
ABSTRACT
The Ubiquitin-Proteasome System plays a central role in signal transduction associated with stress, in the skin in particular by the control of NF-κB pathways. Under normal conditions, the inhibitory protein IκB is phosphorylated by kinases, then ubiquitinated and ends up at the proteasome to be degraded. The present short review discusses recent progress in the inhibition of NF-κB activation by proteasome inhibitors prevents the degradation of protein IκB, which accumulates in the cytosol, and there by the activation of NF-κB. Moreover, would not only limit the expression of adhesion molecules and cytokines involved in metastatic processes, but also increase the sensitivity of cancer cells to apoptosis. Considering this fact, the activity of NF-κB is regulated by the phosphorylation and proteasome-dependent degradation of its inhibitor Iκb. In this scenario, the use of a proteasome inhibitor might be an effective strategy in the treatment of skin cancer with constitutive activation of NF-κB.
1. Introduction
The Ubiquitin-Proteasome Pathway (UPP) is the main metabolic pathway responsible for the controlled degradation of intracellular proteins in higher eukaryotes. In mammals, it is responsible for the elimination of the majority of previously ubiquitinylated cytosolic and nuclear proteins.Citation1
In 2004 a Nobel Prize was awarded to Avram Hershko, Irwin Rose, and Aaron Ciechanover for “Discovery of ubiquitin-mediated protein degradation”.Citation2 It is an essential player in the regulated degradation of intracellular proteins and directly plays a major role in maintaining and regulating a wide variety of cellular processes, such as differentiation, proliferation, cell cycle, apoptosis, gene transcription, cell signaling, antigen presentation, inflammation, and all aspects of the cellular metabolic networks including physiological or pathological conditions.Citation3,Citation4 Moreover, any deregulation of this system may cause different various pathologies, such as cancer, neurodegenerative, and many autoimmune diseases.Citation5–7 The proteasome is at the heart of this system, which is a major protease in eukaryotes.
The proteolytic core of this complex, called the 20S proteasome, has the shape of a hollow cylinder enclosing the catalytic sites in an internal cavity.Citation8
The clinical success of bortezomib, a 20S proteasome inhibitor, has demonstrated the therapeutic value of inhibiting the proteasome to fight against cancer. Indeed, this inhibitor is now used routinely against Multiple Myeloma, and the inhibition of the proteasome is tested in clinical trials against multiple other cancers.Citation9 Notably, aberrant proteasome regulation is clinically linked to a wide array of human cancers including skin cancer. Generally, skin cancer is a malignant skin tumor and one of the more severe types of cancer, if not diagnosed and treated early, it may lead to disfigurement and even death. The global incidence of skin cancer has increased significantly in recent decades.Citation10,Citation11 In fact, the cause may be multifactorial, including increased exposure to Ultraviolet (UV), inflammatory agents, environmental carcinogens, tumor promoters, family history, and other several moles.Citation12
Skin cancers are named for the cell from which it originates, and become malignant. Typically, there are three most popular types of skin cancers: basal and squamous cell carcinoma, and melanoma.Citation13 The first two types are less commonly known as non-melanoma skin cancer and each type originates from keratinocytes. Therefore, melanoma is the most type aggressive due to the malignant transformation of melanocytes.Citation14 Because of their critical roles in intracellular protein breakdown, these quantitative and functional changes can have essential pathogenic consequences, by activating of the nuclear factor kappa B (NF-κB) a crucial regulator of immune and inflammatory pathways, in cellular proliferation and differentiation, and present therefore tumor-promoting and tumor-suppressing function in different tissues and carcinogenic models. In particular in epidermal keratinocytes.Citation15–17
In addition, inflammatory dermatoses are marked by high levels of active, phosphorylated NF-κB. Hence, NF-κB is a key mediator involved in skin cancer pathogenesis. The activation of the NF-κB pathway is dependent mainly on the degradation of the proteasome, which is induced by the phosphorylation of the inhibitor of NF-κB proteins (IκB). That maintains NF-κB dimers in an inactive form in the cytosol by physical interaction with inhibitor kappa B (IκB) proteins. Therefore, proteasome inhibition interrupts cellular pathways essential for skin cancer survival, namely the deregulated apoptotic pathway involving activated nuclear factor-κB (NF- κB).Citation18,Citation19
2. Ubiquitin-proteasome pathway (UPP)
The ubiquitin-proteasome pathway degrades various cellular proteins with exquisite specificity.Citation20 To ensure proper destruction of damaged or misfolded proteins or proteins that are no longer needed, the components of this system must function in a highly coordinated manner through different steps including ubiquitination and degradation of the target protein. Ubiquitin is a 76-amino-acid polypeptide present both in the nucleus and in the cytoplasm of eukaryotic cells. Human ubiquitin and that of a yeast share 96% identity for their protein sequence.Citation11,Citation21
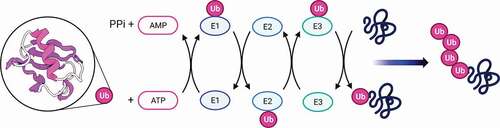
Structural analyzes have shown that it has a compact globular structure with free C-terminal end and lysine residues located on the surface of the molecule.Citation1 The conjugation of ubiquitin to the substrate requires the sequential intervention of at least three types of factors.Citation22 Ubiquitin is first activated by the activating enzyme (ubiquitin-activating enzyme) called E1 which forms a thioester bond with the targeted protein. This bond consist of group (-COOH) of the C-terminal glycine residue of ubiquitin, and its formation involves ATP.Citation23 Then, ubiquitin is transferred to the thiol group of the active cysteine of a conjugation enzyme or E2.Citation24 E2s (around 25–30 in humans) confer the first level of specificity in the recognition of substrates. Some of them can directly transfer ubiquitin to target proteins. However, in the vast majority of cases, a third component, called ubiquitin ligase or E3, is necessary and plays the main function in the recognition of substrates.Citation25,Citation26 Moreover, the conjugation reaction of ubiquitin takes place several times, the ubiquitin is conjugated to the previous ubiquitin molecules, which results in the addition to the substrate of a true poly-ubiquitin chain, which serves as a signal of degradation ().
Figure 1. Ubiquitination cascade: An E1 ubiquitin activating enzyme binds ubiquitin (Ub) that is transferred to E2 ubiquitin-conjugating enzyme. Subsequently, E3 ubiquitin ligase recruits the target protein and mediates the transition of ubiquitin to the protein
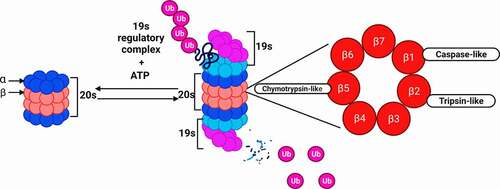
The Proteasome 26S is a large and dynamic protein complex with highly conserved structure and function in eukaryotes.Citation27 It made up of two subcomplexes: a 20S catalytic core and one or two terminal 19S regulatory particle(s) at each end of the 20S core.Citation28,Citation29 The 19S subunit binds to the polyubiquitin chain, and use ATP to unfold the protein substrate and translocate it into the 20S core particle.Citation30 Indeed, 19S subunits confer the capacity to recognize and bind polyubiquitinated substrates, which bind to each the 20S proteasome at each end. Then, the protein passes through the 20S center, where it is degraded into small oligopeptides, below 25 amino acids. Commonly, the 19S subunits overlap the 20S center.
In addition, the 20S nucleus can function on its own to cause degradation of ubiquitin-independent proteins. The core 20S is formed by 4 heptameric rings, the two alpha rings sandwich both beta rings the beta rings each contains three active sites for protein degradation: chymotrypsin-like (β5), trypsin-like (β2), and caspase-like (β1) ().
Figure 2. Proteasome structure and protein degradation. The 20S catalytic core binds to the 19S regulatory complex to form the 26S proteasome structure. Proteins that are tagged with ubiquitin bind to the 19S complex and are degraded at the proteolytic β subunits, using 3 key catalytic activities; chymotrypsin-like, caspase-like and trypsin-like
3. Proteasome inhibitors-induced apoptosis
Apoptosis is the main form of cell death, whether it is in regulating the number of cells during development or in removing damaged cells.Citation30 The ubiquitin-proteasome system is an important regulator of cell sensitivity to apoptosis. It acts as a regulator of the levels of many proteins involved in apoptosis control, including some members of the Bcl-2 family, inhibitors of apoptosis proteins and certain caspase. Many anti-cancer agents currently in use exert their anti-cancer effects through inducing apoptosis, and apoptosis resistance leads to drug resistance and tumor progression. However, there is an increasing need to find effective treatments to combat malignant tumors. Owing to the ubiquitous presence of the proteasome in cellular processes,Citation31,Citation32 and abnormally high proteasomal levels were found in several cancer types.Citation33 High proteasomal activity seems important for the survival of cancer cells, because this is very likely to comfort protect against apoptosis pathways and rid the cell of damaged proteins.Citation34 Wherefore, the proteasome is considered a prime target in the search for anti-cancer treatments, normal cells respond to proteasome inhibitors by simply stopping their cell cycle, while cancer cells tend more toward apoptosis.Citation30,Citation35
Proteasome inhibitors constitute a new clan of anti-cancer therapeutics. In preclinical cancer models, proteasome inhibitors have shown antitumor effects related to the induction of apoptosis and the sensitivity to pro-apoptotic effects of malignant cells and tumors.Citation36 The contribution of ubiquitin in apoptosis surely implied that the proteasome, the degradation machinery for proteins targeted by ubiquitination. The mechanism(s) through which these agents induce apoptosis is unclear, although specific mechanisms seem to be essential in different cells. Indeed, the inhibition of the proteasome, if severe, can interfere with the cell cycle and homeostasis of cell proteins.
