
ABSTRACT
Gemcitabine (Gem) is currently used as the first-line therapy for liver and pancreatic cancer but has limited efficacy in most cases. Dexamethasone (Dex) have been applied as a chemoprotectant and chemosensitizer in cancer chemotherapy. This study further explored the potential of combination of Gem and Dex and tested the hypothesis that glucocorticoid receptor signaling is essential for the synergistic antitumor activity. In the HepG2 and AsPC-1 xenograft models, the combination treatment showed a significantly synergistic antitumor activity. Immunohistochemistry of post-treatment tumors showed a significant decrease in proliferation and angiogenesis as compared to either of the treatments alone. Dex alone and the combination with Gem inhibited the expression of glucocorticoid receptor. The combination of Dex and Gem showed synergistic cytotoxicity in cell lines in vitro. The antiproliferative synergism is prevented by used glucocorticoid receptor (GR) small interfering RNA, demonstrating that the glucocorticoid receptor is required for the antiproliferative synergism of Gem and Dex. The inhibition of glucocorticoid receptor signaling pathway and induction of apoptosis via activation of caspases 3, 8 and 9, PARP, contributed to the synergistic effect of this combination therapy. These results demonstrate that Dex could potentiate the antitumor efficacy of Gem. The synergistic antitumor activity of the combination of Dex and Gem was through glucocorticoid receptor signaling. Taken together, a combination of Dex and Gem shows a significant synergistic antitumor activity and lesser toxicity both in vitro and in vivo and could be a combination chemotherapy for the treatment of highly expression of glucocorticoid receptor patients.
Introduction
Anticancer drugs are limited by the potential toxicity to normal tissues and by the development of drug resistance. Combination therapy using two or more chemotherapeutic drugs is one of the most common strategies used in current oncology to overcome these obstacles. Gemcitabine (Gem), a deoxycytidine analogue, is extensive used for chemotherapy for patients with various tumors.Citation1–Citation6 Gem is a prodrug and metabolized inside cells into the active phosphate type, which incorporates into nucleic acids and terminates DNA synthesis.Citation7,Citation8 Since the limited efficacy in most cases, Gem has the desired characteristic needed to obtain synergistic activity.
Corticosteroids including dexamethasone (Dex) have been applied in clinical cancer treatment for many years. Dex has hematoprotective activity and could be used to prevent chemotherapy-induced toxicity, and also used as chemosensitizer in cancer chemotherapy.Citation9,Citation10 Moreover, Dex exerts the anti-angiogenic effects and inhibits the tumor growth.Citation11,Citation12 Our group and others have reported that Dex monotherapy shown anticancer activity against multiple cancer cell lines in vitro (i.e. breast, metastatic brain tumor, melanoma, renal, glioma, prostate and ovarian cancer) and in xenograft models.Citation13–Citation20 A similar result also reported the combination of Dex and Gem showed a synergistic effect in breast carcinoma MCF7 xenograft model.Citation21 Combination of Dex and Gem, might be possible to achieve a better therapeutic effect than either drug alone and to overcome resistance to one or both drugs. The detailed mechanisms by which Dex enhances antitumor effects of Gem have not been fully understood. Dex treatment will improve sensitivity to chemotherapy – induced cell death through inhibition of glucocorticoid receptor (GR) – mediated cell survival pathways.Citation22 Therefore, Dex exerts synergistic effect with Gem through activating the glucocorticoid receptor (GR) signaling, which is one of the possible the mechanism.
This study aimed to evaluate the therapeutic effects of Gem and Dex, alone or in combination, and elucidate the molecular mechanisms underlying their synergistic antitumor activity, with major focus on glucocorticoid receptor (GR) signaling with apoptosis.
Material and method
Cell cultures
Human hepatocellular carcinoma HepG2 and pancreatic ductal adenocarcinoma AsPC-1 cells were obtained from Cell Center of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College. HepG2 cells were maintained in high-glucose DMEM (Thermo Fisher Scientific, Logan, UT, USA), and AsPC-1 cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA) and penicillin/streptomycin under 5% CO2 at 37°C in a humidified atmosphere.
In vitro cytotoxicity assays
In vitro cytotoxicities of Gem or Dex alone or the combination in cancer cell lines were determinated by using CCK-8 assay. Briefly, cells were seeded at 4000 cells/wells in 96-well plates (Costar, Cambridge, Massachusetts, USA). After overnight incubation, triplicate wells were treated with varying concentrations of Gem with or without Dex for 48 h. Dex was purchased from Sigma-Aldrich and reconstituted according to the manufacturer’s instruction. Pharmaceutical-grade Gem (APP Pharmaceuticals) was used. Cells treated with PBS served as control.CCK-8 solution (MedChem Express, Monmouth Junction, NJ, USA) was added to each well. After incubation for 3 h, the optical density (OD) values were measured with a Multiskan GO microplate reader (Thermo Fisher Scientific, Inc. USA) at 450 nm.
The coefficient of drug interaction (CDI)
Drug interactions in vitro were analyzed according to CDI.Citation23 CDI was calculated according to the formula: CDI = AB/(A × B), where A and B are the ratio of the survival values of respective single agent and the control; AB is the ratio of survival values of two drugs combination and the control. A synergistic effect was considered to be a two-drug combination for CDI<1 and a significantly synergistic effect of a two-drug combination for CDI<0.7. Combination analysis in vivo was also calculated by CDI as previous report.Citation24
Detection of cell apoptosis
The mode of cell death was investigated by the Muse Annexin V & Dead Cell Assay on the Muse Cell Analyzer (Biovision Inc., CA, USA). Treated cells were harvested by trypsinization and washed once with cold PBS. Then, cell suspensions were incubated with Muse Annexin V & Dead Cell reagent at room temperature in the dark. Samples were analyzed according to the instructions of the manufacturer.
