
ABSTRACT
C-X-C motif chemokine ligand 5 (CXCL5) is initially identified to recruit neutrophils by interacting with its receptor, C-X-C motif chemokine receptor 2 (CXCR2). Our prior work demonstrated that the expression levels of CXCL5 and CXCR2 were higher in the papillary thyroid carcinoma (PTC) tumors than that in the non-tumors. This study was performed to further investigate how this axis regulates the growth of PTC cells. B-CPAP cells (BRAFV600E) and TPC-1 cells (RET/PTC rearrangement) expressing CXCR-2 were used as in vitro cell models. Our results showed that the recombinant human CXCL5 (rhCXCL5) promoted the proliferation of PTC cells. rhCXCL5 accelerated the G1/S transition, upregulated the expression of a group of S (DNA synthesis) or M (mitosis)-promoting cyclins and cyclin-dependent kinases (CDKs), and downregulated CDK inhibitors in PTC cells. The CDS region of homo sapiens CXCL5 gene was inserted into an eukaryotic expression vector to mediate the overexpression of CXCL5 in PTC cells. The phosphorylation of c-Jun N-terminal kinases (JNK) and p38, and the nuclear translocation of c-Jun were enhanced by CXCL5 overexpression, whereas attenuated by CXCR2 antagonist SB225002. Additionally, CXCL5/CXCR2 axis, JNK and p38 pathway inhibitors, SB225002, SP600125 and SB203580, suppressed the growth of PTC cells overexpressing CXCL5 in nude mice, respectively. Collectively, our study demonstrates a growth-promoting effect of CXCL5-CXCR2 axis in PTC cells in vitro and in vivo.
Introduction
The worldwide incidence of thyroid carcinomas (TCs) is steadily rising over the last decade, and the papillary thyroid carcinoma (PTC) accounts for approximately 85% of all TCsCitation1. Although PTC is a well-differentiated tumor and can be removed by surgical resectionCitation2, its recurrence risk still threatens the PTC patients’ life post the surgery. Identification of molecules essential for tumor cell survival and metastasis will provide novel insights in PTC therapy.
C-X-C motif chemokine ligand 5 (CXCL5) gene (the alias for CXCL5 gene is neutrophil-activating protein 78) encodes a protein consisted of 114 amino acidsCitation3. It is initially identified as a C-X-C chemokine that recruits neutrophils by binding to C-X-C motif chemokine receptor 2 (CXCR2)Citation4. Our group prior demonstrated that the exogenous CXCL5 augmented the mobility and induced the epithelial-mesenchymal transition of two PTC cell lines expressing CXCR2Citation5. Several previous studies are consistent with our work showing a similar pro-metastatic role of CXCL5-CXCR2 axis in various types of cancers, such as prostate cancer and breast cancerCitation6,Citation7. However, the effects of this axis on tumor cell proliferation differ. While the exogenous CXCL5 of 1 nM promoted the proliferation of prostate cancer cellsCitation6, an even higher concentration of CXCL5 (10 nM) hardly affected the proliferation of breast cancer cellsCitation7. Although the discrepant phenomena may result from different purities and activities of the recombinant human CXCL5 (rhCXCL5) used in these two studies, it is plausible that CXCL5-CXCR2 axis has inconsistent effects on cell proliferation in different types of cancers.
Mitogen-activated protein kinase (MAPK) cascades regulate multiple cellular activities involved in cancer progression, including cell proliferation and cell cycle, with c-Jun N-terminal kinases (JNKs) and p38 as the two major molecule hubsCitation8. Earlier studies revealing the roles of JNKs in the tumorigenesis are related to their ability to regulate the stress-induced cell apoptosis and antitumor therapy-induced cytotoxic effectsCitation9. Paradoxically, the phosphorylated JNKs were found to be overexpressed in human PTCs, and this elevation was associated with poor prognosisCitation10. A broad spectrum inhibitor for JNK pathways, SP600125, can suppress the growth of TPC-1 cells, a PTC cell line harboring RET/PTC rearrangementCitation11. In addition, it has been reported that the proliferation decline of PTC cells is often accompanied by the deactivation of p38 pathwayCitation12. Although both pro-survival and pro-death effects of JNK and p38 pathways have been reported in cancer cells before, the aforementioned studies suggest that the activation of these two MAPK pathways may contribute to the survival of PTC cells. Interestingly, both JNK and p38 pathways in the lung tissues of CXCL5 knockout mice were deactivated by comparison with the corresponding wild type miceCitation13. The study at least implies that the CXCL5-CXCR2 axis is required for maintaining the signaling transduction of these two MAPK pathways.
In this study, we propose that CXCL5 can promote the proliferation of PTC cells by activating JNK and p38 MAPK pathways. The underlying mechanisms were explored in PTC cell lines harboring mutated oncoprotein BRAFV600E (B-CPAP) and RET/PTC rearrangement (TPC-1).