Normal cells respond to proteasome inhibitors by simply stopping their cell cycle, while cancer cells tend toward apoptosis.Citation37 Additionally, endoplasmic reticulum stress and oxidative stress in cancer cells can be caused by inhibition of the function of the proteasome.Citation38 In fact, proteasome inhibitors have antitumor activity in both solid malignant and hematologic tumors. These compounds block the activation of NF-κB, resulting in apoptosis, reduced production of antigenic cytokines, and inhibition of tumor cell adhesion to stroma.Citation39
These inhibitors are divided into five groups: peptide aldehydes, peptide vinyl sulfones, peptide boronates, peptide epoxyketones, and β-lactones (lactacystin and its derivatives)Citation40 (), have been widely used as research tools to enable researchers to discover many key functions of UPP.
Table 1. Structures and principal mechanisms of action of some commonly used proteasome inhibitors
Bortezomib or PS-341 is an inhibitor of the proteasome, capable of specifically binding the proteasome and reversibly inhibiting its chymotrypsin-like activity. This compound was approved by the United States Food and Drug Administration (USFDA). It has also been approved for the treatment of multiple myeloma and mantle cell lymphoma, and has been successfully used over a decade.Citation47–49 In this context, Bortezomib has been used to inhibit the proteasome in several types of skin cancer, in a study about tumor growth retardation tests, PS-341 had antitumor activity counter to primary Lewis lung carcinoma. Moreover, PS-341 in combination with 5-fluorouracil, cisplatin, paclitaxel, and doxorubicin can cause cumulative tumor growth retardation for primary subcutaneous tumors.Citation50 Hence, PS-341 can indirectly inhibits the activation of NF-κB. PS-341 was able to induce apoptosis in four murine and two human squamous cell carcinoma cell lines and inhibits the development of murine and human squamous cell carcinoma tumors in mice.Citation51 Compared with normal hematopoietic cells, multiple myeloma cells have increased NF-κB activity. PS-341 blocked the nuclear translocation of NF-κB and demonstrated antitumor activity counter to multiple myeloma cells, chemo-resistant, and chemo-sensitive, both in vitro and in vivo multiple myeloma xenografts, on their own and in bone marrow microenvironment models.Citation52 All of these studies have involved proteasome inhibition as an effective means of treating cancer cells.
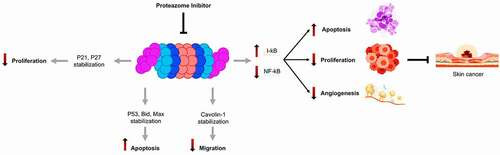
Consequently, Proteasome inhibition is therefore expected to affect survival differentially, depending on cell type and proliferation status. Proteasome inhibitors are known to induce rapid and selective cell death in transformed cells, but not normal cells, in particular by stabilizing the factors p53 and p27.Citation53,Citation54 These factors allow the arrest of the cell cycle in G1 phase, the induction of apoptosis, and the inhibition of NF-κB especially in the skin.Citation55 They thus increase apoptosis and reduction of cell proliferation and angiogenesisCitation56 ().
Figure 3. Consequences of proteasome inhibition
Finally, even though proteasomal inhibition causes proteotoxic stress with an increase in free radicals, the effect of DNA damaging agents is amplified, particularly in resistant cells.Citation57 Moreover, one study has shown that even under proteasome inhibitor-induced proteotoxic stress, that is, under substantial depletion of free ubiquitin. At the center of this unexpected phenotype is an excess of the E3 ubiquitin ligase ring finger protein 168 (RNF168), which allows more efficient utilization of the remaining free ubiquitin. Higher RNF168 levels make cells more resistant to combined treatment with ionizing radiation and proteasome inhibition, suggesting that such aberrant RNF168-mediated signaling may represent an adaptation to chronic proteotoxic and genotoxic stress for tumor cells.Citation58
4. Nuclear factor-kappa B (NF-κB) Signaling
One of the first pathways described to be affected by proteasome inhibition is the activation of the NF-κB signaling pathway.Citation59 This transcription factor is formed from two heterodimers p50 and p65. The p50 subunit comes from the degradation by the ubiquitin proteasome system of its precursor p105.Citation60 Functional nuclear NF-κB is needed for growth control during upward cell migration and differentiation of epidermal cells, which play a central role in inhibiting skin carcinogenesis.Citation61 NF-κB is found in almost all types of animal cells, under normal conditions (quiescent cells), it is maintained in the cytoplasm in inactive form, and associated with its IκB inhibitor which prevents its migration into the nucleus. Under the effect of extracellular signals (stress, radiation, cytotoxic agents), the inhibitory protein IκB is phosphorylated by a kinase, then ubiquitinylated and degraded by the proteasome. It involved several processes of cancer cells including proliferation, inflammation, development, angiogenesis, invasion, and metastasis. Through transcriptional regulation of many genes, NF-κB controls various immune and inflammatory responses. Therefore, it suppresses apoptosis, induces angiogenesis, cell proliferation, and migration, and inhibits NF-κB translocation to the nucleus for gene activation ().
Figure 4. Regulation NF-κB functions by the 26S proteasome complex
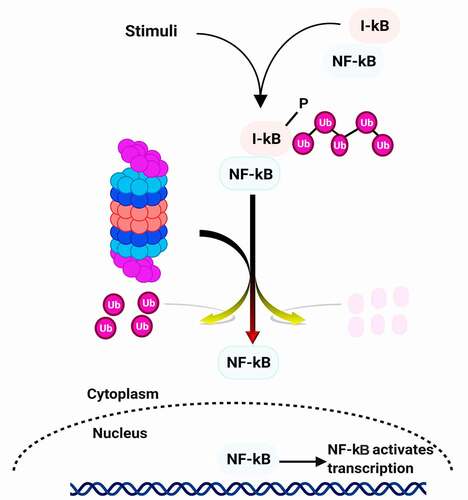
External stimuli (such as ionizing and ultraviolet irradiation, pathogens, stress, free radicals, and cytokines) induce the phosphorylation of IκB. This phosphorylation triggers polyubiquitination of IκB, which is degraded by the 26S proteasome complex. The degradation of the IκB proteasome leads to nuclear translocation of NF-κB and induction of transcription of target genes. Therefore, the 26S proteasome system is included in regulating NF-κB and related key intracellular and intercellular events. Consequently, the misregulation of NF-κB lead to various types of cancer. By the way, several cancers such as breast cancer, skin cancer, myeloma, and leukemia show the constitutive activity of NF-κB.Citation18,Citation62–64
Subsequently, the use of proteasome inhibitors can represent an effective way to treat skin cancer through constitutive activation of NF-κB.Citation65 In a number of preclinical studies, bortezomib has also been tested and showed antitumor activity against a variety of solid tumor types, including head and neck squamous cell carcinoma (HNSCC). Indeed, Bortezomib blocked NF-κB activation and proliferation, induced apoptosis in HNSCC cell lines, and inhibited development and angiogenesis in syngeneic murine SCC and human HNSCC xenograft models.Citation66–68
Since tumor cells often use NF-κB to develop resistance to anticancer drugs and radiation, inhibition of NF-κB activation appears to be a promising option to improve conventional cancer therapies efficacy.Citation69 However, cancer genomic has only recently proven useful in identifying the critical steps that drive NF-κB activation and revealing the key role of NF-κB in carcinogenesis. One difficulty that may have delayed the realization that NF-κB usually triggered in cancer is that in most cases NF-κB is activated in malignant cells not by intrinsic mutations, but in response to inflammatory microenvironmental stimuli.Citation70,Citation71 In other cases, the oncogenic mutations are located in genes encoding upstream regulators of NF-κB activity.Citation72 However, in the DMBA/TPA induced models, the inhibition of NF-κB in keratinocytes increases the risk of squamous cell carcinoma (SCC).Citation73 These results suggest that suppression of NF-κB may affect cell cycle arrest in the presence of DNA damage or oncogenic stress. Interestingly, NF-κB and JNK are two of the major signaling pathway downstream of TNFR1 that regulate each other. When NF-κB is inhibited, JNK signaling is unleashed, leading to excessive oxidative stress and DNA damage.Citation74,Citation75 This may be one of the mechanisms by which NF-κB acts as a tumor suppressor in both chemically induced skin cancer and liver cancer.
