
ABSTRACT
We developed a DNA aptamer, Ap52, against the shared tumor-specific MAGE-A3111-125 peptide antigen that was used to target multiple types of cancer cells. Here we report the in vivo study of mice implanted with pancreatic tumor cells AsPC-1, which demonstrates accumulation of phosphorothioate-modified Ap52 (ThioAp52) at the xenograft tumor following either intravenous or in situ injection. When complexed with antitumor drug doxorubicin (Dox), ThioAp52 achieves targeted delivery to four types of cancer cells, including breast, oral, pancreatic, and skin. Image analysis shows that ThioAp52-Dox complex selectively enters cancer cells, while free Dox is taken up by all cell lines. The cytotoxicity of ThioAp52-Dox for cancer cells is enhanced as compared to that for the corresponding normal/noncancerous cells. These results indicate that this aptamer against shared tumor-specific antigen can be a potential delivery vehicle for therapeutics to treat multiple cancers.
Introduction
Cancer therapy is the major human health issue around the world. At present, surgical removal coupled with radiation and chemotherapy remains the standard treatment. However, chemotherapeutics often associated with serious side effects due to the simultaneous damage of cancer and normal tissues.Citation1,Citation2 Thus, drug delivery in a cancer-specific manner is the main challenge for chemotherapy today. In order to reduce the unwanted effects associated with nonspecific bio-distribution of drugs, targeted delivery is being sought for enhancing cytotoxicity against tumor and reducing adverse effects on normal tissues.Citation3–5
Shared tumor-specific antigens, or often called cancer/testis antigens (CTAs), induce immune responses when expressed in tumor cells.Citation6,Citation7 Two CTAs, melanoma-associated antigen A3 (MAGE-A3) and New York esophageal squamous cell carcinoma 1 (NY-ESO-1), were ranked among the top 10 targets for cancer therapy.Citation8 More than 20 epitopes of MAGE-A3 have been identified,Citation9 and several epitopes are promiscuous and naturally processed.Citation10 These antigens are considered good candidates for targeting a large population of cancer patients.Citation11
Aptamers are single-stranded oligonucleotides selected from randomized libraries through systematic evolution of ligands by exponential enrichment (SELEX) processes.Citation12–14 A host of aptamers has been identified that recognizes the targets on various cancer cells, including prostate-specific membrane antigen (PSMA),Citation15,Citation16 transmembrane glycoprotein MUC1,Citation17,Citation18 highly expressed proteins nucleolin,Citation19,Citation20 etc. Because of high affinity and specificity to the target, aptamer-based drug delivery system has been developed to transport anti-cancer agents to tumor sites, in order to reduce nonspecific toxicity and to enhance penetration.Citation14,Citation21 Recently we developed a DNA aptamer, Ap52, against the shared tumor-specific MAGE-A3111–125 peptide antigen, and have shown its capability to target seven types of cancer cells.Citation11
Doxorubicin (Dox) is an anthracycline antineoplastic chemotherapeutic showing efficacy on a variety of cancers, including leukemia,Citation22 lymphoma,Citation23 bladder,Citation24 breast,Citation25 gastric,Citation26 ovarian,Citation27 prostate,Citation28 and many other solid tumors.Citation29,Citation30 Due to the intrinsic fluorescence of Dox and the ease of its intercalation within DNA structure, aptamer-Dox complex becomes a conveniently visible probe for targeting.Citation31–34
Here we report the in vivo targeting capacity of Ap52 in xenograft pancreatic tumor model. A phosphorothioate-modified Ap52 (ThioAp52) accumulated at the tumor site following either intravenous or in situ injection. ThioAp52-Dox complex was then generated and tested in four types of cancer and corresponding normal cells: breast, oral, pancreatic, and skin. The mechanism for the uptake of ThioAp52-Dox and of free Dox appeared to be different in cancer vs. normal cells according to image analysis and binding assay. ThioAp52-Dox complex enhanced cellular cytotoxicity in cancer cells as compared to corresponding normal/noncancerous cells. Based on these results, we conclude that this aptamer against shared tumor-specific antigen can be used as a delivery system for cancer therapy.