Materials and methods
Cell culture and transfection
B-CPAP and KTC-1 cells were obtained from the Cell bank of Chinese Academy of Sciences (Shanghai, China), whilst TPC-1 cells were obtained from Zhong Qiao Xin Zhou Biotechnology Co.,Ltd. The cell lines were authenticated with short tandem repeat analysis, and had no mycoplasma infection detected in our lab. Cells were allowed to grow in RPMI medium (Gibco) plus 10% FBS (HyClone) under a standard cell culture condition.
The complete CDS region of homo sapiens CXCL5 (NM_002994) was amplified and inserted into the eukaryotic expression vector pEGFP-N1 between XhoI (upstream) and BamHI (downstream). The construction of CXCL5 overexpression (OE) plasmid was validated via DNA sequencing. Empty pEGFP-N1 vector (NC plasmid) was used as control. B-CPAP and TPC-1 cells were transfected with CXCL5 OE or NC plasmid by using Lipofectamine™ 3000 transfection reagent (#L3000075; Invitrogen) according the supplier’s instruction. For stable transfection, B-CPAP cells were first transfected with a plasmid and then selected with G418 (400 μg/mL) for over 2 weeks. Cells with the resistance to G418 were used in the following xenograft tumor assay.
Cell viability and cell cycle
PTC cells were seeded in the 96-well plates (5,000 cells per well), and after the attachment, cells were treated with 1, 5, 10 nM rhCXCL5 (#RPA860Hu01; USCN Business Co., Ltd.) up to 96 hrs, or with 5, 10 μM ribociclib (a CDK4/6 inhibitor) up to 120 hrs (Selleck Chemicals, Houston, TX, USA). Cell vitality was determined with a CCK-8 kit (#C0037; Beyotime) according to the manufacturer’s protocols. In addition, PTC cells were transfected with CXCL5 OE or NC plasmid, and 48 hrs later, these cells were treated with SB225002 of 5 μM (#SML0716; Sigma) for another 24 hrs before CCK-8 assay. Flow cytometry assay using propidium (#C1052; Beyotime) to stain the double-stranded DNA was performed to determine the cell cycle according to the manufacturer’s protocols.
Real-time quantitative PCR
Using the total RNAs as templates, cDNAs were processed via Super M-MLV reverse transcriptase (#PR6502; BioTeke). The relative mRNA expression levels of CXCL5 gene were analyzed on the ExicyclerTM 96 by using the SYBR Green I (#SY1020; Solarbio) and calculated via 2−ΔΔCt (with β-actin as control). The primer sequences for CXCL5 gene were as following: F-5ʹ cgctgctgtgttgagaga 3ʹ/R-5ʹ ggctaccacttccaccttg 3ʹ.
Antibodies
Anti-CXCL5 antibody (#PAA860Hu01) and anti-CXCR2 antibody (#BA0732-2) were obtained from Wuhan USCN Business Co., Ltd. and Boster, respectively. Anti-phosphorylated cyclin dependent kinase (CDK)-1Thr161 antibody (#ab183554) was from Abcam. Anti-cyclinD1 (#2922), anti-cyclinB1 (#4138), anti-CDK-4 (#12790), anti-phosphorylated JNK1/2Thr183/Tyr185 (#4668) and anti-JNK1/2 (#9252) antibodies were purchased from Cell Signaling Technology, Inc. Anti-cyclinE1 (#11554-1-AP), anti-CDK inhibitor 1A/p21 (CDKN1A) (#10355-1-AP), anti-CDKN1B/p27 (#25614-1-AP), and anti-proliferating cell nuclear antigen (PCNA) (#10205-2-AP) antibodies were obtained from SANYING Proteintech. Anti-c-JUN (#bs-20067R), anti-phosphorylated p38Thr180/Tyr182 (#bs-0636R) and anti-p38 (#bs-0637R) antibodies were obtained from Bioss.
Western blot
Total proteins were isolated by using RIPA buffer containing 1% phenylmethanesulfonyl fluoride (#P0013B; Beyotime). Equal protein samples were first separated with 10 % SDS-PAGE, and then were transferred to PVDF membranes (#IPVH00010; Millipore). After blocking with 5% (M/V) skim milk, the membranes were incubated with one of the primary antibodies at 4℃ overnight. The dilutions for anti-CXCL5 and anti-CXCR2 were 1: 300 and 1: 200, respectively. The dilutions for anti-cyclinD1, anti-CDK4 antibody, anti-cyclinE1, anti-CDKN1A, anti-CDKN1B, anti-cyclinB1, anti-p-CDK1, anti-p-JNK1/2, anti-JNK1/2 and anti-PCNA were 1: 1000. The dilutions for anti-c-jun, p-p38 and p38 were 1: 500. Then, the membranes were incubated with a secondary antibody, and the blot signals were visualized with ECL reagent (#P0018; Beyotime). For some experiments, the cells were first transfected with CXCL5 OE or NC plasmid, and 48 hrs later, cells were treated with 10 μM SP600125 (#S1876; Beyotime) or 20 μM SB203580 (#S1863; Beyotime) for another 24 hrs.