On the other hand, the NF-κB activation pathway involves the activation of NF-κB-inducing kinase (NIK), which plays a crucial role in the regulation of immunity and inflammation. In addition, NIK is critical for maintaining cellular health by controlling basic cellular processes such as differentiation, growth, and cell survival. Thus, impaired expression or regulation of NIK has been associated with various diseases. For example, NIK deficiency causes severe immunodeficiency, whereas NIK overexpression is seen in inflammatory diseases, metabolic disorders, and cancer development and progression. Abnormalities of NIK activation have been observed in a variety of solid cancers, including melanoma, which is characterized by dysregulation of several signaling and tumor suppressor/oncogene pathways, including BRAF.Citation76 Melanoma cells have higher NIK expression and activity than normal epidermal melanocytes due to AKT and extracellular signal-regulated kinase (ERK), and silencing of NIK decreases melanoma cell survival through decreased activation of the non-canonical NF-κB pathway and expression of survival-promoting genes.Citation76,Citation77
Therefore, E3 ubiquitin ligases play important in a variety of biological processes, including cell cycle progression, DNA replication, signal transduction, and development.Citation78 E3 ligases are altered in many human cancers and have therefore emerged as attractive targets for anticancer therapy. In particular, aberrant regulation of E3 ligases is clinically associated with a wide range of human cancers, including skin cancer.Citation79 Some cell line studies have been conducted to uncover possible molecular mechanisms. Some E3 ligase components are overexpressed in order to activate E3 ligases during skin carcinogenesis. As a result, E3 ligases may be appealing targets for skin cancer treatment. Indeed, MLN4924, a small molecule inhibitor of the NEDD8-activating enzyme that inhibits SKP1-CUL1-F- Box-Protein (SCF) E3 ligases by preventing Cullin-1 neddylation,Citation79,Citation80 is currently in Phase I clinical trials for the treatment of several human malignancies, including melanoma.Citation79,Citation80 Several preclinical studies have shown that MLN4924 effectively suppresses human cancer cell growth by inducing apoptosis, which has been observed primarily in leukemia and lymphoma, as well as in several solid tumor lines by accumulating IκBα to inactivate NF-κB.Citation81
Other inflammatory pathways except NF-κB, such as the transcription factors STAT1 and STAT3 appear to play opposing roles in tumorigenesis. Indeed, STAT3 promotes cell survival/proliferation, motility, and immune tolerance and is considered an oncogene, whereas STAT1 primarily elicits antiproliferative and pro-apoptotic responses and enhances antitumor immunity. Their activation is reciprocally controlled, and disruption of their balanced expression or phosphorylation levels can trigger cytokine/growth factor signals from proliferative to apoptotic or from inflammatory to anti-inflammatory.Citation82 In one study, STAT2 protein levels were reported to be higher in melanoma tissue than in normal skin tissue. Therefore, STAT2 protein levels are also related to melanoma cell proliferation, implying that STAT2 activity is important in the progression of melanoma.Citation83 Indeed, STAT3 does, play an important role in a variety of biological activities such as cell proliferation, migration, and survival. However, in studies using keratinocyte-specific Stat3-deficient mice, STAT3 has been shown to play an important role in skin homeostasis, including keratinocyte migration, wound healing, and hair follicle growth. The use of constitutive and inducible keratinocyte-specific STAT3-deficient mouse models revealed that Stat3 is required for both the initiation and promotion phases of multistep skin carcinogenesis. Other study has shown a transgenic mouse model with a “gain of function” mutant of STAT3 (STAT3 C) expressed in the basal layer of the epidermis, have revealed a new role for STAT3 the progression of skin cancer. Nevertheless, using similar STAT3-deficient and -gain-of-function mouse models have indicated a similar role in UVB radiation-mediated skin carcinogenesis.Citation84
Interferon regulatory factor 6 (IRF6) is a transcription factor that belongs to the IRF family. IRF6 regulates craniofacial development and epidermal proliferation. IRF6 has been identified as a component of a regulatory feedback loop that regulates epidermal cell proliferation. IRF6 is transcriptionally activated by p63 and induces its proteasome-mediated down-regulation, limiting keratinocyte proliferative.Citation85
5. The Ubiquitin-Proteasome pathway and inflammation in skin cancer
One of the main challenges of current cancer treatment is the intrinsic or acquired resistance of advanced tumors to antitumoral therapy.Citation86 A vast number of studies on the involvement of the proteasomal pathway in skin carcinogenesis have been performed to highlight mechanisms of proteasomal involvement skin cancer carcinogenesis.Citation87
Since the ubiquitin-proteasome pathway may be implicated in the degradation of related proteins, UPP is important in the development of skin cancer. There were various studies focused on apoptotic pathways and proteasome in cutaneous carcinoma.Citation87 Where we will focus on the three most prevalent skin types cancer, basal cell carcinoma, squamous cell carcinoma, and melanoma.
Furthermore, most pathogens produce an acute inflammatory response that results in complete clearance of the irritant in a suitable host, inadequate resolution of inflammation and an uncontrolled inflammatory response can produce chronic inflammation that predisposes the host to various diseases, including cancer. Inflammation contributes to tumorigenesis by causing DNA damage and chromosomal instability. They promote tumor development by increasing tumor cell proliferation and resistance to apoptosis. Inflammation also stimulates angiogenesis and tissue remodeling, both of which contribute to tumor cell invasion and metastasis.Citation71,Citation88,Citation89 All of these altered biochemical processes are influenced by chemical mediators of inflammation in the tumor microenvironment. Tumors are heterogeneous, complex structural entities in which cancer cells are embedded in an extracellular matrix and vascular network surrounded by a variety of innate and adaptive immune cells, and also some stromal cells.Citation88,Citation90 This diverse cellular network communicates through direct contact or through the production of cytokines and chemokines and exerts both autocrine and paracrine effects on tumor growth and progression.Citation91 In the skin, there is a clear connection between tissue destruction, inflammation, and tumorigenesis. However, the most common types of skin cancer, squamous cell carcinoma (SCC) and basal cell carcinoma (BCC), have a significant inflammatory component.Citation92 A number of clinical conditions, including discoid lupus erythematosus, dystrophic epidermolysis bullosa, and chronic wounds are associated with skin inflammation. When the skin is injured, it sets off a chain of events that includes inflammation, new tissue formation, and tissue remodeling, all of which lead to wound healing. Active inflammation, tissue destruction, and attempted repair occur simultaneously in chronic inflammation.Citation61
5.1. Skin structure and function
Skin is histologically and physiologically a complex organ,Citation55 an important barrier that protects the body from the damage of the environment. This is made of three large layers: epidermis, dermis, and hypodermis (). The epidermis is an external and continually regenerative, stratified epithelium devoid of blood or nerve supplies of approximately 5–100 μm thickness. This is made up of a variety of distinct cell types, the primary components being keratinocytes and melanocytes. Keratinocytes, which make up 95% of the epidermis and organized into four layers. The inner layer is the stratum germinativum (stratum basale, basal layer), from which columnar-shaped keratinocytes divide to migrate to the next layer. The stratum spinosum is composed of polygonal-shaped; keratinocytes are beginning to become somewhat flattened. Further differentiation of cells leads to the stratum granulosum (granular layer), which contains basophilic granules. The stratum lucidum is a thin coating of translucent cells. It is found only in the thick skin of the palms, soles, and digits. Of the feet, the outermost layer is the stratum corneum, such as the palms of the hands and the soles of the feet the outermost layer is the stratum corneum (horny layer), which contains keratin and dead cells that confer to the skin its barrier function. Melanocytes are cells of neural crest embryogenic origin whose primary function is to produce melanin.Citation61
Figure 5. Schematic presentation of skin structure, types of cells and skin cancers
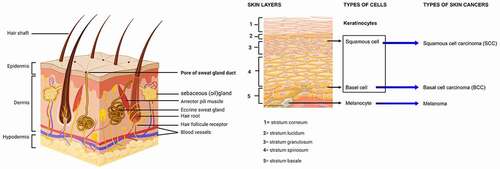
The skin has multiple functions, the most significant of which is to form a physical barrier to the environment, allowing and restricting the inward and outward passage to the exterior of water, electrolytes, and various substances, offering protection against ultraviolet rays (UVR), toxic agents, mechanical attacks, and micro-organisms. The other functions involve the sensation and synthesis of vitamin D, temperature regulation, and the protection of vitamin B folate.Citation61 The half-life of around 80% of cellular proteins is often controlled in various skin cells by the proteasome. Therefore, proteasomes are often involved in skin diseases and may be used for therapeutic approaches. However, the different proteasome inhibitors by different structures may have different chemical mechanisms, and they provide a window for therapeutic intervention.
