
ABSTRACT
Tumor metastasis is the leading cause of cancer mortality, often characterized by abnormal cell growth and invasion to distant organs. The cancer invasion due to epithelial to mesenchymal transition is affected by metabolic and oxygen availability in the tumor-associated micro-environment. A precise alteration in oxygen and metabolic signaling between healthy and metastatic cells is a substantial probe for understanding tumor progression and metastasis. Molecular heterogeneity in the tumor microenvironment help to sustain the metastatic cell growth during their survival shift from low to high metabolic-oxygen-rich sites and reinforces the metastatic events. This review highlighted the crucial role of oxygen and metabolites in metastatic progression and exemplified the role of metabolic rewiring and oxygen availability in cancer cell adaptation. Furthermore, we have also addressed potential applications of altered oxygen and metabolic networking with tumor type that could be a signature pattern to assess tumor growth and chemotherapeutics efficacy in managing cancer metastasis.
Introduction
Cancer is a group of diseases allied with reprogrammed metabolic, respiratory, and one or more gene function disorders. The abnormal growth pattern and cancer invasion are two significant hurdles for cancer management. In cancer, an invasion process often characterizes the metastasis, a direct extension, colonization, and proliferation of cancer cells into a new tissue host. Clinically the metastasis has already been classified as stage-four cancer, with a low survival rate and 90% of cancer deaths.Citation1 Metastasis is a reversible, evolutionary, and migratory survival process in which epithelial cells transform into mesenchymal cells, i.e., epithelial to mesenchymal transformation (EMT) or mesenchymal cells transform into epithelial cells, i.e., mesenchymal to epithelial transformation (MET). Several metastases (EMT/MET) inducing trans-differentiation factors such as chemokines, growth factors, hypoxia, nutrient deprivation, cellular signaling, cell-cell, and extracellular matrix (ECM) interaction have linked cancer prognosis to metastatic cells.
Metastatic inducers vary in different cancers, with metabolic (e.g., glucose, lactate, pyruvate, glutamine, and acetyl-CoA) and oxygen reprogramming (presence and absence of oxygen), the two fundamental hallmark processes that have a reported role in inducing metastasis. For instance, Marin-Valencia et al. reported the high glucose oxidation and glutamine content in glioblastoma (GBM) cells compared to tumor surrounding healthy cells and correlated the glioma cells infiltration to oxygen-rich brain tissues.Citation2 Notably, oxygen and metabolic availability regulate several steps () in the metastasis cascade, including extravasation, migration, angiogenesis, lymphomagenesis, invasion, survival, and colonization of metastatic cells.Citation3 In general, metastatic cells based on primary cancer types tend to colonize toward a pre-defined metastatic niche of secondary organs such as the brain, lungs, liver, and bone. For instance, high nutritional and oxygen abundance in the brain, and low oxidative stress, and increased oxygen availability in the lungs, develop the metastatic niche at these organs. Similarly, a high metabolic rate in the liver makes it more susceptible as an organ to support secondary tumor growth. Furthermore, continuous mineral and nutritional availability and hypoxic conditions in the bone microenvironment add to circulatory cancer cells’ deposition.Citation4,Citation5 A precise alteration in oxygen and metabolic signaling has been found between the healthy, cancer, and metastatic cells. Significantly, in the GBM tumor microenvironment (TME), metabolic heterogeneity favors the growth of two distinct types of cells, the fast-growing cells by aerobic glycolysis and the slow-growing cancer cells through oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO).Citation6
Figure 1. In the tumor microenvironment, together metabolites and cellular oxygen partial pressure (PO2) regulates different steps of metastasis. Where sufficient metabolites concentration favors the proliferation of cancer cells, their shortage induce the migration of cancer cells to metabolite and oxygen rich organs. Under the oxygen shortage metabolite rewiring maintains the viability of cancer cells, or even presence and absence of oxygen also regulates the metabolite rewiring of tumor microenvironment. During the cancer cell migration, altered metabolism and oxygen environment regulates autophagy for the maintenance of cancer cell metabolite requirement, immune evasion and resistance for anoikis to facilitate migratory behavior of cancer cells. Similarly cellular redox homeostasis maintains oxidative stress of cancer cells and vasculogenesis regulates the metabolites and oxygen shortage and also facilitate the migratory route of circulatory cancer cells. Additionally, altered oxygen and metabolism also maintains the drug resistance and dormancy in cancer cells followed by reoccurrence of cancer
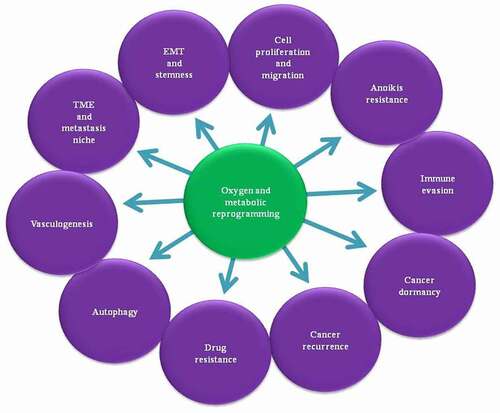
Metastasis is a nonrandom process with scarce nutrients and oxygen at the TME, inducing metabolic and oxidative stress. Thus, cancer cells migrate toward oxygen and nutrient-rich organs such as the lungs and the brain or trigger metabolic and oxygen reprogramming to substitute the deficient metabolites function to full fill the bio-energetic and bio-synthetic demand of cancer cells. Healthy cells generate a significant part of the energy from oxygen-dependent OXPHOS. In contrast, cancer cells shift their metabolism from OXPHOS to aerobic glycolysis (Warburg effect) to fulfill the high energy demand and anabolic biosynthesis for fast-growing cancer during oxygen scarcity (hypoxia). Additionally, metabolic rewiring also protects cancer cells from drug-induced toxicity and maintains cancer viability and cancer recurrence.