Immunofluorescence
After being fixed with 4 % paraformaldehyde, cells on the glass slides were permeabilizated with 0.1% tritonX-100 (#ST795; Beyotime), and blocked with normal goat serum. Thereafter, cells were incubated with anti-c-Jun (1:200) or anti-PCNA (1: 200) overnight, and then with Cy3-labled goat anti-rabbit secondary antibody (1: 200; #A0516; Beyotime) for 60 min in the dark. Cell nuclei were stained with DAPI (#C1002; Beyotime).
Xenografted tumor
The animal procedures complied with the Guide for the Care and Use of Laboratory Animals, and were approved by the Ethics Committee of Dalian Medical University. B-CPAP cells were resuspended in 0.1 mL 1× PBS, mixed with 0.1 mL Metrigel and then injected into the axillary skin of nude mice. Nude mice injected with B-CPAP cells overexpressing CXCL5 were given CXCR2 antagonist SB225002 (intraperitoneal injection of 10 mg/kg for three times a week), JNK pathway inhibitor SP600125 (intraperitoneal injection of 5 mg/kg for three times a week) or p38 inhibitor SB203580 (subcutaneous injection of 4 mg/kg for five times a week) for four weeks. The xenografted tumors were isolated and photographed on day 28 post the injection of cancer cells. The tumor volume was measured every three day before sacrificing on day 28.
Statistical analysis
Data were expressed as mean ± standard deviation or standard error, and analyzed with soft SPSS 20.0 package (IBM, Armonk, NY, USA). All experiments were repeated for three times. One-way ANOVA followed by Bonferroni multiple test was used to evaluate the statistical differences over three groups. Two-way ANOVA followed by Bonferroni multiple test was performed to compare the differences from different time points. Statistical differences between groups were considered significant when the p values < 0.05.
Results
Exogenous rhCXCL5 promotes proliferation and G1 to s transition of PTC cells
The expression level of CXCR2 was determined in TPC-1, B-CPAP and KTC-1 cells with western blot analysis. We found that the basal expression of CXCR2 was TPC-1 > B-CPAP > KTC-1 ()). TPC-1 is a cell line of RET/PTC rearrangement, while B-CPAP and KTC-1 are cell lines carrying BRAFV600E. Therefore, TPC-1 and B-CPAP cells with higher CXCR2 level and respectively representing RET/PTC rearrangement and BRAFV600E were used in the following study.
Figure 1. Exogenous rhCXCL5 promotes PTC cell proliferation (a) The basal expression of CXCR2 in B-CPAP, TPC-1 and KTC-1 cells were determined with Western blot analysis. (b-c) B-CPAP cells and (d-e) TPC-1 cells were treated with 1, 5, 10 nM rhCXCL5 protein for 24, 48, 72 or 96 hrs before CCK-8 (n = 5), immunofluorescence and Western blot assays using anti-PCNA antibody (bars, 50 μm; 48 hrs post rhCXCL5 treatment). **P < 0.01 and *P < 0.05 (1 nM rhCXCL5 versus Control), ##P < 0.01 and #P < 0.05 (5 nM rhCXCL5 versus Control), §§P < 0.01 (10 nM rhCXCL5 versus Control). PTC, papillary thyroid carcinoma; rhCXCL5, recombinant human C-X-C motif chemokine ligand 5; CXCR2, C-X-C motif chemokine receptor 2; PCNA, proliferating cell nuclear antigen.
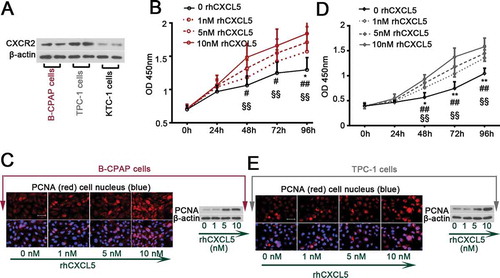
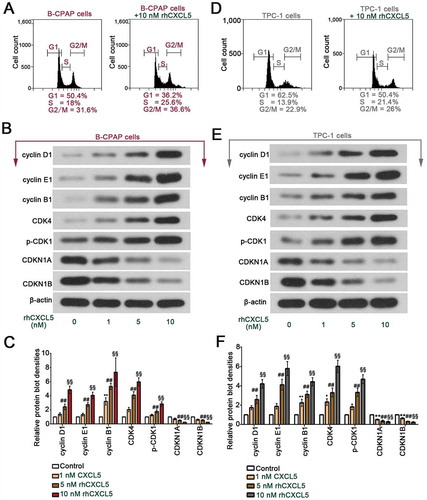
B-CPAP and TPC-1 cells, grown in complete culture medium, were treated with 1, 5 or 10 nM rhCXCL5. Results from IF staining and western blot for PCNA and from CCK-8 assay indicated that rhCXCL5 promoted the PTC cell proliferation in a dose-dependent manner (-e)). Data from flow cytometry assay confirmed the pro-proliferative effects of CXCL5 by showing that more PTC cells entered S phase when treated with 10 nM rhCXCL5 ( & )). Further, the expression levels of a group of cell cycle-associated proteins in B-CPAP and TPC-1 cells were determined with western blot. rhCXCL5 upregulated the expression of cyclins (cyclin D1, E1 and B1) and enhanced the activity of cyclin kinases (CDK4 and CDK1), and downregulated the expression of cyclin dependent kinase inhibitors (CDKN1A and CDKN1B) () & ()). Furthermore, 5 or 10 μM ribociclib, a CDK4/6 inhibitor, was used to treat B-CPAP and TPC-1 cells for −120 hrs. Ribociclib reduced cell proliferation as revealed by CCK-8 data (sFigure 1). B-CPAP cells responded to ribociclib more quickly than TPC-1 cells: 24 hrs versus 48 hrs post the ribociclib treatment (sFigure 1).