Immunoblotting analysis
Cells were allowed to adhere overnight in media containing 10% FCS. The following day, cells were treated with different treatment and cultured for an additional 24 h, followed by lysis in buffer containing PhosSTOP phosphatase inhibitor (Roche Life Science) and Complete EDTA protease inhibitor (Roche Life Science). Protein concentrations were measured using the BCA Assay Kit (Thermo Scientific) and 2× Laemmli buffer supplemented with 5% 2-mercaptoethanol was added to an equivalent volume of protein lysate. Proteins (20 μg per lane) were resolved by SDS-PAGE and then transferred to polyvinylidene difluoride membranes (Bio-Rad). The membranes were washed three times in 0.1% Tween 20 in TBS (TBST), and then incubated with blocking solution (5% bovine serum albumin (BSA; Fisher Scientific) in TBST) for 1 h at room temperature. Primary antibodies were diluted in blocking solution and incubated with the membrane overnight incubation at 4°C. Antibodies and concentrations were as follows: monoclonal rabbit anti-glucocorticoid receptor (GR) XP antibody (Cell Signaling), rabbit monoclonal anti-Nrf2 antibody (Cell Signaling), mouse anti-PARP-1 antibody, mouse anti-Caspase-3 antibody (Santa Cruz Biotechnology), anti-Capase-8, anti-Capase-9 (Cell Signaling Technology), mouse anti-GAPDH antibody (Zhongshan Inc.). After additional washing, membranes were incubated with peroxidase-coupled antibody (Zhongshan Inc.) at RT for 2 h which used for detecting the primary antibody binding. Protein bands were visualized with an enhanced chemiluminescence kit (Merck Millipore).
Sirna transfection
The GR siRNA was purchased commercially (Santa Cruz Biotechnology, Santa Cruz, CA). The negative control siRNA was used as non-targeting control for all siRNA experiment. Transfection experiments with and siRNAs were performed with Lipofectamine RNAiMAX (Life Technologies) in six-well plates, according to the manufacturer’s instructions (Invitrogen). Following 48 h transfection, the cells subject to be detected by Western Blot or other assays.
Mice xenograft model
Female BALB/c athymic mice (6–8 weeks old, Weitonglihua, Beijing, China) were kept under conventional conditions and provided with pelleted rodent chow and water ad libitum. The study protocols were carried out in accordance with the regulations of Good Laboratory Practice for Non-clinical laboratory studies of drugs issued by the National Scientific and Technologic Committee of People’s Republic of China.
Suspensions of HepG2 and AsPC-1 cells (5 × 106 in 200 μL of PBS) were inoculated into the mice. After 7 days of tumor transplantation, mice (6 per group) were randomized into five groups: PBS, Dex, Gem, Dex plus Gem (50 mg/kg), Dex plus Gem (100 mg/kg). The mice were treated with PBS as control, 50 or 100 mg/kg of Gem intraperitoneally for once/week, or 2 mg/kg Dex orally for 5 days/week for 3 consecutive weeks. Tumor size was measured every 3–4 days and tumor volume was calculated with the following formula: V (mm3) = 0.5 × a× b2, where a, and b represent the long and the perpendicular short diameters of the tumor, respectively.
At the end of the experiment, the mice were sacrificed and the tumors were dissected, weighed, and photographed. The inhibition rate of tumor growth was calculated as [1 − weight (treated)/tumor weight (control)] × 100%.
Histopathological examination
Tumor xenograft samples were fixed in 10% neutral-buffered formalin for 24 h immediately after necropsy and then embedded in paraffin. IHC studies were performed using 5 μm thick formalin-fixed deparaffinized sections. Xenograft tumor tissue was evaluated by IHC for Ki67, CD31, GR expression and apoptosis by using anti-Ki67 antibody, anti-CD31 anitbody (Zhongshan Inc.), anti-GR-XP antibody (Cell Signaling) and In Situ Cell Death Detection Kit (Roche) and the percentage of positively-staining xenografted tumor cells estimated.
Statistical analysis
Data analyses were performed with SPSS software. Data are presented as mean ± SEM. Probability (P) values of <0.05 was considered statistically significant and were calculated using the Student t test as appropriate.
Results
Combination of dex and gem inhibits tumor growth in human hepatocellular cancer hepg2 and pancreatic cancer aspc-1 xenografts
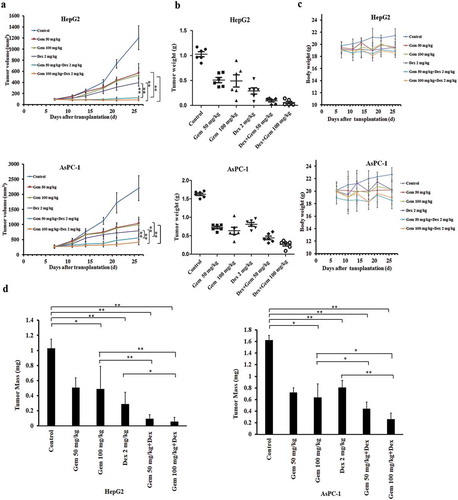
gWe examined the antitumor activity of the combination of Gem and Dex against the hepatocellular cancer HepG2 and pancreatic cancer AsPC-1 xenografts in athymic mice. As shown in ), treatment with either drug alone moderately affected tumor growth as compared with vehicle control, whereas combination treatment much more effectively reduced tumor volume and tumor weight (, ,b)). The combination only slightly reduced the body weights ()), implying the good safety and tolerable toxicity of the experimental regimens. In HepG2 xenograft, the inhibition rate of Gem (50, 100 mg/kg), based on tumor weight change, was 50.10% and 54.55%, respectively. When combined with Dex (2 mg/kg), the inhibition rate was significantly improved up to 91.01% and 94.32%, respectively. The CDI of the combination is 0.63 and 0.43 (). This indicated that the combination therapy with Dex (2 mg/kg) and Gem (50 mg/kg and 100 mg/kg) showed a synergistic inhibitory effect on HepG2 tumor growth. Consistently, single-drug treatment with Dex or Gem moderately reduced tumor sizes, whereas the combination treatment with Dex and Gem significantly reduced both tumor sizes and tumor weight in AsPC-1 xenograft model (, ,b)). The combination only slightly reduced the body weight ()). In AsPC-1 xenograft, the inhibition rate of Gem (50, 100 mg/kg), based on tumor weight change, was 55.52% and 60.86%, respectively. When combined with Dex (2 mg/kg), the inhibition rate was climbed up to 72.87% and 83.79%, respectively. The CDI of the combination (2 mg/kg Dex and 100 mg/kg Gem) was 0.83 (). This indicated that the combination therapy with Dex (2mg/kg) and Gem (100 mg/kg) showed a synergistic effect on AsPC-1 tumor growth inhibition.
