
ABSTRACT
Enhancer of zeste homolog 2 (EZH2) is an important member of the epigenetic regulatory factor polycomb group proteins (PcG) and is abnormally expressed in a wide variety of tumors, including osteosarcoma. Scientists consider EZH2 as an attractive target for the treatment of osteosarcoma and have found many potential EZH inhibitors, such as GlaxoSmithKline 343 (GSK343). It has been reported that GSK343 can be used as an inhibitor in different types of cancer. This study demonstrated that GSK343 not only induced apoptosis by increasing cleaved Casp-3 and poly ADP-ribose polymerase (PARP) expression, but also induced autophagic cell death by inhibiting p62 expression. Apoptosis and autophagic cell death induced by GSK343 were confirmed by the high expression of cleaved caspase-3, LC3-II and transmission electron microscopy. GSK343 inhibited the expression of EZH2 and c-Myc. Additionally, GSK343 inhibited the expression of FUSE binding protein 1 (FBP1), which was identified by its regulatory effects on c-Myc expression. Since c-Myc is a common target of EZH2 and FBP1, and GSK343 inhibited the expression of these proliferation-promoting proteins, a mutual regulatory mechanism between EZH2 and FBP1 was proposed. The knockdown of EZH2 suppressed the expression of FBP1; similarly, the knockdown of FBP1 suppressed the expression of EZH2. These results suggest the mutual regulatory association between EZH2 and FBP1. The knockdown of either EZH2 or FBP1 accelerated the sensitivity of osteosarcoma cells to GSK343. Based on these results, this study clarified that GSK343, an EZH2 inhibitor, may have potential for use in the treatment of osteosarcoma. The underlying mechanisms of the effects of GSK343 are partly mediated by its inhibitory activity against c-Myc and its regulators (EZH2 and FBP1).
Introduction
Enhancer of zeste homolog 2 (EZH2) is an important member of the epigenetic regulatory factor PcG (polycomb group proteins) and a catalytic subunit of polycomb repressive complex 2 (PRC2).Citation1 EZH2 transfers the methyl group of adenosine methionine (SAM) to H3K27 and methylates H3K27.Citation2,Citation3 The Tri-methylation of H3K27 can suppress the expression of target genes, such as RUNX3 and BRCA1.Citation4,Citation5 It has been demonstrated that EZH2 plays an important role in epigenetic regulation.Citation6–Citation8 EZH2 is abnormally expressed in a wide variety of tumors, and its expression is unusually high in osteosarcoma.Citation9–Citation11 Osteosarcoma is the most common malignant tumor of mesenchymal tissues, often occuring in the long bone metaphysis of adolescents, and is a differentiation-defective disease caused by blocked osteoblast differentiation and/or epigenetic alterations.Citation12 Scientists consider EZH2 as an attractive target for the treatment of osteosarcoma and have found many potential EZH inhibitors, such as 3-deazaneplanocin A (DZNeP), GlaxoSmithKline (GSK)126, EI1 and GSK343.Citation13–Citation16 The chemical name of GSK343 is N-[(6-methyl-2-oxo-4-propyl-1H-pyridin-3-yl) methyl]-6-[2-(4-methylpiperazin-1-yl) pyridin-4-yl]-1-propan-2-ylindazole-4-carboxamide (C31H39N7O2, MW: 541.69). GSK343 is a methionine competitive histone lysine methyl-transferase inhibitor that directly and selectively inhibits the activity or expression of EZH2.Citation16,Citation17 It has been reported that GSK343 suppresses glioblastoma, ovarian cancer, liver cancer and colorectal cancer.Citation18–Citation21 By performing in silico and experimental analyses, Liu et al found that GSK343 inhibited hepatocellular carcinoma through the induction of metallothionein 1 and 2A.Citation22 In our previous study, we found that GSK343 not only inhibited EZH2 expression, but also inhibited the expression of FUSE binding protein 1(FBP1).Citation23
FBP1 activates the transcription of the c-Myc gene by binding to the far-upstream element (FUSE).Citation24–Citation26 FBP1 is overexpressed in 80% of HCC and ovarian cancer cases,Citation27–Citation29 physically interacts with p53 and suppresses its transcriptional activity.Citation30,Citation31 FBP1 expression in tumor cells is linked to poor patient survival.Citation27,Citation32 Recently, we demonstrated that FBP1 physically interacted with EZH2 in osteosarcoma cells and that GSK343 treatment inhibited the expression of EZH2 and FBP1.Citation23
In order to enhance our understanding of GSK343 as a molecule for the clinical treatment of osteosarcoma, this study examined the effect of GSK343 on the programmed cell death of human osteosarcoma Saos2 cells, and aimed to elucidate its potential underlying mechanisms. It is well known that c-Myc is the downstream target of FBP1, while EZH2 and c-Myc are a pair of mutually activating proteins.Citation33,Citation34 Therefore, this study also aimed to clarify whether FBP1 and EZH2 share mutual regulatory roles, and whether they play an important role in GSK343-induced programmed cell death.