Figure 2. Exogenous rhCXCL5 promotes the G1 to S transition in PTC cells (a) B-CPAP cells and (D) TPC-1 cells were treated with or without 10 nM rhCXCL5 protein for 48 hrs before flow cytometry assay (Propidium staining). The levels of a group of cell cycle-associated proteins in (b-c) B-CPAP cells and (e-f) TPC-1 cells were detected with western blot (with β-actin as control; n = 3). **P < 0.01 (1 nM rhCXCL5 versus Control), ##P < 0.01 (5 nM rhCXCL5 versus Control), §§P < 0.01 (10 nM rhCXCL5 versus Control). PTC, papillary thyroid carcinoma; rhCXCL5, recombinant human C-X-C motif chemokine ligand 5; CXCR2, C-X-C motif chemokine receptor 2.
MAPK pathways contribute to PTC cell proliferation induced by CXCL5-CXCR2 activation
Forty-eight hours after the CXCL5 OE or NC plasmid transfection, the mRNA and protein expression levels of CXCL5 gene were determined via real-time PCR and western blot, respectively. The results indicated a significant elevation of CXCL5 in cells transfected with CXCL5 OE plasmid () & ()). In addition, SB225002, a CXCR2 selective inhibitor, suppressed the proliferation induced by CXCL5 overexpression ( & )). In response to SB225002 of 5 μM, B-CPAP cells were more sensitive than TPC-1 cells ( & )).
Figure 3. Forced overexpression of CXCL5-induced cell proliferation and activation of JNK and p38 pathways are inhibited by CXCR2 inhibitor SB225002.
The complete CDS region of homo sapiens CXCL5 (NM_002994) was amplified and inserted into the eukaryotic expression vector pEGFP-N1 (CXCL5 OE plasmid). B-CPAP cells and TPC-1 cells were transfected with CXCL5 OE plasmid or negative control (NC) plasmid by using Lipofectamine™ 3000 transfection reagent, and 48 hrs later, the mRNA and protein expression levels of CXCL5 were respectively determined with (a & d) real-time quantitative PCR and (b & e) western blot. Additionally, forty-eight hours post the transfection, CXCR2 antagonist SB225002 (5 μM) was used to treat PTC cells for another 24 hrs. (c & f) Cell viability was determined with CCK-8 as well (n = 3). (g-h & j-k) The levels of nuclear c-Jun, phosphorylated or total JNK1/2, and phosphorylated or total p38 were determined with western blot analysis. (i & l) Immunofluorescence assay using anti-c-Jun antibody was performed to probe the expression of c-Jun in PTC cells (bars, 50 μm). All data were expressed in mean ± standard deviation (SD) (n = 3). **P < 0.01; *P < 0.05. CXCL5, C-X-C motif chemokine ligand 5; CXCR2, C-X-C motif chemokine receptor 2; JNK, c-Jun N-terminal kinases
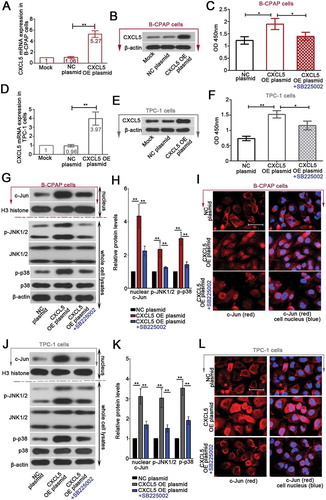
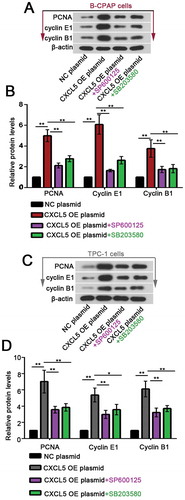
Next, B-CPAP cells and TPC-1 cells overexpressing CXCL5 were treated with 5 μM SB225002 for additional 24 hrs. We found that the overexpression of CXLC5-induced nuclear accumulation of c-jun and phosphorylation of JNK1/2 at 183Thr/185Tyr and p38 at 180Thr/182Tyr in B-CPAP cells and TPC-1 cells were suppressed by SB225002 (. Furthermore, JNK pathway inhibitor (SP600125; 10 μM) or p38 pathway inhibitor (SB203580; 20 μM) was used to treat PTC cells transfected with CXCL5 OE plasmid for 24 hrs. The expression levels of the proliferation marker PCNA and the essential cyclins E1 and B1 were analyzed with western blot. Additional data showed that the elevation in PCNA, cyclins E1 and B1 was partly suppressed by SP600125 or SB225002 (). These results suggested that the pro-proliferative effects of CXCL5-CXCR2 axis at least involved JNK or p38 MAPK pathway activation.