Results and discussion
In vivo targeting of ThioAp52
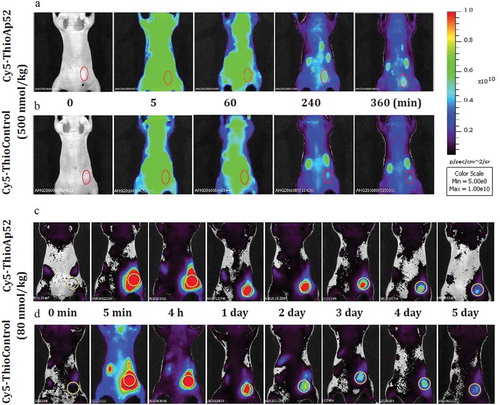
Our previous study showed targeting of Ap52, the DNA aptamer against shared tumor-specific antigen MAGE-A3111–125, to seven types of cancer cells, which was particularly effective for SK-MEL-28 melanoma cells and AsPC-1 pancreatic cancer cells.Citation11 Furthermore, phosphorothioate modifications of Ap52 enhanced the serum stability of the aptamer. To evaluate the effect of in vivo targeting and bio-distribution of ThioAp52, nude mice bearing xenograft tumor, induced with AsPC-1 cells, were administered Cy5-labeled ThioAp52 (Cy5-ThioAp52) or Control (Cy5-ThioControl) probes using intravenous injection. As shown in , after 4 to 6 h following injection, the fluorescence intensity in tumor tissue of mouse treated with Cy5-ThioAp52 remained stronger than that treated with Cy5-ThioControl probe. At 6 h after injection, the calculated tumor to background fluorescence intensity ratio in mice injected with Cy5-ThioAp52 was 1.54-fold that in mice injected with Cy5-ThioControl, and the difference is statistically significant (p< .05, Supplemental Data Figure 1). This result suggests the tumor-targeting capability of Cy5-ThioAp52 in vivo. In the literature there is evidence of intra-tumoral heterogeneity in pancreatic cancer, which is characterized by extensive stromal desmoplasia, thus decreasing blood perfusion and impeding chemotherapy delivery.Citation35,Citation36 Therefore, in situ injection of aptamer was also performed in animal model, and the results are shown in ). The obviously strong fluorescence in the tumor remained for 2–3 d following in situ injection with Cy5-ThioAp52. The ex vivo fluorescence images were also obtained for the tumor tissue and twelve other organs after 6 hours, the fluorescence diminished in tumor and most organs, except in kidney, stomach, cecum, and colon (data not shown). Notably, both intravenous and in situ injections effectively direct Cy5-ThioAp52 to the tumor site in vivo. These results demonstrate that Ap52 can be used to develop reagents for cancer diagnosis and treatment.
Figure 1. Comparison of bio-distribution of tail vein- and in situ-injected Cy5-ThioAp52 and Cy5-ThioControl probe in AsPC-1 xenograft mouse model. In vivo real-time imaging with tail vein-injected Cy5-ThioAp52 (a) or Cy5-ThioControl (b) at 0, 5, 60, 240, and 360 min post-injection, and with in situ-injected Cy5-ThioAp52 (c) or Cy5-ThioControl (d) (yellow circle at the right flank) at 0, 5 min, 4 h, 1, 2, 3, 4 and 5 d post-injection. Color scale applies to all figures. For each treatment group, results obtained with individual mice were consistent, and images from one mouse were shown
ThioAp52-Dox complex formation and binding assay
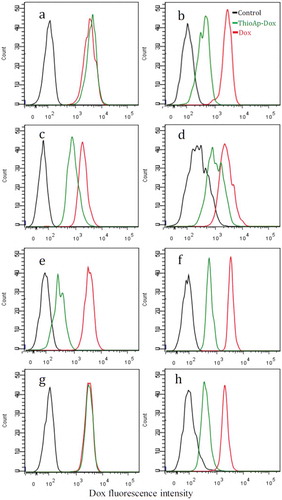
To evaluate the drug targeting capacity, ThioAp52 was then complexed with doxorubicin (Dox). The intrinsic fluorescence of Dox is quenched after binding to DNA.Citation37,Citation38 Maximal quenching of Dox fluorescence occurred at about 1:1.33 molar equivalence of Dox to ThioAp52 (Supplemental Data Figure 2). Consequently, we performed in vitro binding assays using 1:1.5 molar equivalence of Dox to ThioAp52 and treated four types of cancer cells and corresponding normal/noncancerous cells. As seen in , all cells showed similar pattern of relative Dox fluorescence profile after treatment with free Dox (red lines) or without any treatment (culture medium only, black lines). Upon treating with ThioAp52-Dox (green lines), the AsPC-1 pancreatic cancer cells ()) and SK-MEL-28 melanoma cells ()) showed nearly overlapping Dox fluorescence profile with those cells treated with free Dox (red lines), whereas the corresponding normal hTERT-HPNE pancreatic cells ()) and Hs895.Sk skin fibroblasts ()), respectively, showed intermediate fluorescence intensity, higher than that in untreated control (black lines) and yet lower than that in treatment with free Dox (red lines). This indicates that ThioAp52 effectively targeted AsPC-1 and SK-MEL-28 cells. In the other two pairs of cell lines () and )), treatments with ThioAp52-Dox resulted in intermediate Dox fluorescence intensity (green lines), similar to that in the normal pancreatic and skin cells. This may indicate that ThioAp52-Dox binds to the cell, such as the cancer cells Cal-27 ()) and MCF-7 ()) that were shown to express MAGE-A3 and could be targeted by Ap52.Citation11 Further, the low Dox intensity in MCF-7 cells (green line in )) coincides with the low level of MAGE-A3 expression in those cells.Citation11 However, it is also possible that the intermediate Dox fluorescence level is due to the diffusion of Dox released from ThioAp52-Dox complex, since only approximately 60% of ThioAp52 remained stable in serum-containing medium after 6 h (Supplemental Data Table 1). Flow cytometry analysis performed here could not distinguish the free Dox from the complex form. Taken together, these results indicate that ThioAp52-Dox is capable of binding to cancer cells. Whether it also binds to normal/noncancerous cells needs to be further evaluated.