Results
GSK343, GSK126 and DZNeP induce apoptosis in Saos2 and U2OS cells
For apoptosis (type I programmed cell death) assay, Annexin V-FITC/PI double staining and flow cytometry were used. The apoptotic rates were detected after the Saos2 and U2OS cells were treated for 48 h with 0, 5, 10 and 20 μM of GSK343; 0, 1, 2 and 4 μM DZNeP and 0, 5, 10 and 15 μM GSK126. For GSK343, the apoptotic rates of the Saos2 and U2OS cells treated with 10 and 20 μM were significantly higher than those of the controls (0 μM) group (P < .05). The highest apoptotic rate was observed in the 20 μM group (), panels a and b, and 1B, panel a). For DZNeP, significantly higher apoptotic rates were only observed in the U2OS cells treated with 4 μM. No significantly higher apoptotic rates were found in the Saos2 cells (), panels c and d, and B, panel b). For GSK126, the apoptotic rates of the Saos2 and U2OS cells treated with 15 μM GSK126were significantly higher than those of the control (0 μM) group (P < .05) (), panels e and f, and B, panel c).
Figure 1. Effects of GSK343 on the apoptosis of Saos2 cells.
(a and b) Apoptotic Saos2 and U2OS cells were detected by Annexin V/PI assay following treatment with GSK343, DZNeP and GSK126 for 48 h at the indicated concentrations. Apoptotic rates are presented in (b).(c) The expression of apoptosis-associated proteins in Saos2 cells. Saos 2 cells were treated for 48 h with the indicated concentrations of GSK343. Data are presented as the means ± SD (n = 3). *P < .05 compared with the control (0 μM of GSK343). Data shown are representative of a minimum of three independent experiments.
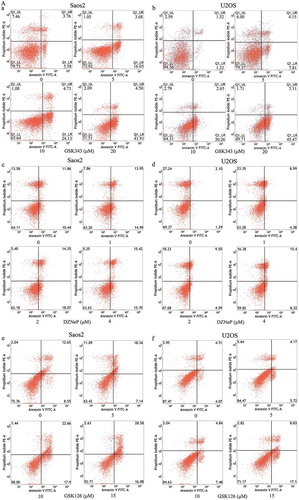
Figure 1. (Continued)
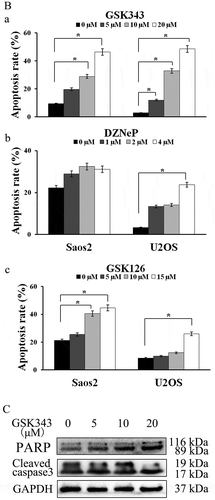
Based on the aforementioned results, further experiments were carried out using GSK343 and Saos2 cells. As apoptotic markers, the expressions of cleaved caspase-3 and ADP-ribose polymerase (PARP) were detected by western blot analysis. With increasing concentrations of GSK343, the expressions of cleaved capase-3 and PARP were markedly increased compared with the control group ()).
GSK343 induces autophagic cell death in Saos2 cells
Autophagy refers to a process through which some proteins and organelles are transported to lysosomes for degradation through the inclusion of autophagosomes.Citation35 Autophagic cell death, also known as type II programmed cell death, is considered a possible tumor suppressor mechanism.Citation36 In order to investigate whether GSK343 induces autophagy, we analyzed the GSK343-induced activation and translocation of LC3-II to the autophagosome. Western blot analysis revealed the accumulation of LC3-II in the autophagosome of Saos2 cells with increasing concentrations GSK343. The majority of LC3-II punctate was found in the cells treated with 20 μM GSK343 (), panels a-d). The formation of autophagic vacuoles was also detected by transmission electron microscopy to evaluate the level of autophagic cell death. Compared with the control group, the group treated with 5 μM GSK 343 exhibited only a mild expansion of the partial mitochondrial intracellular cleft, a mild expansion of the partial rough endoplasmic reticulum, and occasional autophagosomes; the group treated with 10 μM exhibited multiple autophagosomes in the cytoplasm, slight mitochondrial swelling and partial cavitation; the group treated with 20 μM exhibited a large number of autophagosomes, mitochondrial swelling, internal dilatation and partial cavitation (), panels e-h). Autophagosome formation increased gradually with increasing GSK343 concentrations. The greatest degree of autophagosome formation was observed following treatment with 20 μM GSK343.
