivation of host and viral genes by the adenovirus E1B 19K tumor antigen. Oncogene. 1987;2(1):25–35.
- Klein SR, Jiang H, Hossain MB, Fan X, Gumin J, Dong A, Alonso MM, Gomez-Manzano C, Fueyo J. Critical role of autophagy in the processing of adenovirus capsid-incorporated cancer-specific antigens. PloS One. 2016;11(4):e0153814. doi:10.1371/journal.pone.0153814.
- Jiang H., Martin V., Gomez-Manzano C., Johnson D. G., Alonso M., White E., Xu J., McDonnell T. J., Shinojima N., Fueyo J. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010;70(20):7882–7893. doi:10.1158/0008-5472.CAN-10-1604.
- Wang B, Ling S, Lin WC. 14-3-3Tau regulates Beclin 1 and is required for autophagy. PloS One. 2010;5(4):e10409. doi:10.1371/journal.pone.0010409.
- Johnson DG, Degregori J. Putting the oncogenic and tumor suppressive activities of E2F into context. Curr Mol Med. 2006;6(7):731–738.
- Oleinik NV, Krupenko NI, Krupenko SA. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene. 2007;26(51):7222–7230. doi:10.1038/sj.onc.1210526.
- Okada M, Shibuya K, Sato A, Seino S, Watanabe E, Suzuki S, Seino M, Kitanaka C. Specific role of JNK in the maintenance of the tumor-initiating capacity of A549 human non-small cell lung cancer cells. Oncol Rep. 2013;30(4):1957–1964. doi:10.3892/or.2013.2655.
- He XY, Chen JX, Ou-Yang X, Zhang Z, Peng HM. [Construction of let-7a expression plasmid and its inhibitory effect on k-Ras protein in A549 lung cancer cells]. Nan Fang Yi Ke Da Xue Xue Bao = Journal of Southern Medical University. 2010;30(11):2427–2431.
- Tazawa H, Kuroda S, Hasei J, Kagawa S, Fujiwara T. 2017. Impact of Autophagy in Oncolytic Adenoviral Therapy for Cancer. Int J Mol Sci. 18:7. doi:10.3390/ijms18071479.