Figure 2. Binding assay of ThioAp52-Dox complex. Four pairs of cultured cancer cells (a, c, e, g) and corresponding noncancerous cells (b, d, f, h) were assayed with flow cytometry after 6 h treatment of ThioAp52-Dox (5.0 µM, green line), Dox (5.0 µM, red line), or control (culture medium only, black line). Cell lines: a) AsPC-1, b) hTERT-HPNE, c) Cal-27, d) OMF, e) MCF-7, f) MCF-10A, g) SK-MEL-28, h) Hs895.Sk
Uptake of ThioAp52-Dox by skin cells
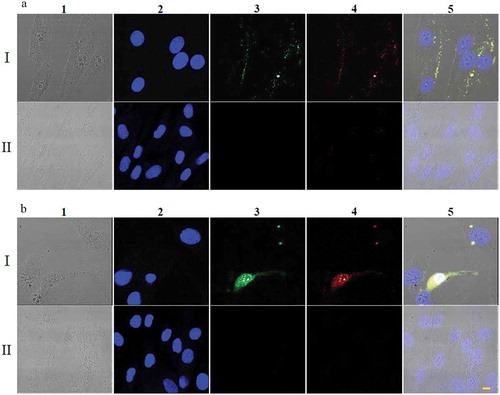
Binding and uptake of the aptamer-drug complex were further analyzed in the pair of SK-MEL-28 melanoma cells and Hs895.Sk skin fibroblasts. ThioAp52 was labeled with FITC and complexed with Dox for side-by-side observation of fluorescence signals. In SK-MEL-28, co-localization of the fluorescence from FITC (i.e., ThioAp52) and that from Dox was seen only along the cell periphery after 2 h of incubation (: A I). This is considered an evidence of specific binding due to ThioAp52 as we previously showed that a nonselective oligonucleotide, No. 45–16, obtained from SELEX failed to show affinity to MAGE-A3111–125 upon validation in SK-MEL-28 cells.Citation11 However, after 6 h of treatment, overlapping FITC-ThioAp52 and Dox fluorescence was found within the cell (: B I). In contrast, treatment of Hs895.Sk cells with FITC-ThioAp52-Dox physical complex failed to produce any fluorescence signals of FITC after either 2 h (: A II) or 6 h-incubation (: B II). These data demonstrate the targeting capacity of ThioAp52-Dox to the surface of SK-MEL-28 cells, which subsequently enters the cell. On the other hand, treatment of free Dox, after either two or 6 h, resulted in Dox signal in the nucleus in both SK-MEL-28 and Hs895.Sk cells (Supplemental Data Figure 3), suggesting that free Dox readily diffuses into both cancer and control cells, as reported in literature.Citation30,Citation34
Figure 3. Uptake of FITC-ThioAp52-Dox by skin cells. Confocal laser scanning microscopy images of SK-MEL-28 (Rows I) and Hs895.Sk cells (Rows II) treated with 2.5 µM FITC-ThioAp52-Dox for 2 h (a) or 6 h (b). Columns 1) transmittance, 2) DAPI (blue), 3) FITC (for FITC-ThioAp52) (green), 4) Dox (red), and 5) superimposed image of 1) ~4). Yellow fluorescence in 5) indicates the localization of FITC-ThioAp52-Dox. Scale bar = 10 µm applies to all figures
Targeting and internalization of ThioAp52-Dox specific to cancer cells
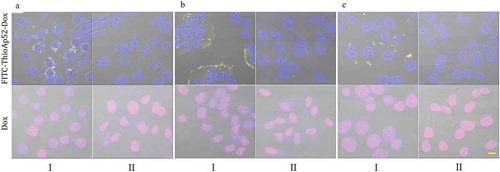
Uptake analysis was then performed in the other three pairs of cancer and corresponding normal/noncancerous cells. After two-hour treatment of free Dox, similar to what observed above, all cells, including cancer and noncancerous, contained Dox fluorescence signal in the nucleus (; see Supplemental Data Figure 4–6 for images of individual fluorescence channels). And yet in a parallel set treating with FITC-ThioAp52-Dox, co-localization of FITC and Dox signals occurred along the periphery of cancer cells (: Columns I), and not in the corresponding noncancerous cells (: Columns II). FITC-ThioAp52-Dox fluorescence signals within cells were also observed after six-hour treatment in cancer cells AsPC-1, Cal-27, and MCF-7 (Supplemental Data Figure 7: Rows A, C, E), but not in the corresponding noncancerous cells (Supplemental Data Figure 7: Rows B, D, F). These results confirm the targeting and internalization of ThioAp52-Dox specific to cancer cells. Weak Dox signal was found in the nuclei of noncancerous cells after treatment with FITC-ThioAp52-Dox for 6 h, which is barely visible in the figures shown above and seemingly lowers than the Dox signal level observed in flow cytometry analysis using ThioAp52-Dox (see ). We therefore examined the stability of FITC-ThioAp52-Dox and found that approximately 70% remained stable, which is higher than the 60% of ThioAp52-Dox, after 6 h in serum-containing culture medium (Supplemental Data Table 1). Thus, Dox signal in noncancerous cells observed here and in flow cytometry analysis could be due to the partial release of Dox from the complex with aptamer over time.