Figure 2. GSK343 induces autophagic cell death in Saos 2 cells.
(a, panels a-d) LC3-II puncta formation in Saos2 cells treated with the indicated concentrations of GSK343 for 48 h, and examined by immunohistochemistry. Images were co-stained with DAPI and observed immediately using a Zeiss confocal microscope (magnification, x400). The arrow indicates LC-II puncta.(a, panels e-h) Autophagic vacuole formation in Saos2 cells treated with the indicated concentrations GSK343 for 48 h, captured by electronic microscopy (magnification, x15,000; scale bar, 500 nm).(b) The expressions of p62, LC3-I and -II in Saos2 cells treated with the indicated concentrations of GSK343 for 48 h, detected by western blot analysis. GAPDH was used as the loading control.(c) Relative expression of p62 against GAPDH at the indicated concentrations of GSK343. (d) LC-II/LC-I ratios in Saos2 cells treated with the indicated concentrations of GSK343 for 48 h. *P < .05 and #P < .01 compared with the control (0 μM of GSK343). Data shown are representative of a minimum of three independent experiments.
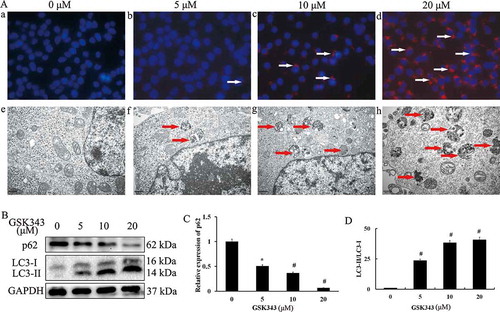
Similar to LC3, p62 is recognized as a marker of autophagic cell death. In order to evaluate the level of autophagic cell death,we measured the ratio of LC3-II/LC3-I and the expression of p62 in Saos2 cells treated with various concentrations of GSK343 for 48 h. With increasing concentrations of GSK343,the protein expression level of p62 gradually decreased (),while LC3-II protein and the LC3-II/I ratio gradually increased (). The reduction in p62 expression and the induction of LC3-II were both GSK343 dose-dependent. A significant reduction in p62 expression and the induction of LC3-II were observed following treatment with 5 μM GSK343.
GSK343 inhibits the expression of c-Myc, EZH2 and FBP1 in Saos2 cells
The expression of EZH2 was detected by western blot analysis. As shown in , the expression level of EZH2 was inhibited by GSK 343 in the Saos2 cells, and this inhibitory effect occurred in a dose-dependent manner. As the target of EZH2 and an oncogene, the expression level of c-Myc in the Saos2 cells was also reduced by GSK343 in a dose-dependent ().
Figure 3. Effects of GSK343 on associated protein expression in Saos2 cells.
(a) The expression of EZH2, FBP1 and c-Myc in Saos2 cells treated with the indicated concentrations of GSK343 and detected by western blot analysis. GAPDH was used as a loading control.(b-d) The relative expressions of p62, FBP1 and c-Myc at the indicated concentrations of GSK343. #P < .01 compared with the control (0 μM of GSK343). Data shown are representative of a minimum of three independent experiments.
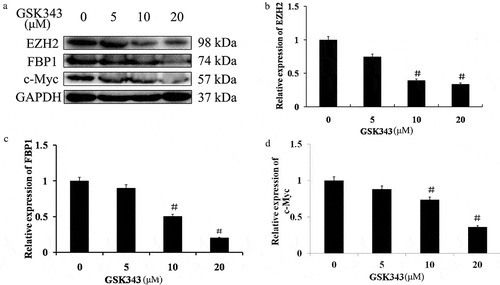
Since c-Myc is up-regulated by FBP1, the expression level of FBP1 was examined. Similar to c-Myc and EZH2, the expression level of FBP1 was also inhibited by GSK343 in a dose-dependent manner ().
The expressions of EZH2, FBP1 and c-Myc were significantly inhibited by 10 and 20 μM GSK343. Since c-Myc is a common target of EZH2 and FBP1, and GSK343 inhibited the expression of these proliferation-promoting proteins, this suggests a mutual regulatory association between EZH2 and FBP1.