Table 1. The antitumor activity of Dex and Gem on the hepatoma cancer HepG2 xenograft model.
Table 2. The antitumor activity of Dex and Gem on the pancreatic cancer AsPC-1 xenograft model.
Figure 1. The antitumor effect of Dex and/or Gem and their combination on HepG2 and AsPC-1 xenograft models. (a) Tumor growing curves of human HepG2 and AsPC-1 xenograft models during the experiment. (b) Tumor weight of human HepG2 and AsPC-1 xenograft models after a 3-week treatment. (c) Body weight change of HepG2 and AsPC-1 xenograft-bearing mice during the experiment. (d) Bar diagram (Mean ± SEM) showing the tumor weight on day 27 after the treatment. Significance was determined using a two-way ANOVA. *P < .05, **P < .01. Dex was given at 2 mg/kg (oral, 5 days/week×3). Gem was given at 50 mg/kg or 100 mg/kg (i.p. once/week×3). The control mice bearing xenografts received an equal volume of saline on the same treatment schedule. (n = 6 in each group). Gem, gemcitabine; Dex, Dexamethasone.
To verify whether the combination therapy increased apoptosis in vivo, the tumor from human HepG2 and AsPC-1 xenograft models were immunostained for Ki67 (cell proliferation marker), CD31 (tumor vessel marker) and TUNEL (apoptosis). As shown in , all xenografted tumors had >95% strongly positive Ki67 expression, regardless of treatment group in HepG2 models (). In AsPC-1 models, Dex and Gem monotherapy moderately decreased the number of proliferative tumor cells. The combination treatment significantly decreased the Ki67 positive rate.
Figure 2. Immunohistochemistry examination of tumor from mouse with by the treatment of Dex, Gem and their combination. Representative expression of Ki-67, CD31 and TUNEL in HepG2 and AsPC-1 xenograft model detected by immunohistochemistry (n = 3 in each group) (×200). Bar diagram (Mean ± SEM) showing the quantitation of average number of Ki-67 and TUNEL positive cells and CD-31 positive vessels as percentage of saline treated control. n = 10; *P < .05, **P < .01.
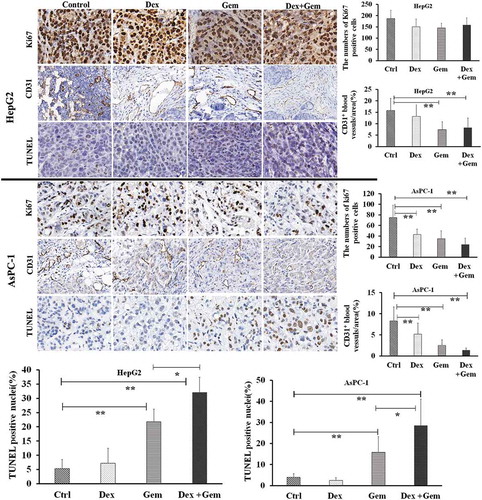
Tumor blood vessels staining revealed a marked reduction in the proportion of CD31-stained vessels in the Dex and Gem treated mouse compared to the control group. The proportion of CD31-stained vessels of the combination treatment was further reduced than those of the monotherapy mouse in AsPC-1 models ().
TUNEL assay showed Dex monotherapy did not increase in the proportion of apoptotic cells and Gem monotherapy moderately increased the numbers of TUNEL-positive cells. Dex plus Gem combination therapy, however, significantly increased the numbers of TUNEL-positive cells as compared with control group, confirming induction of apoptosis. ()
Dex sensitizes HepG2 cells and AsPC-1 cells to gem in vitro
To further verify the synergistic activity of Gem and Dex, we examined the effect of Dex, Gem and its combination on proliferation of HepG2 and AsPC-1 cells. Firstly, in the concentration of 0.1–100 μM, Dex did not affect the viability on both cells ()). Physiology concentration of Dex is about 1 μM.Citation25 Thus, in the following experiment, 1 and 10 μM Dex was used to combine with Gem to test whether Dex could potentiate the effect of Gem.
Figure 3. Effect of Dex on Gem-induced Cytotoxicity in HepG2 and AsPC-1 cells. (a) Cytotoxicity of Dex to HepG2 and AsPC-1 cells determined by CCK-8 assay. (b) Growth inhibition of Gem, with or without Dex to HepG2 and AsPC-1cells, determined by the CCK-8 assay. (c) CDI values were calculated as the quantitative measure of the degree of interaction between Gem and Dex in HepG2 and AsPC-1cells. The cell were treated with Gem (4 concentrations) or Dex (2 concentrations) alone or combined (8 combined groups) for 48 h. The values represent Mean ± SEM for at least three independent populations measured in triplicates.