Figure 4. Blockade of JNK or p38 pathway suppresses CXCL5 overexpression-induced cell proliferation.
Forty-eight hours post the transfection, 10 μM SP600125 (JNK pathway inhibitor) or 20 μM SB203580 (p38 pathway inhibitor) was used to treat PTC cells for another 24 hrs, and the expression levels of PCNA, cyclin E1 and B1 were determined with western blot assay. **P < 0.01; *P < 0.05. CXCL5, C-X-C motif chemokine ligand 5; JNK, c-Jun N-terminal kinases; PCNA, proliferating cell nuclear antigen
Forced overexpression of CXCL5 promotes the growth of B-CPAP xenografted tumors
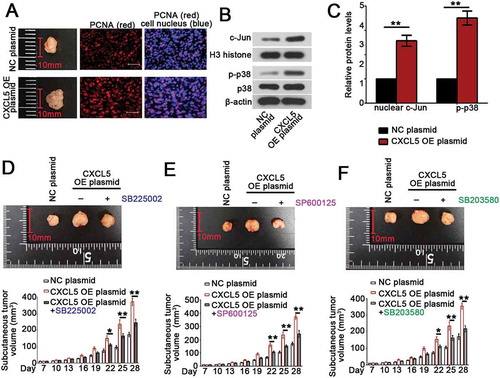
Finally, the in vivo growth of B-CPAP cells was evaluated. As shown in ), the xenografted tumor formed by B-CPAP cells overexpressing CXCL5 was larger than that generated by NC plasmid transfected cells (); left column). More PCNA-positive cells presented within the tumor when CXCL5 was overexpressed (); middle and right columns). CXCL5 overexpression promoted the nuclear translocation of c-Jun and augmented the phosphorylation of p38 in tumor ()). Furthermore, nude mice injected with CXCL5 OE B-CPAP cells were given CXCR2 antagonist SB225002, JNK pathway inhibitor SP600125 or p38 inhibitor SB203580 for four weeks. We found that blockade of CXCL5/CXCR2 axis with SB225002 restricted tumor growth without affecting tumor formation rate ()). Similar anti-tumor effects were also observed in mice treated with SP600125 ()) or p38 inhibitor ()).
Figure 5. Forced overexpression of CXCL5 promotes the growth of B-CPAP xenografted tumors via activation of JNK or p38 pathway.
BCPAP cells were resuspended in 0.1 mL serum-free RPMI medium, mixed with 0.1 mL Metrigel and then injected into the axillary skin of nude mice. (a) The xenografted tumors were isolated and photographed on day 28 post the injection of cancer cells (left column). Immunofluorescence assay using PCNA antibody was conducted to probe PCNA-positive cells within the tumors (bars, 50 μm; middle and right columns). (b) The levels of nuclear c-Jun and phosphorylated or total p38 were determined with western blot analysis, and (c) calculated by comparing to H3 histone or β-actin. (d-f) Nude mice injected with CXCL5 OE BCPAP cells were given CXCR2 antagonist SB225002 (intraperitoneal injection of 10 mg/kg for three times a week), JNK pathway inhibitor SP600125 (intraperitoneal injection of 5 mg/kg for three times a week) or p38 inhibitor SB203580 (subcutaneous injection of 4 mg/kg for five times a week) for four weeks. The xenografted tumors were isolated and photographed on day 28 post the injection of cancer cells. The tumor volume was measured every three day before sacrificing on day 28. All data were expressed in mean ± standard error (n = 6). **P < 0.01; *P < 0.05. CXCL5, C-X-C motif chemokine ligand 5; PCNA, anti-proliferating cell nuclear antigen; OE, overexpression
Discussion
BRAFV600E mutation and RET/PTC rearrangement are the two major oncogenic events that frequently occur in patients with PTCs Citation2. To evaluate the role of CXCL5-CXCR2 axis in the proliferation of PTC cells, B-CPAP (BRAFV600E mutation) and TPC-1 (RET/PTC rearrangement) cells were used as the in vitro cell models. The expression of CXCR2 on the surface of these two cell lines enables the delivery of CXCL-5-mediated cellular signals. We found that, regardless of the discrepancy in genetic background of the two PTC cell lines, both rhCXCL5 treatment and CXCL5 overexpression enhanced the cell proliferation.
Deregulated cell cycle is one of the most consistently observed events in human cancers, including PTCCitation14–16. A previous study from Kuo et al. showed that exogenous CXCL5 induced a decrease in CDKN1B and an increase in CDK4 in prostate cancer cellsCitation17, promoting us to decipher the role of CXCL5-CXCR2 in the cell cycle progress of PTC cells. Our data showed that rhCXCL5 treatment accelerated the transition from G1 phase to S phase (DNA synthesis) in B-CPAP and TPC-1 cells, which was accompanied with an upregulation in cyclins and CDKs, and a downregulation in CDK inhibitors. Cell cycle-associated proteins are considered as attractive targets for treating human cancersCitation18,Citation19. Our work revealing that rhCXCL5 effectively triggers changes in cyclins and CDKs implies that PTC cells may be sensitive to CDK inhibitors. This assumption was then tested by treating PTC cells with ribociclib, a CDK4/CDK6 inhibitor. Interestingly, both B-CPAP and TPC-1 cells exposed to ribociclib displayed a reduced proliferation. Two recent studies have respectively demonstrated that ribociclib and palbociclib (another CDK4/CDK6 inhibitor) can inhibit the proliferation of anaplastic thyroid cancer cellsCitation20,Citation21. These previous studies together with our findings support the rationale for clinical application of the CDK4/6 inhibitor in patients with thyroid cancer.