The mutual regulatory activity of EZH2 and FBP1
Since c-Myc is the downstream target of EZH2 and FBP1,Citation23–Citation26,Citation33,Citation34 and a physical interaction has been identified between EZH2 and FBP1,Citation23 it would be of interest to clarify whether there is a mutual regulatory effect between FBP1 and EZH2. In this study, either EZH2 or FBP1 was silenced, and the expressions of EZH2 or FBP1 were investigated by RT-qPCR and western blot analysis in normal Saos2 cells and in Saos2 cells in which FBP1/EZH2 was knocked down.
The knockdown of EZH2 and FBP1 was found to be successful by comparing lane 3 () or lane 2 () with lane 1 (). By comparing lane 2 with lane 1 of , it is evident that the protein and mRNA expression of EZH2 were significantly inhibited by FBP1 knockdown. The knockdown of FBP1 significantly accelerated the inhibitory effects of GSK343 on EZH2 at both the protein and mRNA expressions, as shown by comparing lanes 5 and 2 of . The knockdown of EZH2 promoted the inhibitory effects of GSK343 on EZH2 expressions both at the protein and mRNA level (lanes 6 and 3 of ).
Figure 4. Mutual regulatory association between EZH2 and FBP1 expression.
(a) The expression of EZH2 and FBP1 in FBP1 or EZH2 normal or knockdown Saos2 cells treated with 0 or 10 μM GSK343, and detected by western blot analysis. GAPDH was used as a loading control.(b and c) The relative protein expressions of EZH2 and FBP1 in FBP1 or EZH2 normal or knockdown Saos2 cells treated with 0 or 10 μM GSK343.(d and e) The relative mRNA expressions of EZH2 and FBP1 in FBP1 or EZH2 normal or knockdown Saos2 cells treated with 0 or 10 μM GSK343. *P < .05 compared with the control (0 μM of GSK343). Data are representative of a minimum of three independent experiments.
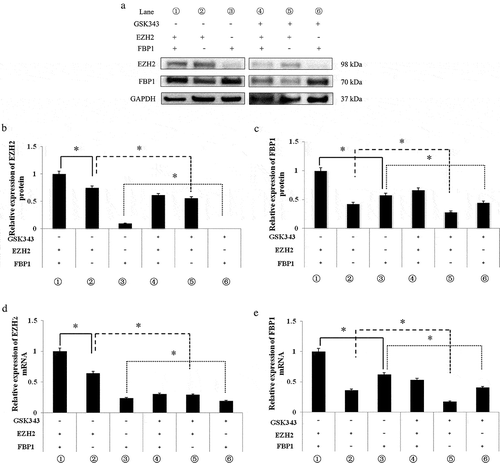
Similarly, the protein and mRNA expressions of FBP1 were significantly inhibited by EZH2 knockdown (compare lanes 1 and 3 of ). The knockdown of EZH2 significantly accelerated the inhibitory effects of GSK343 on FBP1 expression both at the protein and mRNA level, as evidenced by comparing lanes 6 and 3 of . The knockdown of FBP1 promoted the inhibitory effects of GSK343 on the FBP1 expression both at protein and mRNA level (lanes 6 and 3 of ).
These results indicated a mutual regulatory mechanism between FBP1 and EZH2 expression; the knockdown of FBP1 and EZH2 enhanced the sensitivity of Saos2 cells to GSK343.
Discussion
The occurrence of a tumor is a multi-factorial, multi-stage evolutionary process, involving mutations and epigenetic alterations in a number of genes, including oncogenes, tumor suppressor and DNA damage repair genes.Citation37 EZH2 is an important component of the epigenetic regulatory factor, PcG, and a catalytic subunit of PRC2. It has been reported that EZH2 is abnormally and highly expressed in various tumor tissues,Citation9–Citation11 and in recent years, several potential inhibitors of EZH2 have been discovered.Citation13–Citation16 GSK343 is a direct competitive inhibitor of methionine, which can inhibit the expression of EZH2 in tumor cells to achieve an anti-tumor effect.Citation18–Citation21 Currently, relatively few studies have been conducted on the efficacy of GSK343 in solid tumors, such as ovarian cancer, liver cancer and colorectal cancer.Citation19–Citation21
In this study, we found that GSK343 induced apoptosis, also known as type I programmed cell death, in Saos2 cells in a dose-dependent manner, and that treatment with 10 and 20 μM GSK343 significantly induced Saos2 cell apoptosis. The expressions of apoptosis-related proteins, such as cleaved caspase-3 and PARP, were also increased.