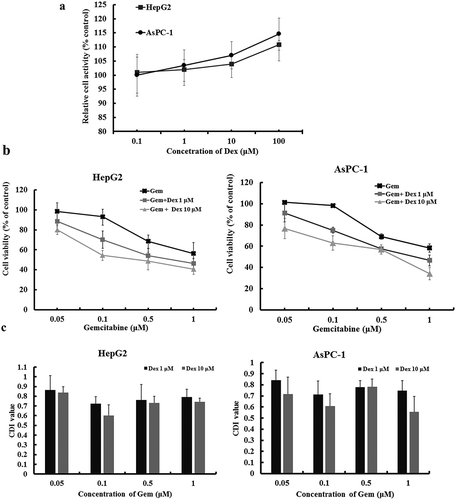
Treatments of HepG2 and AsPC-1 cells with variable concentrations of Gem for 48 h resulted in dose-dependent growth arrest in both cell lines with IC 50 of 0.91 μM and 0.92 μM, respectively ()). When Dex was added with Gem, Dex significantly enhanced Gem-induced growth arrest determined by CCK-8 assays (18.7% to 52.7% decrease of IC 50 in AsPC-1cells and 26.1% to 56.5 decrease of IC 50 in HepG2 cells). The combined effects of Dex and Gem were clearly synergistic as determined by the CDI value. Combination of 10 μM Dex plus 0.1 μM Gem in HepG2 cells and 10 μM Dex plus 1 μM Gem in AsPC-1 cells showed significantly synergism ()).
Gem and dex cooperatively induced apoptosis through caspase pathway
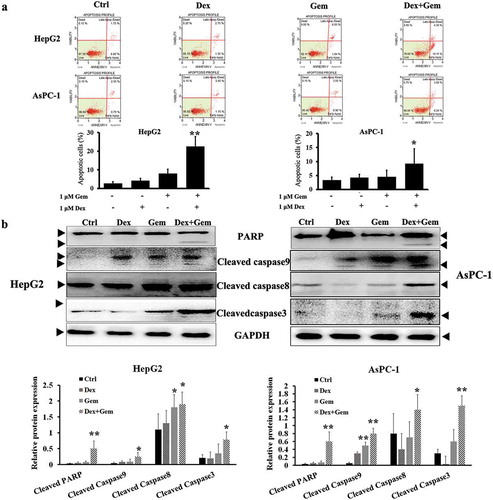
Experiments were then undertaken to determine if the synergistic antitumor effects of Gem and Dex were due to induction of apoptosis. The activated caspase-dependent mitochondrial apoptosis pathways were checked in HepG2 and AsPC-1 cell lines administrated with Gem and Dex for 48 h.
Apoptosis analysis was performed as described in Methods part. Results are shown as mean percent of annexin V+ cells of triplicates from one representative experiment. Although 1 μM Dex or Gem alone induced less apoptosis, Dex potently enhanced Gem-induced apoptosis in HepG2 cells ()). This was accompanied by cooperative cleavage/activation of PARP, caspases 3, 8 and 9 ()). Similar results were also obtained in AsPC-1 cells, though to less extent. Western blot showed the levels of apoptosis-associated proteins including PARP, cleaved caspase-3, cleaved caspase-8 and cleaved caspase-9 in HepG2 and AsPC-1 cells. Cleavage of caspases 9 and 3 was detected by immunoblotting analysis ()).
Figure 4. Apoptosis induction by Dex combined with Gem in HepG2 and AsPC-1 cells. (a) Flow cytometry analysis of apoptosis in HepG2 and AsPC-1 cells. The cells treated with Dex, Gem, and Dex plus Gem, respectively. Cells were harvested, stained with annexin V-FITC/propidium iodide and subjected to flow cytometric analysis. The percentage of annexin V-FITC positive cells were determined by calculating the ratio to the total number of cells. Each bar represents the Mean ± SEM of at least three independent experiments. (b) Western blot showed the levels of apoptosis-associated proteins including PARP, cleaved caspase-3, cleaved caspase-8 and cleaved caspase-9 in HepG2 and AsPC-1 cells. Cells were treated with drugs for 48 h, then caspase-3, 8, 9 and PARP was detected. Bar diagram (Mean ± SEM) showing the fold change in the intensity of cleaved caspase and PARP with respect to GAPDH and normalized to control group. n = 3; *P < .05, **P < .01 vs. Control.
Gem and dex cooperatively downregulated GR expression in vitro and in vivo
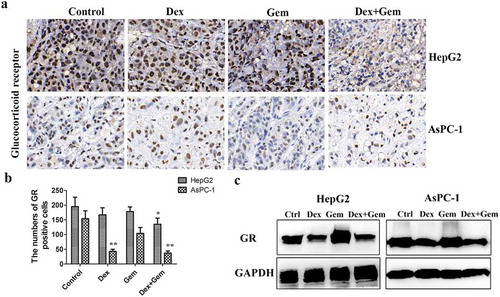
To determine whether in vivo administration of Dex or Gem alters the level of GR expression, the tumor from both HepG2 and AsPC-1 xenograft models after treatment was detected the GR expression by immunohistochemical (,b)). Dex slightly inhibited the expression of GR in HepG2 xenograft and significantly inhibited the expression of GR in AsPC-1 xenograft, whereas the expression of GR was not effected in Gem treatment group in both xenografts. The downregulation of GR expression was also verified in the combination groups. To further confirm these results and we examined the GR expression in cultured cell lines. Both HepG2 and AsPC-1 cells expressed high levels of GR protein. GR protein expression was downregulated by Dex and the combination the Dex and Gem, but not affected by Gem in both of cell lines ()).
Figure 5. The effect of Dex, Gem and their combination on the expression of GR in cancer cells and tumor. (a) GR expression in tumor mass was analysis by immunohistochemistry staining. Dex at 2 mg/kg, Gem at 100 mg/kg. (b). Bar diagram (Mean ± SEM) showing the quantitation of average number of GR positive cells. n = 5; *P < .05, **P < .01 vs. Control. (c). GR expression in cancer cells detected by western blot analysis. The cells were treated by Dex (1 μM), Gem (1 μM) or the combination for 48 h. The equally amount of whole cell lysate was loading. GAPDH was used as a loading control. GR, glucocorticoid receptor.