In addition to treating PTC cells with rhCXCL5, eukaryotic expression vector carrying the complete CDS region of homo sapiens CXCL5 gene was constructed to mediate the overexpression of CXCL5 in PTC cells. Like exogenous rhCXCL5, genetic overexpression of CXCL5 also promoted the growth of B-CPAP and TPC-1 cells. The pro-proliferative effect of CXCL5 was attenuated when CXCR2 was selectively antagonized by SB225002 in vitro and in vivo. The growth-inhibiting effects induced by SB225002 in CXCL5-overexpressing B-CPAP cells were stronger than that in TPC-1 cells. Although this phenomenon may result from the discrepant basal states of the two cell lines, it at least suggests that PTC cells harboring BRAFV600E mutation are more sensitive to CXCL5-CXCR2-induced alterations than cells carrying RET/PTC rearrangement.
In vivo, how CXCL5-CXCR2 axis influences the growth of cancer cells remains controversial. Zhou et al. demonstrated that overexpression of CXCL5 promoted the growth and lung metastasis of hepatocellular carcinoma cells in nude miceCitation22, whilst Speetjens et al. found that re-expression of CXCL5 had opposite effects on colorectal cancer cells in Wag/Rij ratsCitation23. Such controversial phenomena may result from the different immune backgrounds of the experimental animals. Thymic aplasia leads to immunodeficiency in nude mice. In order to avoid the interference from the host immune system, we and Zhou et al.Citation22 chose to inject the cancer cells into nude mice. As a chemokine, CXCL5 can be released by various cell types, including the intratumor cells, and it links the outer microenvironment to the tumor and influences the tumorigenic progress. The intratumoral infiltration of CXCR2-expressing neutrophils, natural killer cells and T cells in response to CXCL5-CXCR2 interaction may trigger antitumor immune responsesCitation23. Unlike the nude mice, Wag/Rij rats have intact immune system. The growth-inhibiting effects of CXCL5 on colorectal cancer cells in Wag/Rij rats may result from an enhanced antitumor immune attack. It is possible that when the infiltration of anti-tumor immune cells induced by CXCL5-CXCR2 interaction overwhelms the net pro-survival effects of this axis within tumor masses, tumor growth will be suppressed in vivo. To verify this hypothesis during thyroid tumorigenesis, further study is needed.
The role of p38 and JNK MAPK pathways in cancer cells is controversialCitation24,Citation25. Both activation and deactivation of p38 and JNK pathways have been reported to contribute to aggressive tumor behaviors of thyroid cancer cellsCitation26–Citation28. Our current results indicated that genetic overexpression of CXCL5 activated p38 and JNK signaling pathways in PTC cells, which was blunted by CXCR2 antagonist SB225002. Blockade of p38 pathway with SB203580 and JNK pathway with SP600125 suppressed CXCL5 overexpression-evoked PTC cell growth in vitro and in vivo. It is generally considered that the function of MAPK pathways at least depends on the nature of stimuli. Our data thus suggest that the signals delivered by CXCL5-CXCR2 axis are more likely to promote cancer cell survival. Besides p38 and JNK pathways, CXCL5 also promotes the activation of ERK1/2 in colorectal cancer cellsCitation29. ERK pathway is one of the three critical MAPK pathways, the activation of which can stabilize and allow the c-Fos protein to interact with c-Jun to form activating protein-1 (AP-1) complexesCitation30. Our results imply an involvement of CXCL5-CXCR2 in the formation of AP-1 dimers. Moreover, it should be noted that the regulation of p38 and JNK signaling by CXCL5/CXCR2 activity is linear. Gene or protein expression profiling using high throughput detection and screening method will provide more information to how this axis influences the development and progress of PTCs.
Exogenous CXCL5 binds to CXCR2 and triggers its intercellular internalization Citation31,Citation32 without significantly affecting its expression in cancer cellsCitation29,Citation33. CXCR2 is the only identified receptor for CXCL5 up to dateCitation4. It is plausible that cells lacking CXCR2 will not respond to addition of rhCXCL5 or CXCL5 overexpression. Several non-synonymous mutations have been identified in CXCR2 protein, including Arg153His, Arg236Cys, Arg248Gln and Ala249LeuCitation34–Citation36. These mutations are located within the CXCR2 intracellular loop, a structure that is important for G-protein interactions and receptor activationCitation34,Citation35. Lys327 residue is essential for the poly-ubiquitination of CXCR2Citation37. However, to our knowledge, none of these CXCR2 mutations have been reported to be associated with carcinogenesis of PTC or any other malignancies. As mutations within the key positions may alter the intracellular signaling transduction of CXCR2, there is a need to further investigate potential nucleotide polymorphisms of CXCR2 gene in PTC clinical samples, and analyze the association of the CXCR2 mutations with the clinical outcome of PTC patients.