Autophagy, also known as type II programmed cell death, is a subcellular degradative process where lysosomes degrade macromolecular proteins and organelles. Autophagy is important for maintaining cell homeostasis and for adaptation to adverse internal and external environments.Citation38 Moderate autophagy is considered to promote cell tolerance during environmental adaptation. However, the persistence of unfavorable factors can lead to excessive autophagy, which subsequently induces and accelerates autophagic cell death.Citation39 LC3 is commonly used to detect autophagic cell death and has type I and II forms. When autophagic cell death occurs, LC3-I and phosphatidyl ethanolamine combine to form LC3-II and localize to the autophagosome membrane prior to fusion with lysosomes; thus to a certain extent, LC3-II expression can reflect autophagic activity.Citation40 p62 protein, an autophagic substrate, is often used in conjunction with LC3 to detect autophagy. This study demonstrated a decrease in the expression of p62 and an increase in the LC3-II/LC3-I ratio with increasing GSK343 concentrations. This suggested that GSK343 promoted autophagic cell death, which was confirmed by transmission electron microscopy and immunofluorescence; the number of autophagosomes increased with increasing concentrations of GSK343, which further verified that GSK343 was able to induce autophagic cell death.
c-Myc is one of the most commonly overexpressed oncogenes in cancers, and is a transcription factor involved in DNA replication and cell proliferation via regulating target gene expression. EZH2 induces c-Myc expression via the inhibition of Myc targeting miR-494.Citation33,Citation34 As feedback, c-Myc contributes to EZH2 upregulation via the repression of EZH2 targeting miR-26a.Citation33,Citation34 Thus, EZH2 and c-Myc activate each other via a feedback loop mechanism. On the other hand, FBP1 up-regulates c-Myc expression by binding to the FUSE domain of the c-Myc operator. Consistent with the results presented in ta study by Ezponda et al,Citation41 we demonstrated that c-Myc expression was decreased following GSK343 treatment, in addition to the expressions of EZH2 and FBP1. These data demonstrated that both EZH2 and FBP1 were suppressed by GSK343. GSK343 may directly or indirectly influence c-Myc expression through EZH2 and/or FBP1. Gene knockdown experiments demonstrated that the knockdown of either EZH2 or FBP1 impaired their expressions. The current study identified that the knockdown of EZH2 decreased FBP1 expression; similarly, the knockdown of FBP1 also decreased EZH2 expression. Furthermore, the knockdown of EZH2 or FBP1 accelerated the reduction of FBP1 and EZH2 expression induced by GSK343 treatment. These results suggest the possibility of the existence of a mutual regulatory association between EZH2 and FBP1.
In consistent to our published results,Citation23 this study confirmed that GSK343 induced programmed cell death in human osteosarcoma cells in a dose-dependent manner. The reduction in EZH2 and FBP1 expressions may, at least in part, be the mechanism underlying the effects of GSK343. Additionally, this study demonstrated the mutual positive relationship between EZH2 and FBP1 basing on the fact that the knockdown of EZH2 or FBP1 suppressed the expression of FBP1 or EZH2.
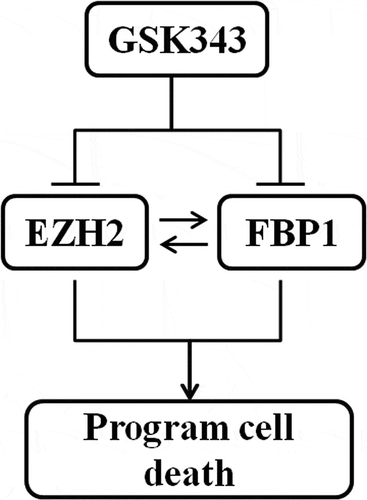
The main results are summarized in .
Figure 5. Hypothetical model of GSK343-induced programmed cell death in osteosarcoma cells.
Materials and methods
Reagents, cells and cell culture
GSK343, GSK126 and DZNeP were provided by Selleck Chemical Co. (Cat. nos. S7164, S7061 and S7120). The Saos2 and U2OS human osteosarcoma cell lines were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Cat. no. TCHu114 and TCHu88). Penicillin and streptomycin were purchased from HyClone (Cat. no. SV30010). The Saos2 cells were cultured in high-glucose DMEM (Gibco, Cat. no. C11965500137) containing 10% FBS (Gibco, Cat. no. 10099–141) 100 U/ml penicillin and 100 U/ml streptomycin at 37°C and 5% CO2.