The synergistic effect of gem and dex is dependent on GR expression
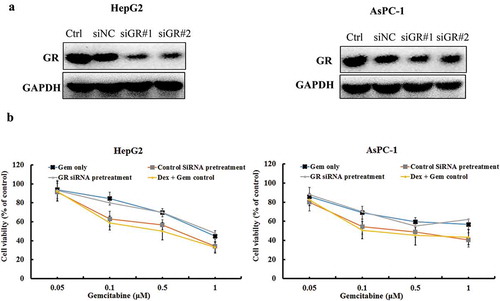
We next investigated the effects of GR blockage on the synergistic effect of Gem and Dex using GR short interfering RNA (GR siRNA). Transfection of GR siRNA significantly downregulated GR expression in both HepG2 and AsPC-1 cells ()). Both of cells were pretreated either control siRNA or GR siRNA for 48 h, then the combination of Dex and Gem was added. The inhibitory proliferative capabilities of combined Dex and Gem was evaluated by CCK-8 assays. In both HepG2 and AsPC-1 cells, compare to the Gem alone group, the combination group was shown enhanced inhibitory activity which shown the synergistic effect. In the control siRNA pretreated group, the cell proliferation was not changed much after administrating the combination of Dex and Gem compared to un-pretreated group. In the GR siRNA pretreated group, the enhanced proliferation inhibition effect of Dex to Gem was dismissed by the transfection of GR siRNA ()). In this condition, the synergistic effect of the combination could not be produced. This indicated that downregulation of endogenous GR may inhibit the synergistic effect of Dex and Gem in both HepG2 and AsPC-1 cells.
Figure 6. The effect of Dex, Gem and their combination on GR expression and proliferation of HepG2 and AsPC-1 cells using GR siRNA. (a) The cells were transfected with control siRNA and GR siRNA for 48 h. GR expression was measured in both cells by western blot analysis. Equally amount of whole cell lysate was loading. GAPDH was used as a loading control. (b) The cells were transfected with Control siRNA and GR siRNA #1 for 48 h. The cells was then treated with Gem, and Dex (1 μM) plus Gem, respectively. The viability rate of cells were detected by CCK-8 assay. The values represent Mean ± SEM for at least three independent populations measured in triplicates.
The combination of dex and gem showed enhanced inhibition expression of Nrf2 and Bcl2
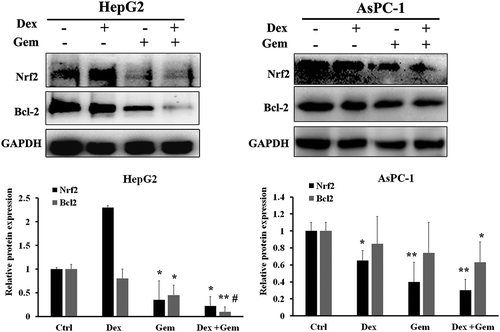
We further investigated whether the combination of Dex and Gem affect the downstream molecular of GR signaling. We examined the downstream signal transduction pathways associated with GR expression in both HepG2 and AsPC-1 cells by immunoblot analysis. Nrf2 and Bcl2 were detected after treatment with Dex, Gem or its combination for 48 h. As shown in , Dex had no effect on the Nrf2 expression in HepG2. The Nrf2 was remarkably suppressed by Gem or Gem plus Dex. Bcl2 was slightly decreased by Dex and further suppressed by Gem. The combination of Dex and Gem remarkably suppressed the Bcl2 in HepG2 cells. Similar results were also shown in AsPC-1 cells. This results indicate Dex enhances inhibition expression of Nrf2 and Bcl2 induced by Gem.
Figure 7. The effect of Dex, Gem and their combination on the expression of Nrf2 and Bcl2 in HepG2 and AsPC-1 cells. Western blot analysis of Nrf2 and Bcl2 after treatment with combination of Dex and Gem in both cells for 48 h. Representative protein bands for Nrf2 and Bcl2 expression levels were decreased at 48 h after treatment. GAPDH was used as a loading control. Bar diagram (Mean ± SEM) showing the fold change in the intensity of proteins with respect to GAPDH and normalized to control group. n = 3; *, P < .05, **, P < .01 vs control, #, P < .05 vs Gem, respectively.
Discussion
Both hepatocellular carcinoma and pancreatic ductal adenocarcinoma are most lethal cancers with the extremely poor prognosis. Although first-line treatment of both diseases can include chemotherapy with Gem, long-term survival rates remain limited.Citation26,Citation27 We hypothesized that the addition of Dex to Gem would further increase cancer cell cytotoxicity, either synergistically or in an additive manner. In this paper, we investigated whether combining Dex could enhance the efficacy of Gem in the treatment of liver and pancreatic cancers. We found the combination therapy of Dex and Gem were highly effective against human hematoma carcinoma HepG2 xenografts and pancreatic carcinoma xenografts AsPC-1 in athymic mice (). The therapeutic efficacy was more potent than that of Dex or Gem alone and shown synergistic effect ( and ). We found that this combination showed a synergistic antiproliferative activity, apoptosis induction and inhibition of angiogenesis (). Conclusively, the research demonstrates that Dex and Gem combination therapy are more effective than gemcitabine alone.
However, there are several opposite conclusions about the Dex and Gem combination. For example, some studies reported glucocorticoids including Dex may contribute to the chemotherapy resistance.Citation28,Citation29 Dex induced cisplatin and Gem resistance toward chemotherapy in lung and breast cancer cells.Citation30 The underlying mechanisms are largely unknown and may be contributed to the glucocorticoids activated GR pathways.Citation31 A recent study reported that Dex mediated progression by membrane effects and binding to GR.Citation32 The pancreatic cancer spheres was treated with Dex for 48 h in vitro, then transplanted into mice. The larger tumor sizes were observed in Dex treatment groups compare to the control which indicated Dex mediated tumor progression. This results is very impressive and also indicated the careful weighing of estimating the glucocorticoids application.