Figure 4. Targeting of FITC-ThioAp52-Dox to cancer cells. Confocal laser scanning microscopy images of three pairs of cultured cancer cells (Columns I) and corresponding normal/noncancerous cells (Columns II) after 2 h treatment of 2.5 µM FITC-ThioAp52-Dox or Dox. Cell lines: A. Pancreatic, I) AsPC-1, II) hTERT-HPNE; B. Oral, I) Cal-27, II) OMF; C. Breast, I) MCF-7, II) MCF-10A. All micrographs are merged images of transmittance, DAPI, FITC, and Dox (See Figure S3-S6 for images of individual channels). Yellow fluorescence in Columns I indicates the localization of FITC-ThioAp52-Dox complex. Scale bar = 10 µm applies to all figures
We then quantitated the uptake of FITC-ThioAp52-Dox by various cells using the fluorescence signals in confocal images. As shown in , the fluorescence levels of Dox and of FITC-ThioAp52 in cancer cells were significantly higher than those in normal/noncancerous cells (p< .0009). The FITC-ThioAp52 to Dox fluorescence ratio in each cell was close to 1 in the four types of cancer cells (AsPC-1, Cal-27, MCF-7, SK-MEL-28). This result is consistent with the observed co-localization of FITC and Dox signals. The ratio in cancer cells is also significantly different from that in the corresponding normal/noncancerous cells (p< .0001). Therefore, we conclude that FITC-ThioAp-Dox selectively targets cancer cells. Taken together, the binding and imaging data provide the proof of concept for Ap52-Dox physical complex to serve as a drug-delivery platform.
Table 1. Uptake analysis of FITC-ThioAp52-Doxa
Cytotoxicity of ThioAp52-Dox complex
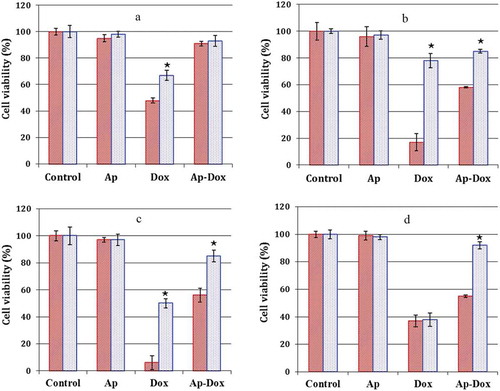
We next examined whether targeted delivery of ThioAp52-Dox to cancer cells resulted in cellular cytotoxicity. Free Dox incubation caused substantial cell death in all cell lines, albeit to varied degrees (, Dox columns). Comparatively, higher cell viability was observed with all cells treated with ThioAp52-Dox, and the effect was pronounced for most noncancerous cells (, Ap-Dox columns). Selective cytotoxicity of ThioAp52-Dox was clearly seen in MCF-7, Cal-27, and SK-MEL-28 cells (p< .004), comparing to the corresponding noncancerous cells (see the Ap-Dox columns in ), respectively). Free Dox showed nearly equipotent cytotoxicity to the Hs895.Sk and SK-MEL-28 cells (), Dox columns), whereas the effect of ThioAp52-Dox was more dramatic on SK-MEL-28 cells than that on Hs895.Sk cells (), Ap-Dox columns, p< .002). Both Ap-Dox and free Dox was relatively less toxic to pancreatic cell pairs, AsPC-1 and hTERT-HPNE ()), comparing to the other cells examined here (: B ~ D). This is consistent with the report concerning varied effects of chemotherapeutic agents on malignant pancreatic cell lines, with AsPC-1 cells having low responses to Dox.Citation39 On the other hand, cytotoxicity of free Dox on MCF-7 and Cal-27 was observed ()). This may be due to the fast growing rate of those cancer cells (data not shown). Collectively, the enhanced cytotoxicity against cancer cells and reduced adverse effects on corresponding normal/noncancerous cells suggest that Ap52-drug conjugate system can be used as a delivery vehicle for chemotherapy.
Figure 5. Cytotoxicity of ThioAp52-Dox. Cell viability assay in four pairs of cancer cells (![]()
In conclusion, we show that Ap52, an aptamer against the shared tumor-specific antigen MAGE-A3111–125, effectively targets tumor site in xenograft pancreatic mouse model, and selectively delivers Dox to multiple types of cancer cells, including breast, oral, pancreatic, and skin, causing cytotoxicity. Thus, Ap52 can be considered a promising reagent for cancer diagnosis and therapy. Since many shared tumor-specific antigens are currently known, a vast number of aptamers may be generated. In addition to developing tools for cancer diagnosis, this unique category of aptamer-drug complex may provide combinatorial manipulation of cell immunity and drug targeting for cancer therapy. Further, aptamers targeting various shared tumor-specific antigens may be used in combination to tackle tumor heterogeneity.