5.2. Ubiquitin-proteasome related pathways in melanoma
Melanoma is considered one of the most aggressive skin cancer.Citation65,Citation93 It is produced by the malignant transformation of melanocytes or neural crest-derived precursors in the skinCitation94 (). Depending on the characteristics, surgical resection, chemotherapy, photodynamic therapy, immunotherapy, biochemotherapy, and targeted therapy are therapeutic options. of the tumor (location, stage, and genetic profile).Citation81,Citation95 Depending on the patient’s health, tumor stage, and location, the therapeutic strategy may include single agents or combination therapies.Citation96 The efficacy of these treatments may be compromised by the development of various resistance mechanisms. Research into the genetic profile of melanocytes and the identification of molecular factors involved in the development of malignant transformation have produced new therapeutic targets.Citation95,Citation96 The etiology of melanoma is complex and heterogeneous and includes environmental, phenotypic, and genetic risk factors. Exposure to ultraviolet radiation is the most important environmental risk factor for melanoma (UVR).Citation97 UVR causes DNA damage through the formation of pyrimidine dimers, photoproducts, gene mutations, oxidative stress, inflammation, and immunosuppression, thus promoting the carcinogenic process.Citation98 It has been widely demonstrated that UVR is involved in the development of nevus and melanoma. UVR can cause both clinical changes (increased pigmentation, scaling, and erythema) and dermoscopic changes in pigmentation (changes in the size and number of globules and dots, regression features such as bluish-gray granules, fuzzy pigment network, and increased vascularity).Citation99 The use of sunscreen may prevent some of the effects of UV radiation on nevi.Citation100 However. Some melanoma risk factors show familial aggregations independently of melanoma, suggesting the existence of specific genetic factors for these phenotypes that likely to interact with genes susceptible to melanoma. Finally, family history is also associated with an increased risk of melanoma.Citation101
Although melanoma accounts for only 1–5% of skin cancer, it caused a large majority of skin cancer deaths.Citation102 At the present time, prevention and early detection of the primary lesion remain the only means of achieving a cure. Indeed, this cancer is particularly recognized for its resistance to current therapies in its metastatic form. Therefore, there is a necessity for new combinatorial treatments, and the potentially valuable antineoplastic agents in this approach proteasome inhibition. This may be a new treatment target for melanoma.Citation103–105
Many preclinical trials in cell culture and animal models have shown the anti-tumor potential of proteasome inhibitors against melanoma.Citation87,Citation105–107 Indeed, Sorla et al.Citation87 analyzed the effects on cell proliferation and the apoptosis mechanism by application four distinct proteasome inhibitors bortezomib, acetyl-leu-norleu-al (ALLN), epoxomicin, and MG-132, in 16 human melanoma-derived cell lines. Results showed that these inhibitors induced both caspase-dependent and independent cell cycle arrest and apoptosis, whereas the apoptosis-inducing factor was a possible caspase-independent executor of cell death.Citation87 This study showed the potential efficacy of proteasome inhibitors as effective cancer therapeutic agents against melanoma and deserves more evaluation. Compared to other tumors, several melanomas showed constitutive activation of the NF-κB pathway.Citation108
Amiri et al.Citation109 focused on the NF-κB, promoting melanoma cell proliferation through inhibiting the apoptotic reactions to chemotherapy. This study has been shown that the proteasome inhibitor bortezomib in combination with the chemotherapeutic agent temozolomide inhibited the growth of melanoma cells in vitro. These data strongly suggest that bortezomib should be further studied especially in combination with chemotherapeutic agents for the treatment of melanoma.Citation109 Therefore, NF-κB is activated by phosphorylation and deactivated by the inhibitor of NF-κB, which is degraded through the ubiquitin-proteasome pathway. NF- κB activation has been directly involved in tumorigenesis of various types of cancer, including cutaneous melanoma.Citation110
Bortezomib and Gossypol (a family inhibitor of Bcl-2) treated melanoma cells have been shown to more effectively induce apoptosis in vivo.Citation111 Moreover, bortezomib and IFN-α work synergistically to overcome in pro-survival proteins Mcl-1 and Bcl-2 overexpression.Citation112 While combination therapies with bortezomib appear to be more successful in cancer treatment, there has evidence that combination strategies often are less effective.Citation32 It was found that apoptosis has involved Bcl-2 family proteins that cause depolarization of the mitochondrial membrane and caspase cascade activation.Citation113
Receptor interacting protein 1 (RIP1) is the one with integrated influence on both cell survival and cell death.Citation114 Liu et al.Citation114 found that RIP1 is often up-regulated and plays a carcinogenic role in human melanoma, while RIP1 promotes the proliferation of melanoma cells by activating NF-κB. It is worth noting that the ubiquitination of RIP1 was also significantly increased in melanoma cells, which was liable for the high expression of RIP1 and constitutive activation of NF-κB. By the RIP1 ubiquitination, the activity of NF-κB and the growth of melanoma were impeded.Citation114 On the other hand, NF-κB activation can regulate the progress of the cell cycle by means of controlling the expression of important cell cycles regulatory proteins such as cyclin D1 and cyclin-dependent kinase 2 (CDK2), and advance contributing to tumor growth. These cell cycle regulatory proteins were demonstrated to be critical in controlling the G1/S transition, by overexpressing cyclin D1 and CDK2 among others, melanoma cells escape the control mechanisms of the cell cycle and can initiate cell proliferation.Citation108
It was reported that microphthalmia-associated transcription factor (MITF) is a key transcription factor for regulating melanocyte cell lineage.Citation115,Citation116 MITF target genes are also involved in melanocyte cell survival and the progression of the cell cycle. Although MITF plays a fundamental role in melanocyte differentiation, it is also amplified and highly active in certain melanoma cells.Citation117 Regulation by pathways linked to ubiquitin, MITF is cleaved by caspases activated during TRAIL (TNF-Related Apoptosis-Inducing Ligand) treatment in human melanoma cells.Citation117 The caspases cleave the MiTF into fragments of 17 and 45 kDa. A point mutation in the caspase cleavage site decreases the number of cells undergoing apoptosis.Citation117,Citation118
It will be important to report a major driver of melanoma is induced by BRAF-(V600) mutations. due to mutations in BRAF gene are the most common mutation associated with melanoma, and among these mutations, BRAF-(V600E) has been detected in approximately 50% of melanoma patients.Citation119,Citation120 BRAF-(V600E) antagonist is now being used successfully in the clinic.Citation121 Knowledge of these genetic alterations has led to the development of personalized and targeted therapeutic strategies that block various pathways of melanoma pathogenesis. Many targeted therapeutics including vemurafenib, dabrafenib, and encorafenib have been approved by the FDA as BRAF inhibitors.Citation122 Chemotherapy is often unsuccessful because resistance to the drug develops after chemotherapy.Citation123,Citation124 Moreover, leaving the patient unresponsive to treatment. Resistance develops to vemurafenib after 2–18 months of treatment.Citation125 Indeed, BRAFIs is also approved for the treatment of advanced metastatic melanoma,Citation126 and can reversibly bind to BRAF (V600E) mutant to inhibit downstream extracellular signal-regulated kinase (ERK). However, Binding of BRAFIs in wild-type cells or cells with UV-induced mutant RAS results in heterodimerization of BRAF kinases, which activates downstream signaling and thus causes opposite effects.Citation127
5.3. Ubiquitin-Proteasome Pathways in cutaneous cell carcinoma
Cutaneous squamous cell carcinoma (cSCC) is the second most common types of skin cancer in the United States.Citation128 Cause a large number of deaths and morbidity among the general population.Citation129,Citation130 Due to the influence of UV exposures and owing to the aging population, the incidence of squamous cell carcinoma is increasing continuously.Citation131 Patients who are immunosuppressed as well face a high risk of cSCC. For example, recipients of organ transplants are as many as 150 times more susceptible to develop cSCC compared to the general population and appear to be more aggressive in these tumors.Citation129,Citation132 Many cSCCs may be effectively treated by surgery. However, there is a necessity for developed therapy for the minority of cSCCs that are responsible for this major health problem.Citation36,Citation67 Given and many medications have serious adverse effects that reduce the sustained clinical benefit. Consequently, there is still a need to develop new strategies in cSCC therapy.Citation133 Since the UPP may be implicated in the degradation of involved proteins, the UPP is important in the development of cSCC. Therefore, proteasome can be considered a promising therapeutic target.Citation47,Citation105
A recent study has shown that proteasome and ubiquitin E1 inhibitors have therapeutic potential for cSCC.Citation129 Indeed, Angela et al.Citation129 were compared the effects of three clinically approved proteasome inhibitors and the ubiquitin E1 inhibitor MLN7243. This was done in normal keratinocytes and in cSCC cell lines derived from epidermolysis bullosa, immunocompetent, and immunosuppressed patients. These agents may selectively kill cSCC cells from primary tumors as well as metastatic ones. Compared to pulses exposed to proteasome inhibitors, selectivity for cSCC is higher than normal keratinocytes.Citation129
The development of cutaneous carcinoma takes places in a multi-step process mainly including initiation, promotion and progression. Mutations and abnormalities of key cell regulators (such as RAS, p53, EGFR, and p16INK4A) are early events in tumorigenesis, were caused by various carcinogens (such as ultraviolet rays), or by inducing epigenetic changes and caused by intracellular oxidative stress.Citation134 However, the treatment of cSCC through topical therapy or intratumoral injection has great potential. Through directly delivered therapy, the dose of the inhibitor and the exposure time of the inhibitor can be controlled to achieve the best tumor selectivity.Citation129,Citation135,Citation136
In a model of spontaneous development of squamous cell carcinoma K5-IκBαDN transgenic mice, the inhibition of NF-κB results in significant inflammation associated with hyperproliferation before SCCs are development.Citation137 Regardless of the differences in SCC development in these models, their phenotype depends on tumor necrosis factor receptor 1 (TNFR1) signaling, because the blockage of TNFR1 makes the epidermis fully normal. The critical role of TNFR1 in SCC development and also has no effect on inflammation or cancer development in K5-IκBαDN, by observing the absence of IL1 signaling.Citation138,Citation139 Therefore, NF-κB blockade in keratinocytes can enhance the development of SCC by enhancing TNFR1-dependent proliferation and inducing TNFR1-dependent chronic inflammation, which may provide additional growth stimulation and genotoxicity. However, it is not clear whether the increased apoptosis caused by NF-κB block triggers inflammation or compensatory hyperplasia.