In our study, we considered the effectiveness and less toxicity when combination Dex with Gem. In the tested two cancer cell lines, HepG2 and AsPC-1 cells. We found that the combination therapy with Dex and Gem (100 mg/kg) showed eminently synergistic effect on tumor growth inhibition of xenograft models. Several recent report our observation of effective tumor growth inhibition with the combination of Dex and Gem in ovarian cancer and breast cancer xenograft models.Citation21,Citation22
We observed that Dex downregulated the GR expression in both of HepG2 and AsPC-1 cells, which means, in this condition, Dex did not activate the GR pathways. GR plays an important role in the regulation of numerous metabolic and proliferative processes, including tumorigenesis through interaction with specific response elements in the target genes.Citation33–Citation35 Glucocorticoids can stimulate directly the mitochondrial transcription through glucocorticoid receptors.Citation36 GR activation by Dex, inhibits taxane-induced tumor cell death in ovarian, breast, cervical and lung cancer cell lines and in xenograft models.Citation37 In this study, we found that the Dex and the combination with Gem downregulated the GR expression () and further inhibited the GR signaling molecular Nrf2 and Bcl-2 ().
Glucocorticoid (GC) modulates Nrf2 activity through GR-Nrf2 interaction.Citation38 Glucocorticoid receptor (GR) signaling events modify activation or apoptotic pathways. Therefore, in the present study, we intended to examine the apoptosis process. Furthermore, we investigated GR signaling and the potential relationship between the GR and Nrf2 and Bcl-2 proteins. The transcription factor Nrf2 is a key regulator of multiple target genes involved in cellular responses to oxidative, electrophilic stress, protein degradation and detoxification. Recently, the potential of Nrf2 to induce Bcl-2 protein has been demonstrated to be associated with decreased apoptosis, cancer cell survival, and drug resistance. Nrf2 overexpression in bladder cancer was associated with cisplatin chemotherapy resistance.Citation39 The chemo-resistance in ovarian, cervical, and lung cancer was derived by the elevated expression and activity of Nrf2.Citation40 Some studies reported the knockout of Nrf2 increased chemosensitivity in cancer models.Citation41,Citation42 On the other hand, Nrf2 involved in cancer cells apoptosis process. Upregulation of Bcl-2 by Nrf2 prevents apoptosis that leads to increased drug resistance.Citation43 The gene silencing of Nrf2 with siRNA induced cells apoptosis, which was accompanied by downregulation of the antiapoptotic protein Bcl-2 and upregulation of the proapoptotic protein Bax.Citation44 Here, we found Nrf2 expression was inhibited by Gem, but not effected by Dex. Although some study reported the Gem increased the Nrf2 expression,Citation45 we found the contrary result in our study. One of the explanations may be the difference like the tested cells and lower concentration of Gem between different studies. According to our results, GR was inhibited by Dex, but not effected by Gem; and Nrf2, as one of the direct interactions proteins with GR, was inhibited by Gem, but not effected by Dex, which exerted dual inhibition to downstream signaling pathways and produced the synergistic effect in the combination of Dex and Gem.
This study focuses on GC-induced mitochondrial apoptotic pathway. The mitochondrial, or intrinsic, apoptotic pathway is regulated by pro-and anti-apoptotic members of the Bcl-2 protein family at the level of the mitochondria.Citation46 Activation of procaspases plays an important role in the classical apoptosis pathway. Caspase-3 can also be activated by caspase-8 after the initiation of the extrinsic apoptotic pathway.Citation47,Citation48 Of these, the cleavage of caspase-3 and its substrate PARP serve as a biochemical hallmark of apoptosis. The synergic effect of Dex and Gem was acquired by GR. As the downstream key molecular, the combination of Dex and Gem inhibit the expression of Nrf2 and Bcl2. We also found the combination of Dex and Gem induced the cleavage of caspase-3, 8, 9 and PARP in both tested cancer cells.
In the present study, there was no anti-proliferation effect of Dex alone in HepG2 and AsPC-1 cells despite of the higher expression of glucocorticoids receptor. Although Dex against HepG2 and AsPC-1 cancer cells in vitro showed no anticancer effect when it was used as monotherapy, Dex exerted substantial direct anticancer effect in vivo. This indicated Dex exert activity was highly dependent on the microenvironment in vivo.
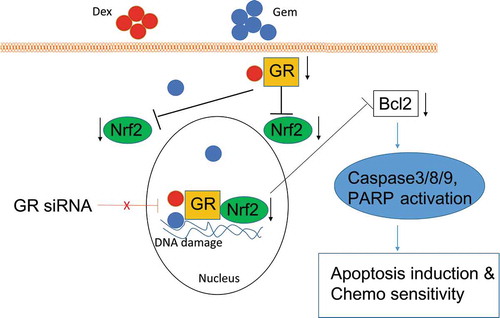
Conclusively, schematic model of our study is shown in . In summary, combination of Dex and Gem produces a synergistic effect, which could be diminished by GR siRNA. This results illuminate the expression of GR is primary to the synergistic effect of combination of Dex and Gem in our tested cancer cell lines. The downstream target moleculars Nrf2 and Bcl2 of GR signaling pathways are inhibited by the combination of Dex and Gem, which play an essential role in regulating apoptosis via induction of caspases cascade, and finally lead to a significant decrease in angiogenesis and inhibition of cancer cell proliferation. This study provides unique insights into synergistic effect of the combination Dex and Gem though the GR signaling.
Figure 8. A model for synergistic effect of Dex and Gem on tumor cells. The synergistic effect of Dex and Gem was diminished by GR siRNA. The downstream target molecular Nrf2 of GR signaling pathways was inhibited by the combination of Dex and Gem, which play an essential role in regulating apoptosis via induction of caspases cascade leading to a significant decrease in angiogenesis and inhibition of cancer cell proliferation.
Disclosure of Potential Conflicts of Interest
The authors declare that they have no conflicts of interest concerning this article.
Additional information
Funding
References
- Petrioli R, Roviello G, Fiaschi AI, Laera L, Marrelli D, Roviello F and Francini E. Gemcitabine, oxaliplatin, and capecitabine (GEMOXEL) compared with gemcitabine alone in metastatic pancreatic cancer: a randomized phase II study. Cancer Chemother Pharmacol. 2015;75:683–690. doi:10.1007/s00280-015-2683-1.