The apoptosis detection kits were purchased from KeyGen Biotech (Cat. No. G003-1-2). The cell total protein extraction kit and BCA protein quantification kit were purchased from Beyotime Biotechnology (Cat. no. P00138) and ThermoFisher Scientific (Cat. no. P0011), respectively. GAPDH antibody (Cat. no. 5174), EZH2 antibody (Cat. no. 5246.), p62 antibody (Cat. no. 39749), PARP (Cat. no. 9542), LC3 (Cat. no. 3868) and cleaved caspase-3 (Cat. no. 9664) antibodies were purchased from Cell Signaling Technology. FBP1 (Cat. no. ab181111.) and c-Myc (Cat. no. ab320752.) antibodies were provided by Abcam.
Flow cytometric assay
An Annexin V-FITC/propidium iodide (PI) double staining assay was used to measure the level of apoptosis in Saos2 cells. The cells were inoculated on a 6-well plate and treated with various concentrations of GSK343 for 48 h. Each group of cells was stained following the manufacturer’s instructions. The number of apoptotic cells was detected by flow cytometry and analyzed using CellQuest™ software (BD Biosciences). The experiment was repeated 3 times.
Western blot analysis
Following treatment with various concentrations of GSK343 for 48 h, the Saos2 cells were harvested. Prior to western blotting, the cells were lysed in modified RIPA buffer [150 mM NaCl, 1% NP-40, 50 mM Tris-Cl (pH 8.0), 0.1% SDS] supplemented with protease and phosphatase inhibitor, PMSF (1 mM) ThermoFisher Scientific (Cat. no. 36978). Following rapid homogenization, the homogenate was incubated in ice for 30 min and centrifuged at 12,000 x g for 15 min at 4°C. The protein concentration was measured by BCA assay. Approximately 40 μg of protein sample per well was loaded on 12% SDS-PAGE and transferred onto PVDF membranes (Merck Millipore, Cat. no. ISEQ00010). The membranes were blocked with 5% nonfat milk in Tris-buffered saline with Tween-20 (TBST) for 1 hour at room temperature, incubated overnight at 4°C with the designated primary antibodies, and incubated with secondary antibodies for 1 h at room temperature. An ECL luminescence assay was used to develop the blots (Pierce, Cat. no. 32209), and relative abundance was quantified by densitometry using Quantity One 4.6.7 software (Bio-Rad).
Quantitative analysis of gene expression by reverse transcription-quantitative polymerase chain reaction PCR (RT-qPCR)
Approximately 2 × 105 cells were transferred to a 6-well plate (final total volume of 4 ml), and cultured for 48 h without or with GSK343. Total RNA was extracted from the cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Cat. no. 15596026) and converted to cDNA using PrimeScript RT Master Mix (Takara, Cat. no. RR036A). qPCR was performed with SYBR Premix ExTaq (Takara, Cat. no. RR420A) in a real-time PCR system (Analytik Jena). All samples were performed in triplicate. GAPDH was used as an internal control. The primer sequences were as follows: FBP1-forward, 5ʹ-TGATTCCAGCTAGCAAGGCA-3ʹ and reverse, 5ʹ-CGGCCCGTCTTGAATCATAA-3ʹ; EZH2-forward, 5ʹ-ACCAGCATTTGGAGGGAGC-3ʹ and reverse, 5ʹ-TGGGAAGCCGTCCTCTTCT-3ʹ; GAPDH forward, 5ʹ-GATTCCACCCATGGCAAATT-3ʹ and reverse, 5ʹ-TCTCGCTCCTGGAAGATGGT-3ʹ. The relative gene expression was quantified using the 2−ΔΔCq method and normalized to the internal reference gene, GAPDH.
Immunofluorescence assay
The Saos2 cells were treated with various concentrations of GSK343 for 48 h, and then fixed in 4% paraformaldehyde for 15 min, washed in PBS, blocked using 10% normal goat serum, and incubated with a rabbit anti-human L3 polyclonal antibody (1:100) overnight at 4°C. Dylight 594-conjugated anti-rabbit IgGs (Abcam, Cat. no. ab96885) were applied at a 1:200 dilution for 1 h at room temperature. The cells were counterstained with DAPI and observed immediately using a ZIESS confocal microscope (Carl Zeiss)
Transmission electron microscopy
The Saos2 cells were treated with various concentrations of GSK343 for 48 h. Following treatment with GSK343, the Saos2 cells were fixed with 2% glutaraldehyde at 4°C for 15 min. The cells were then dehydrated in a graded ethanol series and embedded in Agar 100 epoxy resin. Ultra-thin sections were mounted on Cu grids and stained first with uranyl acetate, followed by lead citrate. Transmission electron microscopy (FEI) was used to detect autophagic vacuoles.