5.4. Ubiquitin-proteasome -related pathways in Basal cell carcinoma (BCC)
Basal cell carcinoma (BCC) is the most common human skin cancer, accounting for 75%, 80% of the skin cancers.Citation140 It is named this way since basal cell carcinoma cells are similar to the cells of the epithelium basal cell layer. Basal cell carcinoma mainly affects the skin of the head and neck chronically exposed to the sun in the fair-complexioned elderly.Citation141 Both intermittent acute, and long-term continuous exposure to UV are high risk factors for cutaneous basal cell carcinoma, other etiological, and known risk factors can be identified, such as lymphoma/leukemia, AIDS and other immunological disorders, radiotherapy, X-rays, arsenic poisoning, infection with the human papilloma virus, etc.Citation142 A study has been conducted in basal cell carcinoma cell lines treated with UVB. The expression of the Bcl-XL protein that participates in the protection of apoptosis was reduced by proteasome-mediated degradation in the presence of a proteasome inhibitor, MG132, before the change of mRNA level in UVB-induced apoptotic basal cell carcinoma cell lines, therefore, these results will provide critical information for developing a strategy to induce apoptosis of skin cancer cells.Citation143
5.4.1. Notch1 emerged as a tumor suppressor in BCC
Notch1 is a highly conserved transmembrane protein and plays a vital role in regulating the determination of cell fate, cell survival, cell differentiation, and oncogenic activity in vertebrates and invertebrates.Citation144 The intracellular domain of Notch1 (Notch1-IC) is degraded in the nucleus through the ubiquitin-proteasome pathway under the intervention of Fbw7 (a Notch1-IC ubiquitinated E3 ligase). After the transcriptional regulation of the target genes. However, deregulated expression of Notch receptors, ligands, and targets is observed in a growing number of solid tumors including skin cancer.Citation144 Moreover, it has been shown that Notch1 emerged as a tumor suppressor in BCC-like skin cancer.Citation145
The key question is whether this tumor suppressing function may be a peculiarity of the mouse skin system, or it may also apply to the human situation.Citation146 It is a significant question, considering the tumor-promoting function widely assigned to Notch signaling and its clinical implications.Citation147 At the transcriptional level, the expression of the Notch1 gene itself is regulated, with less pronounced effects for Notch2. Astonishingly, little is known about the mechanisms for controlling the transcription of Notch receptor genes, although this may be an important mode of regulating Notch activity. A study of the human genome showed that the promoter Notch1 was a possible binding target for p53.Citation148 Given the high frequency of p53 mutations in cutaneous BCC, an interesting possibility was that the downward modulation of Notch1 expression occurring in these tumors at least partly due to compromised p53 function. In fact, in primary human keratinocytes, the endogenous p53 binds to the Notch1 promoter and the removal of p53 in these cells results in a downward modulation of Notch1 expression.Citation149,Citation150 Inversely, increased p53 levels, through either exogenous expression or endogenous protein stabilization (through inhibition of MDM2), contribute to an up-regulation of Notch1 in regular keratinocytes and, to a greater degree, in BCC cells.Citation149
6. Conclusion and outlook
In recent years, basic research has provided a growing body of information on the extent of the ubiquitin-proteasome pathway that clearly represents an important research area in critical cellular processes, such as cell cycle progression and apoptosis regulation. Generally, ubiquitination-dependent degradation plays a significant role in the progression of skin cancer. Therefore, focusing on one pathway is not sufficient and integrative studies will be required to understand entire the network of the ubiquitin-proteasome complex signaling related to skin cancers. On the grounds that the NF-κB pathway is important for transducing developmental, inflammatory and proliferative signals, it is not astonishing that NF-κB signaling involved in skin cancer development.
The inhibition of the proteasome-by-proteasome inhibitors prevents the proteasome from activating NF-κB which results in the one hand survival, angiogenesis, and growth factors are down-regulated, and on the other hand, the apoptosis is up-regulated in multiple tumor cell lines.
These studies will provide important insights into the specificity or generality of the three major skin tumors regulated by the ubiquitin-proteasome pathway. Altogether, inhibition of the activation of NF-κB by inhibiting the proteasome could be very useful for their anti-angiogenic and anti-metastatic properties. It reduces the possibility of adverse side effects that may arise from the general inhibition of NF-κB. The identification of the mechanisms underlying stress-associated signal transduction, in the skin in particular through the control of the NF-κB pathways. Will provide new means to target NF-κB function rather than its activity. The different animal models with NF-κB alterations will be very useful in studies aimed at achieving this goal.
Table of content
1. Introduction
2. Ubiquitin-proteasome pathway (UPP)
3. Proteasome inhibitors-induced apoptosis
4. Nuclear factor-kappa B (NF-κB) Signaling
5. The Ubiquitin-Proteasome pathway and inflammation in skin cancer
5.1. Skin structure and function
5.2. Ubiquitin-proteasome related pathways in melanoma
5.3. Ubiquitin-Proteasome Pathways in cutaneous cell carcinoma.
5.4. Ubiquitin-proteasome -related pathways in Basal cell carcinoma (BCC)
5.4.1. Notch1 emerged as a tumor suppressor in BCC
6. Conclusion and outlook
List of abbreviations
Bcl-2: B-Cell Lymphoma; BTZ: Bortézomib; DNA: Deoxyribo Nucleic Acid; E1: Ubiquitin activating enzyme; E2: Ubiquitin conjugating enzyme; E3: Ubiquitin ligase enzyme; IκB: Inhibitor of Kappa B; NF-κB: Nuclear Factor-Kappa B; UV: Ultraviolet
Acknowledgments
This research has no acknowledgment.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi:https://doi.org/10.1152/physrev.00027.2001.
- Yang H, Chen X, Li K, Cheaito H, Yang Q, Wu G, Liu J, Dou QP. Repurposing old drugs as new inhibitors of the ubiquitin-proteasome pathway for cancer treatment. Semin Cancer Biol. 2021;68:105–122.
- Sulkshane P, Duek I, Ram J, Thakur A, Reis N, Ziv T, Glickman MH. Inhibition of proteasome reveals basal mitochondrial ubiquitination. J Proteomics. 2020;229:103949. doi:https://doi.org/10.1016/j.jprot.2020.103949.
- Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:12–36. doi:https://doi.org/10.2183/pjab.85.12.
- Mlynarczuk-Bialy I, Doeppner TR, Golab J, Nowis D, Wilczynski GM, Parobczak K, Wigand ME, Hajdamowicz M, Biały LP, Aniolek O, et al. Biodistribution and Efficacy Studies of the Proteasome Inhibitor BSc2118 in a Mouse Melanoma Model. Transl Oncol. 2014;7:570–579. doi:https://doi.org/10.1016/j.tranon.2014.07.002.
- Nabavi SF, Atanasov AG, Khan H, Barreca D, Trombetta D, Testai L, Sureda A, Tejada S, Vacca RA, Pittalà V, et al. Targeting ubiquitin-proteasome pathway by natural, in particular polyphenols, anticancer agents: lessons learned from clinical trials. Cancer Lett. 2018;434:101–113. doi:https://doi.org/10.1016/j.canlet.2018.07.018.
- Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. doi:https://doi.org/10.1146/annurev.pharmtox.051208.165340.
- Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009;30:191–296. doi:https://doi.org/10.1016/j.mam.2009.04.001.
- Park JE, Miller Z, Jun Y, Lee W, Kim KB. Next-generation proteasome inhibitors for cancer therapy. Transl Res. 2018;198:1–16. doi:https://doi.org/10.1016/j.trsl.2018.03.002.
- Sun Y, Zheng X, Yuan H, Chen G, Ouyang J, Liu J, Liu X, Xing X, Zhao B. Proteomic analyses reveal divergent ubiquitylation patterns in hepatocellula carcinoma cell lines with different metastasis potential. J Proteomics. 2020;225:103834. doi:https://doi.org/10.1016/j.jprot.2020.103834.
- Ward WH, Farma JM, editors. Cutaneous Melanoma: etiology and Therapy. Brisbane (AU): Codon Publications, 2017;Available from. http://www.ncbi.nlm.nih.gov/books/NBK481860/
- Priya P, Mohan Raj R, Vasanthakumar V, Raj V. Curcumin-loaded layer-by-layer folic acid and casein coated carboxymethyl cellulose/casein nanogels for treatment of skin cancer. Arab J Chem. 2020;13:694–708. doi:https://doi.org/10.1016/j.arabjc.2017.07.010.
- Hayano SM, Whipple KM, Korn BS, Kikkawa DO. Principles of Periocular Reconstruction following Excision of Cutaneous Malignancy. J Skin Cancer. 2012;2012:438502. doi:https://doi.org/10.1155/2012/438502.
- Jg E, Sp S, Gt B, Ds A. Chemoprevention of human skin cancer. Crit Rev Oncol Hematol. 2002;41:269–285. doi:https://doi.org/10.1016/S1040-8428(01)00185-8.
- Zieba BA, Henry L, Lacroix M, Jemaà M, Lavabre-Bertrand T, Meunier L, Coux O, Stoebner P-E. The proteasome maturation protein POMP increases proteasome assembly and activity in psoriatic lesional skin. J Dermatol Sci. 2017;88:10–19. doi:https://doi.org/10.1016/j.jdermsci.2017.04.009.
- Goldminz AM, Au SC, Kim N, Gottlieb AB, Lizzul PF. NF-κB: an essential transcription factor in psoriasis. J Dermatol Sci. 2013;69:89–94. doi:https://doi.org/10.1016/j.jdermsci.2012.11.002.
- Kim C, Pasparakis M. Epidermal p65/NF-κB signalling is essential for skin carcinogenesis. EMBO Mol Med. 2014;6:970–983. doi:https://doi.org/10.15252/emmm.201303541.
- Oeckinghaus A, The GS. NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi:https://doi.org/10.1101/cshperspect.a000034.
- El Yaagoubi OM, Lahmadi A, Bouyahya A, Filali H, Samaki H, El Antri S, Aboudkhil S. Antitumor Effect of Inula viscosa Extracts on DMBA-Induced Skin Carcinoma Are Mediated by Proteasome Inhibition. BioMed Res Int. 2021;2021:e6687589. doi:https://doi.org/10.1155/2021/6687589.
- Myung J, Kim KB, Crews CM. The Ubiquitin-Proteasome Pathway and Proteasome Inhibitors. Med Res Rev. 2001;21:245–273. doi:https://doi.org/10.1002/med.1009.
- Kimura A, Kurata Y, Nakabayashi J, Kagawa H, Hirano H. N-Myristoylation of the Rpt2 subunit of the yeast 26S proteasome is implicated in the subcellular compartment-specific protein quality control system. J Proteomics. 2016;130:33–41. doi:https://doi.org/10.1016/j.jprot.2015.08.021.
- Coux O, Piechaczyk M. Le système ubiquitine/protéasome : un ensemble (de) complexe(s) pour dégrader les protéines. médecine/sciences. 2000;16:623. doi:https://doi.org/10.4267/10608/1705.
- J L, G P, S J. The ubiquitin-like protein HUB1 forms SDS-resistant complexes with cellular proteins in the absence of ATP. EMBO Rep. 2003;4:1169–1174. doi:https://doi.org/10.1038/sj.embor.7400025.