- O’Shaughnessy J, Schwartzberg L, Danso MA, Miller KD, Rugo HS, Neubauer M, Robert N, Hellerstedt B, Saleh M, Richards P, et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2014;32:3840–3847. doi:10.1200/JCO.2014.55.2984.
- Morschhauser F, Depil S, Jourdan E, Wetterwald M, Bouabdallah R, Marit G, Solal-Celigny P, Sebban C, Coiffier B, Chouaki N, et al. Phase II study of gemcitabine–dexamethasone with or without cisplatin in relapsed or refractory mantle cell lymphoma. Ann Oncol. 2007;18:370–375. doi:10.1093/annonc/mdl395.
- Sandler AB, Nemunaitis J, Denham C, von Pawel J, Cormier Y, Gatzemeier U, Mattson K, Manegold C, Palmer MC, Gregor A, et al. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2000;18:122–130. doi:10.1200/JCO.2000.18.1.122.
- von der Maase H, Hansen SW, Roberts JT, Dogliotti L, Oliver T, Moore MJ, Bodrogi I, Albers P, Knuth A, Lippert CM, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol. 2000;18:3068–3077. doi:10.1200/JCO.2000.18.17.3068.
- Pfisterer J, Plante M, Vergote I, Du Bois A, Hirte H, Lacave AJ, Wagner U, Stähle A, Stuart G, Kimmig R, et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol. 2006;24:4699–4707. doi:10.1200/JCO.2006.06.0913.
- de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014;741:8–16. doi:10.1016/j.ejphar.2014.07.041.
- Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(Suppl 5):v7–12. doi:10.1093/annonc/mdj941.
- Wang H, Li M, Rinehart JJ, Zhang R. Dexamethasone as a chemoprotectant in cancer chemotherapy: hematoprotective effects and altered pharmacokinetics and tissue distribution of carboplatin and gemcitabine. Cancer Chemother Pharmacol. 2004;53:459–467. doi:10.1007/s00280-003-0759-9.
- Wang H, Wang Y, Rayburn ER, Hill DL, Rinehart JJ, Zhang R. Dexamethasone as a chemosensitizer for breast cancer chemotherapy: potentiation of the antitumor activity of adriamycin, modulation of cytokine expression, and pharmacokinetics. Int J Oncol. 2007;30:947–953.
- Sun N, Ji H, Wang W, Zhu Q, Cao M, Zang Q. Inhibitory effect of dexamethasone on residual Lewis lung cancer cells in mice following palliative surgery. Oncol Lett. 2017;13:356–362. doi:10.3892/ol.2016.5422.
- Yano A, Fujii Y, Iwai A, Kageyama Y, Kihara K. Glucocorticoids suppress tumor angiogenesis and in vivo growth of prostate cancer cells. Clin Cancer Res. 2006;12:3003–3009. doi:10.1158/1078-0432.CCR-05-2085.
- Arai Y, Nonomura N, Nakai Y, Nishimura K, Oka D, Shiba M, Nakayama M, Takayama H, Mizutani Y, Miki T, et al. The growth-inhibitory effects of dexamethasone on renal cell carcinoma in vivo and in vitro. Cancer Invest. 2008;26:35–40. doi:10.1080/07357900701638418.
- Gong H, Jarzynka MJ, Cole TJ, Lee JH, Wada T, Zhang B, Gao J, Song W-C, DeFranco DB, Cheng S-Y, et al. Glucocorticoids antagonize estrogens by glucocorticoid receptor-mediated activation of estrogen sulfotransferase. Cancer Res. 2008;68:7386–7393. doi:10.1158/0008-5472.CAN-08-1545.
- Lewis KM, Harford-Wright E, Vink R, Ghabriel MN. NK1 receptor antagonists and dexamethasone as anticancer agents in vitro and in a model of brain tumours secondary to breast cancer. Anticancer Drugs. 2013;24:344–354. doi:10.1097/CAD.0b013e32835ef440.
- Villeneuve J, Galarneau H, Beaudet MJ, Tremblay P, Chernomoretz A, Vallieres L. Reduced glioma growth following dexamethasone or anti-angiopoietin 2 treatment. Brain Pathol. 2008;18:401–414. doi:10.1111/j.1750-3639.2008.00139.x.
- Bavaresco L, Bernardi A, Braganhol E, Wink MR, Battastini AM. Dexamethasone inhibits proliferation and stimulates ecto-5ʹ-nucleotidase/CD73 activity in C6 rat glioma cell line. J Neurooncol. 2007;84:1–8. doi:10.1007/s11060-007-9342-2.
- Coimbra M, Rijcken CJ, Stigter M, Hennink WE, Storm G, Schiffelers RM. Antitumor efficacy of dexamethasone-loaded core-crosslinked polymeric micelles. J Control Release. 2012;163:361–367. doi:10.1016/j.jconrel.2012.09.014.
- Nishimura K, Nonomura N, Satoh E, Harada Y, Nakayama M, Tokizane T, Fukui T, Ono Y, Inoue H, Shin M, et al. Potential mechanism for the effects of dexamethasone on growth of androgen-independent prostate cancer. J Natl Cancer Inst. 2001;93:1739–1746. doi:10.1093/jnci/93.22.1739.
- Nishimura K, Nonomura N, Yasunaga Y, Takaha N, Inoue H, Sugao H, Yamaguchi S, Ukimura O, Miki T, Okuyama A, et al. Low doses of oral dexamethasone for hormone-refractory prostate carcinoma. Cancer. 2000;89:2570–2576. doi:10.1002/(ISSN)1097-0142.
- Yuan Y, Zhou X, Ren Y, Zhou S, Wang L, Ji S, Hua M, Li L, Lu W, Zhou T, et al. Semi-mechanism-based pharmacokinetic/pharmacodynamic model for the combination use of dexamethasone and gemcitabine in breast cancer. J Pharm Sci. 2015;104:4399–4408. doi:10.1002/jps.24629.