Materials and methods
Materials
Aptamer Ap52 (5ʹ-A⋆TCCAGAG⋆TGACGCAGCAA⋆GCACTCA⋆ATATTCC⋆CTGGACACG⋆GTGGCTTAG⋆T-3ʹ), its phosphorothioate modifications ThioAp52,Citation11 (⋆ in the above sequence designates the site of modification), Cy5-conjugates, Cy5-ThioAp52, fluorescein isothiocyanate (FITC) conjugates, FITC-ThioAp52, ThioControl probe (5ʹ-A⋆TCCAGAG⋆TGACGCAGCAG⋆TAGAATC⋆GTGGATT⋆GTGGACACG⋆GTGGCTTAG⋆T-3ʹ), and its modifications and Cy5-conjugates were custom-made at Genomics BioSci & Tech (Taipei, Taiwan).Citation11 The cell culture medium components were obtained from Invitrogen, and all other chemicals were purchased from Sigma-Aldrich, unless otherwise indicated. Cell lines AsPC-1 (pancreas adenocarcinoma, ATCC: CRL-1682), Cal-27 (tongue carcinoma, ATCC: CRL-2095), hTERT-HPNE (intermediary cells formed during acinar-to-ductal metaplasia, ATCC: CRL-4023), MCF-7 (breast adenocarcinoma, ATCC: HTB-22), MCF-10A (fibrocystic disease, ATCC: CRL-10317), normal skin cell line Hs895.Sk (fibroblast, ATCC: CRL-7636), and SK-MEL-28 (malignant melanoma, ATCC: HTB-72) were obtained directly from American Type Culture Collection where the cell lines were authenticated with STR analysis and tested negative for contamination of mycoplasma and human pathogenic viruses using DNA-based assays. Frozen stock of OMF (oral mucosal fibroblasts)Citation40 was provided by Dr. Michael Hsiao of Genomics Research Center, Academia Sinica. The alamarBlueTM Cell Viability Assay Reagent was purchased from Thermo Scientific.
Cell culture
Cal-27, MCF-7, and OMF cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), AsPC-1 cells were cultured in Roswell Park Memorial Institute medium (RPMI) and SK-MEL-28 cells were cultured in minimum essential medium. These culture mediums contained 10% FBS and 1× Penicillin-Streptomycin-Glutamine. MCF-10A cells were cultured in DMEM containing 5% horse serum, 20 ng/ml epidermal grow factor (EGF), 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, 20 µg/ml insulin and 1× Penicillin-Streptomycin-Glutamine. The Hs895.Sk cell was cultured in DMEM containing 4 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10% fetal bovine serum (FBS). The hTERT-HPNE cell was cultured in 75% DMEM without glucose and 25% medium M3 base containing 5% FBS, 10 ng/ml EGF, 5.5 mM D-glucose and 750 ng/ml puromycin. All cells were cultured in a 5% CO2-humidified chamber at 37°C to reach 90% confluence before further passage or being used in experiments, unless otherwise indicated.
Formation of aptamer-doxorubicin complexes
Aptamers were first heated at 95°C for 5 min and then cooled immediately on ice for 30 min. The aptamer was incubated at various molar ratios with doxorubicin (Dox) in a “washing buffer” (0.45% glucose, 5 mM MgCl2, in Dulbecco’s Phosphate-Buffered Saline) for 1 h in a black 96-well plate. The fluorescence spectrum of Dox was scanned by a PerkinElmer/EnSpire Multimode Plate Reader (Ex: 480 nm, Em: 500–700 nm, interval: 5 nm). Based on the quenching profile of Dox fluorescence (Supplemental Data Figure 2, also see Results and Discussion), 1:1.5 molar ratio of Dox:Aptamer was subsequently used to ensure that most Dox was incorporated into the aptamer. For confocal microscopy, washing buffer containing 75 μM FITC-labeled ThioAp52 (FITC-ThioAp52) was denatured at 95°C for 5 min and cooled on ice for 30 min. Equal volume of 50 μM Dox solution was then added, and the mixture was incubated at room temperature with shaking at 60 rpm for 1 h. The FITC-ThioAp52-Dox complex solution was diluted 10 times with culture medium before use.
Cell viability assay
In vitro cytotoxicity was assayed with resazurin sodium salt.Citation41 Briefly, 100 μl aliquots of each cell line (1x105 cells/mL) were seeded in 96-well plates (n= 4), allowed to grow for 48 hours, and then treated with 100 μl of fresh culture medium containing one of the following: 1) ThioAp52-Dox (5 μM), 2) Dox (5 μM), 3) ThioAp52 (5 μM), and further incubated for 16, 24 or 48 h. The cells were next washed with the washing buffer twice, and treated with 100 μl MTT solution (10% alamarBlueTM Cell Viability Assay reagent in culture medium). After incubating the plate for 4 h in an incubator at 37°C, 5% CO2, the fluorescence was measured with a Thermo Fluoroskan Ascent FL microplate reader (Ex: 540, Em: 590 nm). Untreated cells were used as a control with 100% viability. The relative cell viability (%) compared to control cells was calculated as (Fsample/Fcontrol) x 100%. All treatments were done in triplicates. Differences between groups of data were analyzed with Student’s t-test.