- Hu Z, Li H, Wang X, Ullah K, Xu G. Proteomic approaches for the profiling of ubiquitylation events and their applications in drug discovery. J Proteomics. 2021;231:103996. doi:https://doi.org/10.1016/j.jprot.2020.103996.
- Hirano H, Kimura Y, Kimura A. Biological significance of co- and post-translational modifications of the yeast 26S proteasome. J Proteomics. 2016;134:37–46. doi:https://doi.org/10.1016/j.jprot.2015.11.016.
- Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta. 2014;1843:182–196.
- Nunes AT, Annunziata CM. Proteasome Inhibitors: structure and Function. Semin Oncol. 2017;44:377–380. doi:https://doi.org/10.1053/j.seminoncol.2018.01.004.
- Monte ERC, Rossato C, Llanos RP, Russo LC, De Castro LM, Gozzo FC, de Araujo CB, Peron JPS, Sant’Anna OA, Ferro ES, et al. Interferon-gamma activity is potentiated by an intracellular peptide derived from the human 19S ATPase regulatory subunit 4 of the proteasome. J Proteomics. 2017;151:74–82. doi:https://doi.org/10.1016/j.jprot.2016.08.003.
- Kim HM, Yu Y, Cheng Y. Structure characterization of the 26S proteasome. Biochim Biophys Acta. 2011;1809:67–79. doi:https://doi.org/10.1016/j.bbagrm.2010.08.008.
- Almond JB, Cohen GM. The proteasome: a novel target for cancer chemotherapy. Leukemia. 2002;16:433–443. doi:https://doi.org/10.1038/sj.leu.2402417.
- Lopitz-Otsoa F, Rodriguez-Suarez E, Aillet F, Casado-Vela J, Lang V, Matthiesen R, Elortza F, Rodriguez MS. Integrative analysis of the ubiquitin proteome isolated using Tandem Ubiquitin Binding Entities (TUBEs). J Proteomics. 2012;75:2998–3014. doi:https://doi.org/10.1016/j.jprot.2011.12.001.
- Shahshahan MA, Beckley MN, Jazirehi AR. Potential usage of proteasome inhibitor bortezomib (Velcade, PS-341) in the treatment of metastatic melanoma: basic and clinical aspects. Am J Cancer Res. 2011;1:913–924.
- Chen L, Madura K. Increased Proteasome Activity, Ubiquitin-Conjugating Enzymes, and eEF1A Translation Factor Detected in Breast Cancer Tissue. Cancer Res. 2005;65:5599–5606. doi:https://doi.org/10.1158/0008-5472.CAN-05-0201.
- Arlt A, Bauer I, Schafmayer C, Tepel J, Müerköster SS, Brosch M, Röder C, Kalthoff H, Hampe J, Moyer MP, et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene. 2009;28:3983–3996. doi:https://doi.org/10.1038/onc.2009.264.
- Crawford LJ, Walker B, Irvine AE. Proteasome inhibitors in cancer therapy. J Cell Commun Signal. 2011;5:101–110. doi:https://doi.org/10.1007/s12079-011-0121-7.
- Harwood CA, Proby CM, Inman GJ, Leigh IM. The Promise of Genomics and the Development of Targeted Therapies for Cutaneous Squamous Cell Carcinoma. Acta Derm Venereol. 2016;96:3–16. doi:https://doi.org/10.2340/00015555-2181.
- Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843:13–25. doi:https://doi.org/10.1016/j.bbamcr.2013.08.012.
- Yadav RK, Chae S-W, Kim H-R, Chae HJ. Endoplasmic Reticulum Stress and Cancer. J Cancer Prev. 2014;19:75–88. doi:https://doi.org/10.15430/JCP.2014.19.2.75.
- Baldi A, Pasquali P, Spugnini EP, editors. Skin Cancer: a Practical Approach. New York:Springer New York, 2014; http://link.springer.com/https://doi.org/10.1007/978-1-4614-7357-2
- Frankland-Searby S, Bhaumik SR. The 26S proteasome complex: an attractive target for cancer therapy. Biochim Biophys Acta BBA - Rev Cancer. 2012;1825:64–76. doi:https://doi.org/10.1016/j.bbcan.2011.10.003.
- Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg Med Chem Lett. 1998;8:333–338. doi:https://doi.org/10.1016/S0960-894X(98)00029-8.
- Gardner RC, Assinder SJ, Christie G, Mason GG, Markwell R, Wadsworth H, McLaughlin M, King R, Chabot-Fletcher MC, Breton JJ, et al. Characterization of peptidyl boronic acid inhibitors of mammalian 20 S and 26 S proteasomes and their inhibition of proteasomes in cultured cells. Biochem J. 2000;346:447–454. doi:https://doi.org/10.1042/bj3460447.
- Hirsch T, Dallaporta B, Zamzami N, Susin SA, Ravagnan L, Marzo I, Brenner C, Kroemer G. Proteasome Activation Occurs at an Early, Premitochondrial Step of Thymocyte Apoptosis. J Immunol. 1998;161:35–40.
- Perel G, Bliss J, Thomas CM. Carfilzomib (Kyprolis): a Novel Proteasome Inhibitor for Relapsed And/or Refractory Multiple Myeloma. Pharm Ther. 2016;41:303–307.
- Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi:https://doi.org/10.1016/S0962-8924(98)01346-4.
- Kessler BM, Tortorella D, Altun M, Kisselev AF, Fiebiger E, Hekking BG, Ploegh HL, Overkleeft HS. Extended peptide-based inhibitors efficiently target the proteasome and reveal overlapping specificities of the catalytic β-subunits. Chem Biol. 2001;8:913–929. doi:https://doi.org/10.1016/S1074-5521(01)00069-2.
- Sugiyama N, Adrian G, Schwartz S, Wennerberg J, Ekblad L. 2020. Proteasome Inhibitors Counteract the Effect of Cisplatin in HPV-Positive Squamous Cell Carcinoma in Vitro. Research Square. DOI:https://doi.org/10.21203/rs.3.rs-30654/v1
- Kisselev AF, Akopian TN, Woo KM, Goldberg AL. The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes Implications for understanding the degradative mechanism and antigen presentation. J Biol Chem. 1999;274:3363–3371. doi:https://doi.org/10.1074/jbc.274.6.3363.
- Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–360. doi:https://doi.org/10.1038/nrc1361.
- Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res Off J Am Assoc Cancer Res. 1999;5:2638–2645.
- Teicher BA, Tomaszewski JE. Proteasome inhibitors. Biochem Pharmacol. 2015;96:1–9. doi:https://doi.org/10.1016/j.bcp.2015.04.008.
- Varga C, Laubach J, Hideshima T, Chauhan D, Anderson KC, Richardson PG. Novel targeted agents in the treatment of multiple myeloma. Hematol Oncol Clin North Am. 2014;28:903–925. doi:https://doi.org/10.1016/j.hoc.2014.07.001.
- Mahmoudian M, Rahimi-Moghaddam P. The anti-cancer activity of noscapine: a review. Recent Patents Anticancer Drug Discov. 2009;4:92–97. doi:https://doi.org/10.2174/157489209787002524.
- Kudo Y, Takata T, Ogawa I, Kaneda T, Sato S, Takekoshi T, Zhao M, Miyauchi M, Nikai H. p27Kip1 accumulation by inhibition of proteasome function induces apoptosis in oral squamous cell carcinoma cells. Clin Cancer Res Off J Am Assoc Cancer Res. 2000;6:916–923.
- Brégégère F, Milner Y, Friguet B. The ubiquitin–proteasome system at the crossroads of stress-response and ageing pathways: a handle for skin care? Ageing Res Rev. 2006;5:60–90. doi:https://doi.org/10.1016/j.arr.2005.09.002.
- Milano A, Iaffaioli RV, Caponigro F. The proteasome: a worthwhile target for the treatment of solid tumours? Eur J Cancer Oxf Engl. 1990;2007(43):1125–1133.
- Sooman L, Gullbo J, Bergqvist M, Bergström S, Lennartsson J, Ekman S. Synergistic effects of combining proteasome inhibitors with chemotherapeutic drugs in lung cancer cells. BMC Res Notes. 2017;10:544. doi:https://doi.org/10.1186/s13104-017-2842-z.
- Chroma K, Mistrik M, Moudry P, Gursky J, Liptay M, Strauss R, Skrott Z, Vrtel R, Bartkova J, Kramara J, et al. Tumors overexpressing RNF168 show altered DNA repair and responses to genotoxic treatments, genomic instability and resistance to proteotoxic stress. Oncogene. 2017;36:2405–2422. doi:https://doi.org/10.1038/onc.2016.392.
- Coux O, Goldberg AL. Enzymes catalyzing ubiquitination and proteolytic processing of the p105 precursor of nuclear factor kappaB1. J Biol Chem. 1998;273:8820–8828. doi:https://doi.org/10.1074/jbc.273.15.8820.
- Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, et al. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB: p52dimers. EMBO J. 2004;23:4202–4210. doi:https://doi.org/10.1038/sj.emboj.7600391.
- Maru GB, Gandhi K, Ramchandani A, Kumar G. The role of inflammation in skin cancer. Basel: Springer; 2014. p. 437–469. doi:https://doi.org/10.1007/978-3-0348-0837-8_17.
- Ueda Y, Richmond A. NF-κB activation in melanoma. Pigment Cell Res Spons Eur Soc Pigment Cell Res Int Pigment Cell Soc. 2006;19:112–124. doi:https://doi.org/10.1111/j.1600-0749.2006.00304.x.
- Roy P, Sarkar UA, The BS. NF-κB Activating Pathways in Multiple Myeloma. Biomedicines. 2018;6:E59. doi:https://doi.org/10.3390/biomedicines6020059.