In summary, our study demonstrates a growth-promoting effect of CXCL5-CXCR2 axis in PTC cells in vitro and in vivo. The activation of JNK and p38 pathways is involved in this process.
Supplemental Material
Download Zip (87.5 KB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Kitahara CM, Sosa JA. The changing incidence of thyroid cancer. Nat Rev Endocrinol. 2016;12:646–653. doi:10.1038/nrendo.2016.110.
- Xing M, Haugen BR, Schlumberger M. Progress in molecular-based management of differentiated thyroid cancer. Lancet. 2013;381:1058–1069. doi:10.1016/S0140-6736(13)60109-9.
- Chang MS, McNinch J, Basu R, Simonet S. Cloning and characterization of the human neutrophil-activating peptide (ENA-78) gene. J Biol Chem. 1994;269:25277–25282.
- Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008;267:226–244. doi:10.1016/j.canlet.2008.04.050.
- Cui D, Zhao Y, Xu J. Activated CXCL5-CXCR2 axis promotes the migration, invasion and EMT of papillary thyroid carcinoma cells via modulation of beta-catenin pathway. Biochimie. 2018. doi:10.1016/j.biochi.2018.02.009.
- Begley LA, Kasina S, Mehra R, Adsule S, Admon AJ, Lonigro RJ, Chinnaiyan AM, Macoska JA. CXCL5 promotes prostate cancer progression. Neoplasia. 2008;10:244–254.
- Hsu YL, Hou MF, Kuo PL, Huang YF, Tsai EM. Breast tumor-associated osteoblast-derived CXCL5 increases cancer progression by ERK/MSK1/Elk-1/snail signaling pathway. Oncogene. 2013;32:4436–4447. doi:10.1038/onc.2012.444.
- Peluso I, Yarla NS, Ambra R, Pastore G, Perry G. MAPK signalling pathway in cancers: olive products as cancer preventive and therapeutic agents. Semin Cancer Biol. 2017. doi:10.1016/j.semcancer.2017.09.002.
- Dhanasekaran DN, Reddy EP. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer. 2017;8:682–694. doi:10.18632/genesandcancer.155.
- Wang X, Chao L, Zhen J, Chen L, Ma G, Li X. Phosphorylated c-Jun NH2-terminal kinase is overexpressed in human papillary thyroid carcinomas and associates with lymph node metastasis. Cancer Lett. 2010;293:175–180. doi:10.1016/j.canlet.2010.01.007.
- Grassi ES, Vezzoli V, Negri I, Labadi A, Fugazzola L, Vitale G, Persani L. SP600125 has a remarkable anticancer potential against undifferentiated thyroid cancer through selective action on ROCK and p53 pathways. Oncotarget. 2015;6:36383–36399. doi:10.18632/oncotarget.5799.
- Liao T, Qu N, Shi RL, Guo K, Ma B, Cao YM, Xiang J, Lu Z-W, Zhu Y-X, Li D-S, et al. BRAF-activated LncRNA functions as a tumor suppressor in papillary thyroid cancer. Oncotarget. 2017;8:238–247. doi:10.18632/oncotarget.10825.
- Balamayooran G, Batra S, Cai S, Mei J, Worthen GS, Penn AL, Jeyaseelan S. Role of CXCL5 in leukocyte recruitment to the lungs during secondhand smoke exposure. Am J Respir Cell Mol Biol. 2012;47:104–111. doi:10.1165/rcmb.2011-0260OC.
- Brzezinski J, Migodzinski A, Gosek A, Tazbir J, Dedecjus M. Cyclin E expression in papillary thyroid carcinoma: relation to staging. Int J Cancer. 2004;109:102–105. doi:10.1002/ijc.11673.
- Lamba Saini M, Bouzin C, Weynand B, Marbaix E. An appraisal of proliferation and apoptotic markers in papillary thyroid carcinoma: an automated analysis. PLoS One. 2016;11:e0148656. doi:10.1371/journal.pone.0148656.
- Kim NY, Kim JH, Pyo JS, Jin Cho W. Clinicopathological significance of loss of p27kip1 expression in papillary thyroid carcinoma. Int J Biol Markers. 2017;32:e255–e9. doi:10.5301/jbm.5000239.
- Kuo PL, Chen YH, Chen TC, Shen KH, Hsu YL. CXCL5/ENA78 increased cell migration and epithelial-to-mesenchymal transition of hormone-independent prostate cancer by early growth response-1/snail signaling pathway. J Cell Physiol. 2011;226:1224–1231. doi:10.1002/jcp.22445.
- Murphy CG, Dickler MN. The role of CDK4/6 inhibition in breast cancer. Oncologist. 2015;20:483–490. doi:10.1634/theoncologist.2014-0443.
- Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5:6512–6525. doi:10.18632/oncotarget.2270.