Construction of FBP1-knockdown lentivirus and generation of stable FBP1-knockdown cells
To knock down FBP1, a pSi-LVRH1GP vector with a puromycin resistance cassette (GeneCopoeia, Cat. no. LP-HSH021663) was used to express a short hairpin (sh)RNA to inhibit FBP1 expression. The control vector expressed a scrambled sequence (5ʹ-GCT TCG CGC CGT AGT CTT A-3ʹ) and was designated as pSi-LVP-FBP1-C. The FBP1 knockdown vector expressed a sequence of 1671–1691 (5ʹ-GCA GGA ACG GAT CCA AAT TCA-3ʹ) of FBP1 and named as pSi-LVP-FBP1-KD.
To knockdown EZH2, a psi-LVRU6MH vector with a hygromycin resistance cassette (GeneCopoeia, Cat. no. LPP-HSH005050) was used to express shRNA to knockdown EZH2 expression. The control vector expressed a scrambled sequence and was designated as pSi-LVH-EZH2-C. The EZH2 knockdown vector expressed a 695–715 sequence (5ʹ-GCCCTTGGTCAATATAATGAT-3ʹ) of EZH2 and was named pSi-LVH-EZH2-KD.
The Saos2 cells were transfected with pSi-LVP-FBP1-C/pSi-LVH-EZH2-C or pSi-LVP-FBP1-KD/pSi-LVH-EZH2-KD. First, 2 × 105 Saos2 cells were seeded in 2 ml antibiotic-free DMEM medium supplemented with FBS, and cells were incubated until they reached 60–80% confluence. After washing the cells once with 2 ml antibiotic- and FBS-free DMEM medium, 900 μl of the same medium was added to the cells, alongside 1 μg pSi-LVP-FBP1-C/pSi-LVH-EZH2-C or pSi-LV-FBP1-KD/pSi-LVH-EZH2-KD diluted in 100 μl medium. After 6 h, the medium was exchanged for DMEM supplemented with FBS and antibiotics. Puromycin or hygromycin were used for selection. Following a 48-h incubation, the medium was exchanged with DMEM containing 2.0 μg/ml puromycin (Invitrogen; Thermo Fisher Scientific, Cat. no. A1113803) or 200 μg/ml of hygromycin B (Invitrogen; Thermo Fisher Scientific, Cat. no. 10687010). Western blotting was used to confirm the knockdown efficiency. The pSi-LV-FBP1-C/pSi-LVH-EZH2-C-transfected cells were designated as FBP1-C/EZH2-C, and the pSi-LV-FBP1-KD/pSi-LVH-EZH2-KD-transfected cells were designated as FBP1-KD/EZH2-KD.
Statistical analysis
All statistical analyses were performed using SPSS 17.0 statistic software (SPSS, Inc. Chicago, IL, USA). The data are expressed as the means ± SD. Significance differences were determined by one-way analysis of variance (ANOVA), and P<0.05 was considered to be statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The flow cytometry experiment was supported by Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University.
Additional information
Funding
References
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349.
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043.
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196.
- Kodach LL, Jacobs RJ, Heijmans J, van Noesel CJ, Langers AM, Verspaget HW, Hommes DW, Offerhaus GJ, van Den Brink GR, Hardwick JC. The role of EZH2 and DNA methylation in the silencing of the tumour suppressor RUNX3 in colorectal cancer. Carcinogenesis. 2010;31:1567–1575.
- Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19:1371–1378.
- Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14:183–193.
- Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29.
- Tsang DP, Cheng AS. Epigenetic regulation of signaling pathways in cancer: role of the histone methyltransferase EZH2. J Gastroenterol Hepatol. 2011;26:19–27.
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629.
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611.
- Lv YF, Yan GN, Meng G, Zhang X, Guo QN. 2015. Enhancer of zeste homolog 2 silencing inhibits tumor growth and lung metastasis in osteosarcoma. Sci Rep. 5:12999. doi: 10.1038/srep12999..
- Tang N, Song WX, Luo J, Haydon RC, He TC. Osteosarcoma development and stem cell differentiation. Clin Orthop Relat Res. 2008;466:2114–2130.
- Crea F, Fornaro L, Bocci G, Sun L, Farrar WL, Falcone A, Danesi R. EZH2 inhibition: targeting the crossroad of tumor invasion and angiogenesis. Cancer Metastasis Rev. 2012;31:753–761.
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109:21360–21365.
- Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlanger KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS. 2012;3:1091–1096.