Flow cytometry
Cellular uptake of the ThioAp52-Dox complex was confirmed using flow cytometry (FACSAria IIu flow cytometer, BD Biosciences). Briefly, 5 × 104 cells were seeded onto 12-well plates (n= 4) for 48 hours, followed by incubation for 6 hours with the complex (1:1.5 molar ratio of Dox to the aptamer), with final Dox concentration at 5.0 μM. Cells were washed with the washing buffer twice, trypsinized, centrifuged at 1200 rpm for 5 minutes, and re-suspended in the washing buffer. The flow cytometry analysis was performed in triplicate for each cell line, and the results were consistent. The untreated cells were used as controls.
Confocal microscopy
Cells were seeded onto eight-well glass slides (Millicell EZ SLIDE, Merck Millipore) and cultured for 48 hours. Adherent cells were incubated with 2.5 µM FITC-ThioAp52-Dox in a total volume of 200 µl culture medium (see above for methods of preparation) for 2, 6, 24, or 48 hr. After washing to remove unbound complex, cells were fixed with 3.7% formaldehyde, and the nuclei were counterstained with 4ʹ,6-diamidino-2-phenylindole dihydrochloride (DAPI). Images of cells were collected with DIC or fluorescence from DAPI (Ex: 405 nm, Em: 430–460 nm), FITC (Ex: 488 nm, Em: 520–550 nm), or Dox (Ex: 488 nm, Em: 570–700 nm) under a confocal microscope (TCS-SP5-MP-SMD, Leica). For quantitation of FITC-ThioAp52-Dox fluorescence signals, mean fluorescence intensity per cell was calculated from total intensity divided by sum processed pixel in a cell, using Leica Application Suite X software package. One hundred cells were measured in each sample. Differences between groups of data were analyzed with Student’s t-test.
The tumor model and in vivo imaging
A pancreatic cancer xenograft model was established using male 6-to-8-wk-old BALB/c nude mice. Human AsPC-1 (5 x 106 cells) was inoculated into the right flank of mice subcutaneously, and tumor formation was observed. Mice whose tumor size reached >5 mm in diameter were injected with Cy5-ThioAp52 or Cy5-ThioControl probe either through tail vein at an equivalent dosage of 500 nmol/kg (n = 3 for each probe), or via intra-tumoral injection at 80 nmol/kg (n = 2 for each probe). Post-probe injection, whole-body fluorescence imaging was conducted with an In Vivo Imaging System (IVIS spectrum, Caliper Life Sciences) with excitation and emission wavelengths of 640 and 680 nm, respectively. At the end of experiment, all animals were sacrificed under euthanasia with pure carbon dioxide. Following euthanasia, tumor and major organs were harvested and imaged with IVIS. Fluorescence signal of regions of interest (ROIs) from tumor (Tu) and background (BG) regions was recorded in digital format, and the fold increase in signal intensity was calculated as Tu/BG. Data were presented as the mean ± standard deviation (SD), and differences between groups were analyzed with unpaired t test. All data analysis was performed using GraphPad Prism version 6.0 software. These experiments were conducted at National Laboratory Animal Center (NLAC), National Applied Research Laboratories (an AAALAC International fully accredited facility). The animals were maintained under specific pathogen-free conditions. The animal study protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of NLAC (IACUC2015M18). All procedures and methods were conducted in accordance with the approved protocol and the relevant Guide for the Care and Use of Laboratory Animals (National Research Council, US).
Nuclease resistance assay
Nuclease resistance assay of aptamers was conducted by incubation in 10% FBS diluted with DMEM at 37°C.Citation42 Half microgram of ThioAp52 or FITC-ThioAp52 was vacuum-dried and then dissolved in 300 μl 10% FBS/DMEM (final concentration 7.5 µM). ThioAp52-Dox or FITC-ThioAp52-Dox complex was freshly prepared in the washing buffer as described above and diluted 10 times with 10% FBS/DMEM to reach a final concentration of 7.5 µM. At 0, 2, 6, 24, and 48 h during the incubation, 50 μl samples were collected and stored at – 20°C for at least 20 min. The samples were vacuum-dried and then dissolved in 20 µl gel loading buffer. Ten microliters of the mixture was separated with 18% polyacrylamide gel. The degradation of aptamer and aptamer complexes was analyzed based on gel-staining intensity using Image J software.
Disclosure of interest
No potential conflicts of interest are disclosed.
Supplemental Material
Download MS Word (8.9 MB)Acknowledgments
We thank Drs Michael Hsiao, Chia-Ning Shen, and Kuo-Ping Chiu of our Center for providing frozen stocks of some of the cell lines, and National Laboratory Animal Center, NAR Labs, Taiwan, for technical support in animal studies.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Shafei A, El-Bakly W, Sobhy A, Wagdy O, Reda A, Aboelenin O, Marzouk A, El Habak K, Mostafa R, Ali MA, et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed Pharmacother. 2017;95:1209–1218. doi:10.1016/j.biopha.2017.09.059.
- McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther. 2017;31(1):63–75. doi10.1007/s10557-016-6711-0.
- Sheikh SE, Marsh C, Dakhlallah D. Aptamer technology in the targeted delivery of doxorubicin. Theranostics Can Res. 2017;1:1. doi:10.31031/NACS.2017.01.000502.