Apart from cancer cells, oxygen and metabolites also regulate different cell types such as fibroblast, macrophages, and neutrophils in the TME. From a therapeutic standpoint, both oxygen and metabolites play a significant role in cancer management. Furthermore, cancer cells have unique adaptability characteristics, which help them survive in adverse conditions. This review focused on the crucial role of oxygen and metabolites in metastatic progression and the role of metabolic rewiring and oxygen availability in cancer cell adaptation. We have also addressed the different metabolic alterations and associated potential applications as markers and therapeutic targets for managing cancer metastasis.
Maintenance of tumor microenvironment and metastasis niche
Cancer cells are terminally more advanced and develop their microenvironment to maintain proliferation, plasticity, and continuous metabolites and oxygen availability. Generally, metabolites and oxygen scarcity induces apoptosis in a healthy cell, but tumor cells continuously proliferate within the TME. Cancer cells induce hypoxia-inducible factor (HIF) and metabolites reprogramming to maintain an adequate oxygen and metabolites level in the TME. Furthermore, in response to hypoxia-induced apoptosis, HIF-1 activates PDK1 mediated aerobic glycolysis and suppresses the mitochondrial TCA cycle and OXPHOS.Citation7 In the TME, most cellular glucose is utilized in aerobic glycolysis; however, cellular glutamine maintains the TCA intermediates.Citation8 Notably, altered metabolism favors tumor growth and develops a metastasis niche; as Stephen Paget explained in his “seed and soil” hypothesis, the metastasis niche favors the proliferation, migration, colonization, survival, and plasticity of cancer cells (). At the metastatic niche, the metabolic rewiring also maintains cancer cell plasticity and self-renewal property. It increases the expression of EMT-inducing factors SMAD, ZEB, Notch, TGF-β, and Wnt.Citation9 The mitochondrial oxidation and high oxygen consumption favor the proliferative nature of epithelial cells. Moreover, the high glycolysis, lipogenesis, elevated pentose phosphate pathway (PPP), and glutamine-mediated TCA intermediate synthesis have been associated with the migratory potential of mesenchymal cells’ .Citation8
Figure 2. Tumor microenvironment facilitate the proliferation and migration of cancer cells to pre-metastatic niche. Cancer microenvironment also maintains the plasticity and dormancy (G0/G1) in cancer cells. Physiological stress or drug environment induces the dormancy in cancer cells and dormant cancer cells initiate the growth of secondary tumor or reoccurrence of cancer
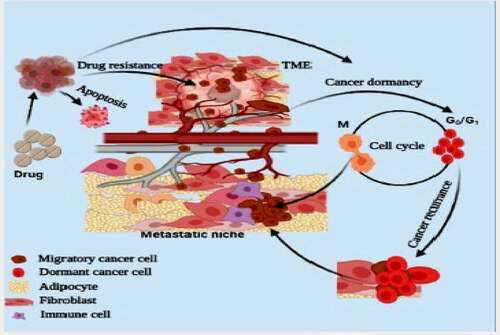
To maintain metabolite homeostasis, cancer cells increase the expression of metabolites transporters such as glucose (e.g., GLUT), fatty acid (e.g., CD36, FABP4, and FATP), aspartate (e.g., SLC1A3), and glutamine transporter (e.g., SLC1A5). Besides metabolic transporters, cancer cells also increase the expression of ionic transporter for pH homeostasis. During aerobic glycolysis, lactate dehydrogenase (LDH) generates lactate, leading to acidification of TME. Through monocarboxylate transporters (MCT1/4), cancer cells continuously export lactate from the cells to suppress lactate-mediated cancer cell acidification.Citation10 MCT1 mediated bidirectional lactate transport maintains ambient lactate concentration in the cancer cells.Citation10,Citation11 Furthermore, cancer-associated fibroblast (CAFs), lymphocytes (e.g., neutrophils and macrophages), and the vascular system create a tumor-friendly microenvironment, favoring the metastatic cascade initiation. Stromal cells at the cancer site support developing TME and act as sources of metabolites, growth factors, and ECM for proliferating cancer cells. For instance, bone adipocytes act as a source of metabolites and growth factors for growing cancer mass,Citation12 and during glutamine starvation, they maintain glutamine homeostasis.Citation13 Thus, to maintain adequate intracellular fatty acid concentration, cells increase the expression of fatty acid transporters.Citation14