- Lee HJ, Lee WK, Kang CW, Ku CR, Cho YH, Lee EJ. A selective cyclin-dependent kinase 4, 6 dual inhibitor, Ribociclib (LEE011) inhibits cell proliferation and induces apoptosis in aggressive thyroid cancer. Cancer Lett. 2018;417:131–140. doi:10.1016/j.canlet.2017.12.037.
- Lopes-Ventura S, Pojo M, Matias AT, Moura MM, Marques IJ, Leite V, Cavaco BM. The efficacy of HRAS and CDK4/6 inhibitors in anaplastic thyroid cancer cell lines. J Endocrinol Invest. 2018. doi:10.1007/s40618-018-0947-4.
- Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, Huang X-W, Fan J, Zhou J. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology. 2012;56:2242–2254. doi:10.1002/hep.25907.
- Speetjens FM, Kuppen PJ, Sandel MH, Menon AG, Burg D, van de Velde CJ, Tollenaar RAEM, de Bont HJGM, Nagelkerke JF. Disrupted expression of CXCL5 in colorectal cancer is associated with rapid tumor formation in rats and poor prognosis in patients. Clin Cancer Res. 2008;14:2276–2284. doi:10.1158/1078-0432.CCR-07-4045.
- Vasilevskaya I, O’Dwyer PJ. Role of Jun and Jun kinase in resistance of cancer cells to therapy. Drug Resist Updat: Rev Commentaries Antimicrobial Anticancer Chemo. 2003;6:147–156.
- Glassmann A, Winter J, Kraus D, Veit N, Probstmeier R. Pharmacological suppression of the Ras/MAPK pathway in thyroid carcinoma cells can provoke opposite effects on cell migration and proliferation: the appearance of yin-yang effects and the need of combinatorial treatments. Int J Oncol. 2014;45:2587–2595. doi:10.3892/ijo.2014.2668.
- Yu W, Imoto I, Inoue J, Onda M, Emi M, Inazawa J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene. 2007;26:1178–1187. doi:10.1038/sj.onc.1209899.
- Gunda V, Sarosiek KA, Brauner E, Kim YS, Amin S, Zhou Z, Letai A, Parangi S. Inhibition of MAPKinase pathway sensitizes thyroid cancer cells to ABT-737 induced apoptosis. Cancer Lett. 2017;395:1–10. doi:10.1016/j.canlet.2017.02.028.
- Knauf JA, Sartor MA, Medvedovic M, Lundsmith E, Ryder M, Salzano M, Nikiforov YE, Giordano TJ, Ghossein RA, Fagin JA. Progression of BRAF-induced thyroid cancer is associated with epithelial-mesenchymal transition requiring concomitant MAP kinase and TGFbeta signaling. Oncogene. 2011;30:3153–3162. doi:10.1038/onc.2011.44.
- Zhao J, Ou B, Han D, Wang P, Zong Y, Zhu C, Liu D, Zheng M, Sun J, Feng H, et al. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3beta/beta-catenin pathways. Mol Cancer. 2017;16:70. doi:10.1186/s12943-017-0629-4.
- Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi:10.1038/ncb822.
- Feniger-Barish R, Belkin D, Zaslaver A, Gal S, Dori M, Ran M, Ben-Baruch A. GCP-2-induced internalization of IL-8 receptors: hierarchical relationships between GCP-2 and other ELR(+)-CXC chemokines and mechanisms regulating CXCR2 internalization and recycling. Blood. 2000;95:1551–1559.
- Fan GH, Yang W, Wang XJ, Qian Q, Richmond A. Identification of a motif in the carboxyl terminus of CXCR2 that is involved in adaptin 2 binding and receptor internalization. Biochemistry. 2001;40:791–800.
- Qiu WZ, Zhang HB, Xia WX, Ke LR, Yang J, Yu YH, Liang H, Huang X-J, Liu G-Y, Li W-Z, et al. The CXCL5/CXCR2 axis contributes to the epithelial-mesenchymal transition of nasopharyngeal carcinoma cells by activating ERK/GSK-3beta/snail signalling. J Exp Clin Cancer Res. 2018;37:85. doi:10.1186/s13046-018-0722-6.
- Auer PL, Teumer A, Schick U, O’Shaughnessy A, Lo KS, Chami N, Carlson C, de Denus S, Dubé M-P, Haessler J, et al. Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet. 2014;46:629–634. doi:10.1038/ng.2962.
- Salchow K, Bond ME, Evans SC, Press NJ, Charlton SJ, Hunt PA, Bradley ME. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br J Pharmacol. 2010;159:1429–1439. doi:10.1111/j.1476-5381.2009.00623.x.
- Kato H, Tsuchiya N, Tokunaga K. Single nucleotide polymorphisms in the coding regions of human CXC-chemokine receptors CXCR1, CXCR2 and CXCR3. Genes Immun. 2000;1:330–337. doi:10.1038/sj.gene.6363682.
- Leclair HM, Dubois SM, Azzi S, Dwyer J, Bidere N, Gavard J. Control of CXCR2 activity through its ubiquitination on K327 residue. BMC Cell Biol. 2014;15:38. doi:10.1186/s12860-014-0038-0.