- Mansouri L, Papakonstantinou N, Ntoufa S, Stamatopoulos K, Rosenquist R. NF-κB activation in chronic lymphocytic leukemia: a point of convergence of external triggers and intrinsic lesions. Semin Cancer Biol. 2016;39:40–48. doi:https://doi.org/10.1016/j.semcancer.2016.07.005.
- Madonna G, Ullman CD, Gentilcore G, Palmieri G, Ascierto PA. NF-κB as potential target in the treatment of melanoma. J Transl Med. 2012;10:53. doi:https://doi.org/10.1186/1479-5876-10-53.
- Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl Bancroft C, Sausville E, Adams J, Elliott P, Van Waes C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2001;7:1419–1428.
- Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421. doi:https://doi.org/10.1016/S1535-6108(04)00120-5.
- Lun M, Zhang PL, Pellitteri PK, Law A, Kennedy TL, Brown RE. Nuclear factor-kappaB pathway as a therapeutic target in head and neck squamous cell carcinoma: pharmaceutical and molecular validation in human cell lines using Velcade and siRNA/NF-kappaB. Ann Clin Lab Sci. 2005;35:251–258.
- Baud V, Is KM. NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi:https://doi.org/10.1038/nrd2781.
- Ahmed F, Haass NK. Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Front Oncol. 2018;8:173. doi:https://doi.org/10.3389/fonc.2018.00173.
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi:https://doi.org/10.1038/nri1703.
- Gilmore TD. NF-κB and Human Cancer: what Have We Learned over the Past 35 Years? Biomedicines. 2021;9:889. doi:https://doi.org/10.3390/biomedicines9080889.
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi:https://doi.org/10.1038/nature01283.
- Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–542. doi:https://doi.org/10.1016/j.cell.2004.10.017.
- Kamata H, Honda S-I, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi:https://doi.org/10.1016/j.cell.2004.12.041.
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi:https://doi.org/10.1038/nature00766.
- Thu YM, Su Y, Yang J, Splittgerber R, Na S, Boyd A, Mosse C, Simons C, Richmond A. NF-κB inducing kinase (NIK) modulates melanoma tumorigenesis by regulating expression of pro-survival factors through the β-catenin pathway. Oncogene. 2012;31:2580–2592. doi:https://doi.org/10.1038/onc.2011.427.
- Dang F, Nie L, Wei W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021;28:427–438. doi:https://doi.org/10.1038/s41418-020-00648-0.
- Jia L, Sun Y, E Ubiquitin SCF. Ligases as Anticancer Targets. Curr Cancer Drug Targets. 2011;11:347–356. doi:https://doi.org/10.2174/156800911794519734.
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi:https://doi.org/10.1038/nature07884.
- Xie C-M, Wei W, Sun Y. Role of SKP1-CUL1-F-Box-Protein (SCF) E3 Ubiquitin Ligases in Skin Cancer. J Genet Genomics. 2013;40:97–106. doi:https://doi.org/10.1016/j.jgg.2013.02.001.
- Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis. JAK-STAT. 2012;1:65–72. doi:https://doi.org/10.4161/jkst.20045.
- Lee C-J, An H-J, Cho ES, Kang HC, Lee JY, Lee HS, Cho -Y-Y. Stat2 stability regulation: an intersection between immunity and carcinogenesis. Exp Mol Med. 2020;52:1526–1536. doi:https://doi.org/10.1038/s12276-020-00506-6.
- Macias E, Rao D, DiGiovanni J. Role of Stat3 in Skin Carcinogenesis: insights Gained from Relevant Mouse Models. J Skin Cancer. 2013;2013:e684050. doi:https://doi.org/10.1155/2013/684050.
- Hixon K, Rhea L, Standley J, Canady FJ, Canady JW, Dunnwald M. Interferon Regulatory Factor 6 Controls Proliferation of Keratinocytes From Children With Van der Woude Syndrome. Cleft Palate-Craniofacial J Off Publ Am Cleft Palate-Craniofacial Assoc. 2017;54:281–286. doi:https://doi.org/10.1597/15-275.
- Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi:https://doi.org/10.1038/nrc1588.
- Sorolla A, Yeramian A, Dolcet X, Santos De AMP, Llobet D, Schoenenberger JA, JM C, Soria X, Egido R, Llombart A, et al. Effect of proteasome inhibitors on proliferation and apoptosis of human cutaneous melanoma-derived cell lines. Br J Dermatol. 2008;158:496–504. doi:https://doi.org/10.1111/j.1365-2133.2007.08390.x.
- Kundu JK, Surh Y-J. Emerging avenues linking inflammation and cancer. Free Radic Biol Med. 2012;52:2013–2037.
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi:https://doi.org/10.1038/nature07205.
- Grivennikov SI, Greten FR, Immunity KM. Inflammation, and Cancer. Cell. 2010;140:883–899. doi:https://doi.org/10.1016/j.cell.2010.01.025.
- Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014;345:164–173. doi:https://doi.org/10.1016/j.canlet.2013.08.014.
- Neagu M, Constantin C, Caruntu C, Dumitru C, Surcel M, Inflammation: ZS. A key process in skin tumorigenesis. Oncol Lett. 2019;17:4068–4084.
- Wang Q, Pan F, Li S, Huang R, Wang X, Wang S, Liao X, Li D, Zhang L. The prognostic value of the proteasome activator subunit gene family in skin cutaneous melanoma. J Cancer. 2019;10:2205–2219. doi:https://doi.org/10.7150/jca.30612.
- Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev. 2012;26:1131–1155. doi:https://doi.org/10.1101/gad.191999.112.
- Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics. CA Cancer J Clin. 2016;66:271–289. doi:https://doi.org/10.3322/caac.21349.
- Merlino G, Herlyn M, Fisher DE, Bastian BC, Flaherty KT, Davies MA, Wargo JA, Curiel-Lewandrowski C, Weber MJ, Leachman SA, et al. The State of Melanoma: challenges and Opportunities. Pigment Cell Melanoma Res. 2016;29:404–416. doi:https://doi.org/10.1111/pcmr.12475.
- Olsen CM, Whiteman DC. Clinical Epidemiology of Melanoma. In: Balch CM, Atkins MB, Garbe C, Gershenwald JE, Halpern AC, Kirkwood JM, McArthur GA, Thompson JF, Sober AJ, editors. Cutaneous Melanoma. Cham: Springer International Publishing; 2020. p. 425–449. doi:https://doi.org/10.1007/978-3-030-05070-2_47.
- Prasad RR, Paudel S, Raina K, Agarwal R. Silibinin and non-melanoma skin cancers. J Tradit Complement Med. 2020;10:236–244. doi:https://doi.org/10.1016/j.jtcme.2020.02.003.
- Khan AQ, Travers JB, Kemp MG. Roles of UVA radiation and DNA damage responses in melanoma pathogenesis. Environ Mol Mutagen. 2018;59:438–460.
- Carrera C, Puig-Butillè JA, Aguilera P, Ogbah Z, Palou J, Lecha M, Malvehy J, Puig S. Impact of sunscreens on preventing UVR-induced effects in nevi: in vivo study comparing protection using a physical barrier vs sunscreen. JAMA Dermatol. 2013;149:803–813. doi:https://doi.org/10.1001/jamadermatol.2013.398.
- Simone DE, Valiante M, Silipo V. Familial melanoma and multiple primary melanoma. G Ital Dermatol Venereol. 2017;152:262–265.
- Trufant J, Jones E. Cham: Springer International Publishing; 2019. p. 171–208. doi:https://doi.org/10.1007/978-3-030-18065-2_17.
- Grazia G, Penna I, Perotti V, Anichini A, Tassi E. Towards combinatorial targeted therapy in melanoma: from pre-clinical evidence to clinical application (review). Int J Oncol. 2014;45:929–949. doi:https://doi.org/10.3892/ijo.2014.2491.
- Obrist F, Manic G, Kroemer G, Vitale I, Trial Watch: GL. Proteasomal inhibitors for anticancer therapy. Mol Cell Oncol. 2015;2:e974463. doi:https://doi.org/10.4161/23723556.2014.974463.
- Sidor-Kaczmarek J, Cichorek M, Spodnik JH, Wójcik S, Moryś J. Proteasome inhibitors against amelanotic melanoma. Cell Biol Toxicol. 2017;33:557–573. doi:https://doi.org/10.1007/s10565-017-9390-0.
- Reuland SN, Goldstein NB, Partyka KA, Smith S, Luo Y, Fujita M, Gonzalez R, Lewis K, Norris DA, Shellman YG. ABT-737 synergizes with Bortezomib to kill melanoma cells. Biol Open. 2012;1:92–100. doi:https://doi.org/10.1242/bio.2011035.
- Selimovic D, Porzig BBOW, El-Khattouti A, Badura HE, Ahmad M, Ghanjati F, Santourlidis S, Haikel Y, Hassan M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell Signal. 2013;25:308–318. doi:https://doi.org/10.1016/j.cellsig.2012.10.004.
- Amiri KI, Richmond A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev. 2005;24:301–313. doi:https://doi.org/10.1007/s10555-005-1579-7.
- Amiri KI, Horton LW, LaFleur BJ, Sosman JA, Richmond A. Augmenting chemosensitivity of malignant melanoma tumors via proteasome inhibition: implication for bortezomib (VELCADE, PS-341) as a therapeutic agent for malignant melanoma. Cancer Res. 2004;64:4912–4918. doi:https://doi.org/10.1158/0008-5472.CAN-04-0673.
- Triozzi PL, Eng C, Singh AD. Targeted therapy for uveal melanoma. Cancer Treat Rev. 2008;34:247–258. doi:https://doi.org/10.1016/j.ctrv.2007.12.002.