- Gilad Y, Firer M, Gellerman G. Recent innovations in peptide based targeted drug delivery to cancer cells. Biomedicines. 2016;4(2):E11. doi10.3390/biomedicines4020011.
- Baudino TA. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol. 2015;12(1):3–20. doi10.2174/1570163812666150602144310.
- Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–836. doi10.1038/nri3084.
- Knight JC. Genomic modulators of the immune response. Trends Genet. 2013;29(2):74–83. doi10.1016/j.tig.2012.10.006.
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15(17):5323–5337. doi:10.1158/1078-0432.CCR-09-0737.
- Cancer Research Institute. Peptide database; [accessed 2016 Jul 27]. https://www.cancerresearch.org/scientists/events-and-resources/peptide-database.
- Meek DW, Marcar L. MAGE-A antigens as targets in tumour therapy. Cancer Lett. 2012;324(2):126–132. doi10.1016/j.canlet.2012.05.011.
- Wang CY, Lin BL, Chen CH. An aptamer targeting shared tumor-specific peptide antigen of MAGE-A3 in multiple cancers. Int J Cancer. 2016;138(4):918–926. doi10.1002/ijc.29826.
- Vorobyeva M, Vorobjev P, Venyaminova A. Multivalent aptamers: versatile tools for diagnostic and therapeutic applications. Molecules. 2016;21(12):E1613. doi10.3390/molecules21121613.
- Mercier MC, Dontenwill M, Choulier L. Selection of nucleic acid aptamers targeting tumor cell-surface protein biomarkers. Cancers (Basel). 2017;9(6):E69. doi10.3390/cancers9060069.
- Catuogno S, Esposito CL, de Franciscis V. Aptamer-mediated targeted delivery of therapeutics: an update. Pharmaceuticals (Basel). 2016;9(4):E69. doi10.3390/ph9040069.
- Ristau BT, O’Keefe DS, Bacich DJ. The prostate-specific membrane antigen: lessons and current clinical implications from 20 years of research. Urol Oncol. 2014;32(3):272–279. doi10.1016/j.urolonc.2013.09.003.
- Almasi F, Mousavi Gargari SL, Bitaraf F, Rasoulinejad S. Development of a single stranded DNA aptamer as a molecular probe for LNCap cells using cell-SELEX. Avicenna J Med Biotechnol. 2016;8(3):104–111. PMCID: PMC4967543.
- Hwang JY, Kim ST, Han HS, Kim K, Han JS. Optical aptamer probes of fluorescent imaging to rapid monitoring of circulating tumor cell. Sensors (Basel). 2016;16(11):E1909. doi10.3390/s16111909.
- Hu Y, Duan J, Zhan Q, Wang F, Lu X, Yang XD. Novel MUC1 aptamer selectively delivers cytotoxic agent to cancer cells in vitro. PLoS One. 2012;7(2):e31970. doi10.1371/journal.pone.0031970.
- Girvan AC, Teng Y, Casson LK, Thomas SD, Jüliger S, Ball MW, Klein JB, Pierce WM Jr, Barve SS, Bates PJ. AGRO100 inhibits activation of nuclear factor-kappaB (NF-kappaB) by forming a complex with NF-kappaB essential modulator (NEMO) and nucleolin. Mol Cancer Ther. 2006;5(7):1790–1799. doi10.1158/1535-7163.MCT-05-0361.
- Reyes-Reyes EM, Teng Y, Bates PJ. A new paradigm for aptamer therapeutic AS1411 action: uptake by macropinocytosis and its stimulation by a nucleolin-dependent mechanism. Cancer Res. 2010;70(21):8617–8629. doi10.1158/0008-5472.CAN-10-0920.
- Jiang F, Liu B, Lu J, Li F, Li D, Liang C, Dang L, Liu J, He B, Badshah SA, et al. Progress and challenges in developing aptamer-functionalized targeted drug delivery systems. Int J Mol Sci. 2015;16(10):23784–23822. doi:10.3390/ijms161023784.
- Huang J, Lyu H, Wang J, Liu B. Influence of survivin-targeted therapy on chemosensitivity in the treatment of acute myeloid leukemia. Cancer Lett. 2015;366(2):160–172. doi10.1016/j.canlet.2015.05.033.
- Miyazaki K. Treatment of diffuse large B-cell lymphoma. J Clin Exp Hematop. 2016;56(2):79–88. doi10.3960/jslrt.56.79.
- Galsky MD, Pal SK, Chowdhury S, Harshman LC, Crabb SJ, Wong YN, Yu EY, Powles T, Moshier EL, Ladoire S, et al. Comparative effectiveness of gemcitabine plus cisplatin versus methotrexate, vinblastine, doxorubicin, plus cisplatin as neoadjuvant therapy for muscle-invasive bladder cancer. Cancer. 2015;121(15):2586–2593. doi:10.1002/cncr.29387.
- Zhao M, Ding X, Shen J, Zhang XP, Ding XW, Xu B. Use of liposomal doxorubicin for adjuvant chemotherapy of breast cancer in clinical practice. J Zhejiang Univ Sci B. 2017;18(1):15–26. doi10.1631/jzus.B1600303.