- Stringer-Reasor EM, Baker GM, Skor MN, Kocherginsky M, Lengyel E, Fleming GF and Conzen S.D. Glucocorticoid receptor activation inhibits chemotherapy-induced cell death in high-grade serous ovarian carcinoma. Gynecol Oncol. 2015;138:656–662. doi:10.1016/j.ygyno.2015.06.033.
- Liu L, Xiong X, Shen M, Ru D, Gao P, Zhang X, Huang C, Sun Y, Li H, Duan Y, et al. Co-delivery of triptolide and curcumin for ovarian cancer targeting therapy via mPEG-DPPE/CaP nanoparticle. J Biomed Nanotechnol. 2018;14:1761–1772. doi:10.1166/jbn.2018.2633.
- Gong JH, Liu XJ, Li Y, Zhen YS. Pingyangmycin downregulates the expression of EGFR and enhances the effects of cetuximab on esophageal cancer cells and the xenograft in athymic mice. Cancer Chemother Pharmacol. 2012;69:1323–1332. doi:10.1007/s00280-012-1827-9.
- Spoorenberg SM, Deneer VH, Grutters JC, Pulles AE, Voorn GP, Rijkers GT, et al. Pharmacokinetics of oral vs. intravenous dexamethasone in patients hospitalized with community-acquired pneumonia. Br J Clin Pharmacol. 2014;78:78–83. doi:10.1111/bcp.2014.78.issue-1.
- Gravitz L. Liver cancer. Nature. 2014;516:S1. doi:10.1038/516S1a.
- Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi:10.1056/NEJMra0901557.
- Zhang C, Wenger T, Mattern J, Ilea S, Frey C, Gutwein P, Altevogt P, Bodenmüller W, Gassler N, Schnabel PA, et al. Clinical and mechanistic aspects of glucocorticoid-induced chemotherapy resistance in the majority of solid tumors. Cancer Biol Ther. 2007;6:278–287. doi:10.4161/cbt.6.2.3652.
- Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011;71:6360–6370. doi:10.1158/0008-5472.CAN-11-0362.
- Gassler N, Zhang C, Wenger T, PA S, Dienemann H, KM D, et al. Dexamethasone-induced cisplatin and gemcitabine resistance in lung carcinoma samples treated ex vivo. Br J Cancer. 2005;92:1084–1088. doi:10.1038/sj.bjc.6602453.
- Chen Z, Lan X, Wu D, Sunkel B, Ye Z, Huang J, Liu Z, Clinton SK, Jin VX, Wang Q, et al. Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat Commun. 2015;6:8323. doi:10.1038/ncomms9323.
- Liu L, Aleksandrowicz E, Schonsiegel F, Groner D, Bauer N, Nwaeburu CC, Zhao Z, Gladkich J, Hoppe-Tichy T, Yefenof E, et al. Dexamethasone mediates pancreatic cancer progression by glucocorticoid receptor, TGFbeta and JNK/AP-1. Cell Death Dis. 2017;8:e3064. doi:10.1038/cddis.2017.455.
- de Guia RM, Herzig S. How do glucocorticoids regulate lipid metabolism? Adv Exp Med Biol. 2015;872:127–144.
- Kim IK, Kim BS, Koh CH, Seok JW, Park JS, Shin KS, Bae E-A, Lee G-E, Jeon H, Cho J, et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein co-stimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat Med. 2015;21:1010–1017. doi:10.1038/nm.3922.
- Minchenko DO, Riabovol OO, Tsymbal DO, Ratushna OO, Minchenko OH. Inhibition of IRE1 signaling affects the expression of genes encoded glucocorticoid receptor and some related factors and their hypoxic regulation in U87 glioma cells. Endocr Regul. 2016;50:127–136. doi:10.1515/enr-2016-0014.
- Psarra AM, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta. 2011;1813:1814–1821. doi:10.1016/j.bbamcr.2011.05.014.
- Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001;276:16649–16654. doi:10.1074/jbc.M010842200.
- Alam MM, Okazaki K, Nguyen LTT, Ota N, Kitamura H, Murakami S, Shima H, Igarashi K, Sekine H, Motohashi H, et al. Glucocorticoid receptor signaling represses the antioxidant response by inhibiting histone acetylation mediated by the transcriptional activator NRF2. J Biol Chem. 2017;292:7519–7530. doi:10.1074/jbc.M116.773960.
- Hayden A, Douglas J, Sommerlad M, Andrews L, Gould K, Hussain S, et al. The Nrf2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol Oncol. 2014;32:806–814. doi:10.1016/j.urolonc.2014.02.006.
- Chen J, Solomides C, Simpkins F, Simpkins H. The role of Nrf2 and ATF2 in resistance to platinum-based chemotherapy. Cancer Chemother Pharmacol. 2017;79:369–380. doi:10.1007/s00280-016-3225-1.
- Bialk P, Wang Y, Banas K, Kmiec EB. Functional gene knockout of NRF2 increases chemosensitivity of human lung cancer A549 cells in vitro and in a xenograft mouse model. Mol Ther Oncolytics. 2018;11:75–89. doi:10.1016/j.omto.2018.10.002.
- Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–1243. doi:10.1093/carcin/bgn095.
- Niture SK, Jaiswal AK. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem. 2012;287:9873–9886. doi:10.1074/jbc.M111.312694.
- Niture SK, Jaiswal AK. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic Biol Med. 2013;57:119–131. doi:10.1016/j.freeradbiomed.2012.12.014.
- Xiang Y, Ye W, Huang C, Yu D, Chen H, Deng T, Zhang F, Lou B, Zhang J, Shi K, et al. Brusatol enhances the chemotherapy efficacy of gemcitabine in pancreatic cancer via the Nrf2 signalling pathway. Oxid Med Cell Longev. 2018;2018:2360427. doi:10.1155/2018/2360427.
- Kiraz Y, Adan A, Kartal Yandim M, Baran Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016;37:8471–8486. doi:10.1007/s13277-016-5035-9.
- Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. 2013;5:a008672–a008672. doi:10.1101/cshperspect.a008672.
- Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. doi:10.1038/sj.cdd.4402060.