- Wolter KG, Verhaegen M, Fernández Y, Nikolovska-Coleska Z, Riblett M, Martin De La Vega C, Wang S, Soengas MS. Therapeutic window for melanoma treatment provided by selective effects of the proteasome on Bcl-2 proteins. Cell Death Differ. 2007;14:1605–1616. doi:https://doi.org/10.1038/sj.cdd.4402163.
- Lesinski GB, Raig ET, Guenterberg K, Brown L, Go MR, Shah NN, Lewis A, Quimper M, Hade E, Young G, et al. IFN-alpha and bortezomib overcome Bcl-2 and Mcl-1 overexpression in melanoma cells by stimulating the extrinsic pathway of apoptosis. Cancer Res. 2008;68:8351–8360. doi:https://doi.org/10.1158/0008-5472.CAN-08-0426.
- Wang C, Li S, Wang M. Evodiamine-induced human melanoma A375-S2 cell death was mediated by PI3K/Akt/caspase and Fas-L/NF-κB signaling pathways and augmented by ubiquitin–proteasome inhibition. Toxicol In Vitro. 2010;24:898–904. doi:https://doi.org/10.1016/j.tiv.2009.11.019.
- Ma J, Guo W, Li C. Ubiquitination in melanoma pathogenesis and treatment. Cancer Med. 2017;6:1362–1377. doi:https://doi.org/10.1002/cam4.1069.
- Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–414. doi:https://doi.org/10.1016/j.molmed.2006.07.008.
- Wiedemann GM, Aithal C, Kraechan A, Heise C, Cadilha BL, Zhang J, Duewell P, Ballotti R, Endres S, Bertolotto C, et al. Microphthalmia-Associated Transcription Factor (MITF) Regulates Immune Cell Migration into Melanoma. Transl Oncol. 2018;12:350–360. doi:https://doi.org/10.1016/j.tranon.2018.10.014.
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi:https://doi.org/10.1038/nature03664.
- Nakayama K. Growth and progression of melanoma and non-melanoma skin cancers regulated by ubiquitination. Pigment Cell Melanoma Res. 2010;23:338–351. doi:https://doi.org/10.1111/j.1755-148X.2010.00692.x.
- Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, Yao H, Lazar AJ, Aldape KD, Medeiros LJ, et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn JMD. 2013;15:220–226. doi:https://doi.org/10.1016/j.jmoldx.2012.10.002.
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi:https://doi.org/10.1056/NEJMoa1112302.
- Vasudevan S, Flashner-Abramson E, Alkhatib H, Roy Chowdhury S, Adejumobi IA, Vilenski D, Stefansky S, Rubinstein AM, Kravchenko-Balasha N. Overcoming resistance to BRAFV600E inhibition in melanoma by deciphering and targeting personalized protein network alterations. Npj Precis Oncol. 2021;5:50. doi:https://doi.org/10.1038/s41698-021-00190-3.
- Alqathama A. BRAF in malignant melanoma progression and metastasis: potentials and challenges. Am J Cancer Res. 2020;10:1103–1114.
- Nikolaou VA, Stratigos AJ, Flaherty KT, Tsao H. Melanoma: new insights and new therapies. J Invest Dermatol. 2012;132:854–863. doi:https://doi.org/10.1038/jid.2011.421.
- Kalal BS, Upadhya D, Pai VR. Chemotherapy Resistance Mechanisms in Advanced Skin Cancer. Oncol Rev. 2017;11:326.
- Wu S, Singh RK. Resistance to Chemotherapy and Molecularly Targeted Therapies: rationale for Combination Therapy in Malignant Melanoma. Curr Mol Med. 2011;11:553–563. doi:https://doi.org/10.2174/156652411800615153.
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi:https://doi.org/10.1056/NEJMoa1103782.
- Lacouture M, Sibaud V. Toxic Side Effects of Targeted Therapies and Immunotherapies Affecting the Skin, Oral Mucosa, Hair, and Nails. Am J Clin Dermatol. 2018;19:31–39. doi:https://doi.org/10.1007/s40257-018-0384-3.
- Capalbo C, Belardinilli F, Filetti M, Parisi C, Petroni M, Colicchia V, Tessitore A, Santoni M, Coppa A, Giannini G, et al. Effective treatment of a platinum-resistant cutaneous squamous cell carcinoma case by EGFR pathway inhibition. Mol Clin Oncol. 2018;9:30–34.
- McHugh A, Fernandes K, South AP, Mellerio JE, Salas-Alanís JC, Proby CM, Leigh IM, Saville MK. Preclinical comparison of proteasome and ubiquitin E1 enzyme inhibitors in cutaneous squamous cell carcinoma: the identification of mechanisms of differential sensitivity. Oncotarget. 2018;9:20265–20281. doi:https://doi.org/10.18632/oncotarget.24750.
- Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J Am Acad Dermatol. 2013;68:957–966. doi:https://doi.org/10.1016/j.jaad.2012.11.037.
- Green AC, Olsen CM. Cutaneous squamous cell carcinoma: an epidemiological review. Br J Dermatol. 2017;177:373–381. doi:https://doi.org/10.1111/bjd.15324.
- Fine J-D, Johnson LB, Weiner M, Li K-P SC. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol. 2009;60:203–211. doi:https://doi.org/10.1016/j.jaad.2008.09.035.
- Zhong L, Yang X, Zhu Y, Peng J, Cao Y. Radix Tetrastigma Hemsleyani Flavone Suppresses Cutaneous Squamous Cell Carcinoma A431 Cells via Proteasome Inhibition. Med Sci Monit Int Med J Exp Clin Res. 2019;25:436–442.
- Wang S, Shen P, Zhou J, Lu Y. Diet phytochemicals and cutaneous carcinoma chemoprevention: a review. Pharmacol Res. 2017;119:327–346. doi:https://doi.org/10.1016/j.phrs.2017.02.021.
- Pandey S, Patil A. Recent advances on self modified patch for (trans) dermal drug delivery. Recent Pat Drug Deliv Formul. 2015;9:88–94. doi:https://doi.org/10.2174/187221130901150303113918.
- Fakhari A, Anand Subramony J. Engineered in-situ depot-forming hydrogels for intratumoral drug delivery. J Control Release Off J Control Release Soc. 2015;220:465–475. doi:https://doi.org/10.1016/j.jconrel.2015.11.014.
- Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861–866. doi:https://doi.org/10.1038/nature00820.
- Lind MH, Rozell B, Wallin RPA, van Hogerlinden M, Ljunggren H-G, Toftgård R, Sur I. Tumor necrosis factor receptor 1-mediated signaling is required for skin cancer development induced by NF-kappaB inhibition. Proc Natl Acad Sci U S A. 2004;101:4972–4977. doi:https://doi.org/10.1073/pnas.0307106101.
- Sur I, Ulvmar M, The Two-Faced TR. NF-κB in the Skin. Int Rev Immunol. 2008;27:205–223. doi:https://doi.org/10.1080/08830180802130319.
- Anghaei S, Kamyab-Hesari K, Haddadi S, Jolehar M. New diagnostic markers in basal cell carcinoma. J Oral Maxillofac Pathol. 2020;24:99. doi:https://doi.org/10.4103/jomfp.JOMFP_199_19.
- Feller L, Khammissa RAG, Kramer B, Altini M, Lemmer J. Basal cell carcinoma, squamous cell carcinoma and melanoma of the head and face. Head Face Med. 2016;12:11. doi:https://doi.org/10.1186/s13005-016-0106-0.
- Dika E, Veronesi G, Patrizi A, De Salvo S, Misciali C, Baraldi C, Mussi M, Fabbri E, Tartari F, It’s Time LM. For Mohs: micrographic Surgery For The Treatment Of High-Risk Basal Cell Carcinomas Of The Head And Neck Region. Dermatologic Therapy 2020;33:e13474. doi:https://doi.org/10.1111/dth.13474.
- Park K, Lee J-H. Bcl-XL protein is markedly decreased in UVB-irradiated basal cell carcinoma cell lines through proteasome-mediated degradation. Oncol Rep. 2009;21:689–692.
- Mo J-S, Kim M-Y, Han S-O, Kim I-S, Ann E-J, Lee KS, Seo M-S, Kim J-Y, Lee S-C, Park J-W, et al. Integrin-linked kinase controls Notch1 signaling by down-regulation of protein stability through Fbw7 ubiquitin ligase. Mol Cell Biol. 2007;27:5565–5574. doi:https://doi.org/10.1128/MCB.02372-06.
- Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui C, Clevers H, Dotto GP, Notch RF. functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–421. doi:https://doi.org/10.1038/ng1099.
- Takebe N, Nguyen D, Yang SX. Targeting Notch signaling pathway in cancer: clinical development advances and challenges. Pharmacol Ther. 2014;141:140–149. doi:https://doi.org/10.1016/j.pharmthera.2013.09.005.
- Miele L, Miao H, Nickoloff BJ. NOTCH signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets. 2006;6:313–323. doi:https://doi.org/10.2174/156800906777441771.
- Wei C-L, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi:https://doi.org/10.1016/j.cell.2005.10.043.
- Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, Devgan V, Lieb J, Raffoul W, Hohl D, et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007;21:562–577. doi:https://doi.org/10.1101/gad.1484707.
- Mandinova A, Lefort K, Di Vignano AT, Stonely W, Ostano P, Chiorino G, Iwaki H, Nakanishi J, Dotto GP. The FoxO3a gene is a key negative target of canonical Notch signalling in the keratinocyte UVB response. EMBO J. 2008;27:1243–1254. doi:https://doi.org/10.1038/emboj.2008.45.