- Peng Z, Wang C, Fang E, Lu X, Wang G, Tong Q, Rozhkova EA. Co-delivery of doxorubicin and SATB1 shRNA by thermosensitive magnetic cationic liposomes for gastric cancer therapy. PLoS One. 2014;9(3):e92924. doi10.1371/journal.pone.0092924.
- Staropoli N, Ciliberto D, Botta C, Fiorillo L, Grimaldi A, Lama S, Caraglia M, Salvino A, Tassone P, Tagliaferri P. Pegylated liposomal doxorubicin in the management of ovarian cancer. Cancer Biol Ther. 2014;15(6):707–720. doi10.4161/cbt.28557.
- Fan X, Wang L, Guo Y, Xiong X, Zhu L, Fang K. Inhibition of prostate cancer growth using doxorubicin assisted by ultrasound-targeted nanobubble destruction. Int J Nanomedicine. 2016;11:3585–3596. doi:10.2147/IJN.S111808.
- Brown KT, Do RK, Gonen M, Covey AM, Getrajdman GI, Sofocleous CT, Jarnagin WR, D’Angelica MI, Allen PJ, Erinjeri JP, et al. Randomized trial of hepatic artery embolization for hepatocellular carcinoma using doxorubicin-eluting microspheres compared with embolization with microspheres alone. J Clin Oncol. 2016;34(17):2046–2053. doi:10.1200/JCO.2015.64.0821.
- Melguizo C, Cabeza L, Prados J, Ortiz R, Caba O, Rama AR, ÁV D, Arias JL. Enhanced antitumoral activity of doxorubicin against lung cancer cells using biodegradable poly(butylcyanoacrylate) nanoparticles. Drug Des Devel Ther. 2015;9:6433–6444. doi:10.2147/DDDT.S92273.
- Yu G, Li H, Yang S, Wen J, Niu J, Zu Y. ssDNA aptamer specifically targets and selectively delivers cytotoxic drug doxorubicin to HepG2 cells. PLoS One. 2016;11(1):e0147674. doi10.1371/journal.pone.0147674.
- Champanhac C, Teng IT, Cansiz S, Zhang L, Wu X, Zhoa Z, Fu T, Tan W. Development of a panel of DNA aptamers with high affinity for pancreatic ductal adenocarcinoma. Sci Rep. 2015;5:16788. doi:10.1038/srep16788.
- Trinh TL, Zhu G, Xiao X, Puszyk W, Sefah K, Wu Q, Tan W, Liu C, Ray RB. A synthetic aptamer-drug adduct for targeted liver cancer therapy. PLoS One. 2015;10(11):e0136673. doi10.1371/journal.pone.0136673.
- Bagalkot V, Farokhzad OC, Langer R, Jon S. An aptamer–doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew Chem Int Ed Engl. 2006;45(48):8149–8152. doi10.1002/anie.200602251.
- Cutts SM, Nudelman A, Rephaeli A, Phillips DR. The power and potential of doxorubicin-DNA adducts. IUBMB Life. 2005;57(2):73–81. doidoi:10.1081/15216540500079093..
- Chang MC, Ho YS, Lee PH, Chan CP, Lee JJ, Hahn LJ, Wang YJ, Jeng JH. Areca nut extract and arecoline induced the cell cycle arrest but not apoptosis of cultured oral KB epithelial cells: association of glutathione, reactive oxygen species and mitochondrial membrane potential. Carcinogenesis. 2001;22(9):1527–1535. doi10.1093/carcin/22.9.1527.
- Chaires JB, Herrera JE, Waring MJ. Preferential binding of daunomycin to 5’eq \o(\s\up 6(A),\s\do 2(T))CG and 5’eq \o(\s\up 6(A),\s\do 2(T))GC sequences revealed by footprinting titration experiments. Biochemistry. 1990;29:6145–6153. doi:10.1021/bi00478a006.
- Fueger BJ, Hamilton G, Raderer M, Pangerl T, Traub T, Angelberger P, Baumgartner G, Dudczak R, Virgolini I. Effects of chemotherapeutic agents on expression of somatostatin receptors in pancreatic tumor cells. J Nucl Med. 2001;42(12):1856–1862. PMID:11752085.
- Al-Nasiry S, Geusens N, Hanssens M, Luyten C, Pijnenborg R. The use of Alamar Blue assay for quantitative analysis of viability, migration and invasion of choriocarcinoma cells. Hum Reprod. 2007;22(5):1304–1309. doi10.1093/humrep/dem011.
- Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, Friess H. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Rev Hepatol. 2012;9(8):454–467. doi10.1038/nrgastro.2012.115.
- Koay EJ, Baio FE, Ondari A, Truty MJ, Cristini V, Thomas RM, Chen R, Chatterjee D, Kang Y, Zhang J, et al. Intra-tumoral heterogeneity of gemcitabine delivery and mass transport in human pancreatic cancer. Phys Biol. 2014;11(6):065002. doi:10.1088/1478-3975/11/6/065002.
- Peng CG, Damha MJ. G-quadruplex induced stabilization by 2ʹ-deoxy-2ʹ-fluoro-D- arabinonucleic acids (2ʹF-ANA). Nucleic Acids Res. 2007;35(15):4977–4988. doi10.1093/nar/gkm520.