- Ferraro A, Boni T, Pintzas A. EZH2 regulates cofilin activity and colon cancer cell migration by targeting ITGA2 gene. PloS One. 2014;9:e115276.
- Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9:643–650.
- Yu T, Wang Y, Hu Q, Wu W, Wu Y, Wei W, Han D, You Y, Lin N, Liu N. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget. 2017;8:98348–98359.
- Amatangelo MD, Garipov A, Li H, Conejo-Garcia JR, Speicher DW, Zhang R. Three-dimensional culture sensitizes epithelial ovarian cancer cells to EZH2 methyltransferase inhibition. Cell Cycle. 2013;12:2113–2119.
- Liu TP, Lo HL, Wei LS, Hsiao HH, Yang PM. S- Adenosyl-L- methionine- competitive inhibitors of the histone methyltransferase EZH2 induce autophagy and enhance drug sensitivity in cancer cells. Anticancer Drugs. 2015;26:139–147.
- Hsieh YY, Lo HL, Yang PM. EZH2 inhibitors transcriptionally upregulate cytotoxic autophagy and cytoprotective unfolded protein response in human colorectal cancer cells. Am J Cancer Res. 2016;6:1661–1680.
- Liu TP, Hong YH, Tung KY, Yang PM. In silico and experimental analyses predict the therapeutic value of an EZH2 inhibitor GSK343 against hepatocellular carcinoma through the induction of metallothionein genes. Oncoscience. 2016;3:9–20.
- Xiong X, Zhang J, Liang W, Cao W, Qin S, Dai L, Ye D, Liu Z. Fuse-binding protein 1 is a target of the EZH2 inhibitor GSK343, in osteosarcoma cells. Int J Oncol. 2016;49:623–628.
- Avigan MI, Strober B, Levens D. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J Biol Chem. 1990;265:18538–18545.
- He L, Weber A, Levens D. Nuclear targeting determinants of the far upstream element binding protein, a c-myc transcription factor. Nucleic Acids Res. 2000;28:4558–4565.
- Liu J, Kouzine F, Nie Z, Chung HJ, Elisha-Feil Z, Weber A, Zhao K, The LD. FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. Embo J. 2006;25:2119–2130.
- Rabenhorst U, Beinoraviciute-Kellner R, Brezniceanu ML, Joos S, Devens F, Lichter P, Rieker RJ, Trojan J, Chung HJ, Levens DL, et al. Overexpression of the far upstream element binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology. 2009;50:1121–1129.
- Xiong X, Zhang J, Hua X, Cao W, Qin S, Dai L, Liu W, Zhang Z, Li X, Liu Z. FBP1 promotes ovarian cancer development through the acceleration of cell cycle transition and metastasis. Oncol Lett. 2018;16:1682–1688.
- Zhang J, Xiong X, Hua X, Cao W, Qin S, Dai L, Liang P, Zhang H, Liu Z. Knockdown of FUSE binding protein 1 enhances the sensitivity of epithelial ovarian cancer cells to carboplatin. Oncol Lett. 2017;14:5819–5824.
- Dixit U, Pandey AK, Liu Z, Kumar S, Neiditch MB, Klein KM, Pandey VN. FUSE binding protein 1 facilitates persistent hepatitis C virus replication in hepatoma cells by regulating tumor suppressor p53. J Virol. 2015;89:7905–7921.
- Dixit U, Liu Z, Pandey AK, Kothari R, Pandey VN. 2014. Fuse binding protein antagonizes the transcription activity of tumor suppressor protein p53. BMC Cancer. 14:925. doi: 10.1186/1471-2407-14-925..
- Malz M, Weber A, Singer S, Riehmer V, Bissinger M, Riener MO, Longerich T, Soll C, Vogel A, Angel P, et al. Overexpression of far upstream element binding proteins: a mechanism regulating proliferation and migration in liver cancer cells. Hepatology. 2009;50:1130–1139.
- Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget. 2011;2:669–683.
- Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012;22:506–523.
- Lozy F, Karantza V. Autophagy and cancer cell metabolism. Semin Cell Dev Biol. 2012;23:395–401.
- Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–581.
- Jia L, Sun X, Ding H. Research progress on the relationship between EZH2 gene and tumor. J Clin Neurol. 2012;25:154–156.
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autoaphagy fights disease though cellular self digestion. Nature. 2008;451:1069–1075.
- Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis,autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975.
- Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 2009;452:1–12.
- Ezponda T, Dupéré-Richer D, Will CM, Small EC, Varghese N, Patel T, Nabet B, Popovic R, Oyer J, Bulic M, et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. 2017;21:628–640.