The adaptable behavior of cancer cells enables them to survive in a restrictive environment at the primary tumor site, during the circulation, and at the secondary cancer site.Citation15 Though metabolic reprogramming not only favors cancer cells to survive under oxidative stress, metabolites shortage, immunological response, and presence of drugs, it also enhances the cancer cells’ ability to survive in the altered conditions that make them more aggressive toward their rapid advancement to secondary organs, colonization, and progressive metastasis. A recent study performed by Lin et al. showed that glioma cell’s adaptation to FAO maintains their proliferation, cellular respiration, and glycolysis. During initial tumor growth, cancer cells utilize a higher amount of glucose; however, they switch to FAO during colonization under shortage of glucose at the secondary site.Citation16 For instance, in metastatic breast cancer cells, Weverwijk et al. observed glycolysis to FAO shift under glucose stress conditions.Citation17
Increased vascular network at the TME provides oxygen and metabolites (e.g., lactate, succinate, and glutamine) to growing tumors, including transportation of neutrophils, macrophages, and platelets to the proliferating site. Succinate activates tumor-associated macrophage (TAM) that promotes lung tumorigenesisCitation18 and angiogenesis at the TME.Citation19 For instance, lactate activates endothelial NF-κβ/IL-8 (CXCL8) for angiogenesis,Citation20 and macrophage CCL5 activates invasive and EMT phenotypes in the breast cancer cells.Citation21 The TME acidification also favors macrophage polarization, including suppressing immunological response against growing tumors and colonization of migratory melanoma cells.Citation22 Furthermore, the lactate-mediated ERK/STAT3 pathway facilitates TAM-mediated proliferation, migration, invasion, and colonization of breast cancer cells.Citation23 Both altered metabolism and hypoxic conditions at the TME influence ECM remodeling and release cancer cells from the primary cancer site, while at the pre-metastatic niche, it favors invasion and colonization of migratory cancer cells.
A limited supply of glucose in the cancer cells reduces collagen synthesis, while glutamine-mediated elevated α-ketoglutarate maintains hydroxylation of proline and lysine for collagen synthesis.Citation24 Elevated glycolysis in cancer cells supports ECM development, resulting in colonization. Similarly, in migratory triple-negative breast cancer (TNBC) cells, denser collagen increases glycolysis, while at low collagen density, they shift their metabolism toward mitochondrial oxidation.Citation25 For instance, elevated glucose uptake supports breast cancer cells’ proliferation; thus, to maintain high glucose availability at the TME, breast cancer cells also hijack CAFs glucose uptake, and low glucose availability at the TME promotes cancer cell metastasis.Citation26 However, it is not clear how the variation in glucose absorption or other metabolites regulates cancer metastasis to date.
Nevertheless, Sullivan et al. showed that ECM digestion increases the GLUT1 mediated glucose uptake, favoring cancer cells’ proliferation and migration.Citation27 Additionally, Sai et al. also reported a critical role of the pre-metastatic niche in homing and colonization of migratory lung cancer cells in the bone marrow microenvironment.Citation28 During the homing of migratory cancer cells, both ECM and chemoattractant molecules on fibroblast cells’ surface support cancer cells’ interaction with host cells. Migratory mesenchymal cells interact with fibroblast cells of the post metastatic niche and activate them for colonization. At the metastasis niche, oral cancer cells and CAFs secrete the ECM remodeling enzymes such as MMP, LOX, and TIMP-1, which play a crucial role in cell transformation and migration.Citation29
Cell proliferation and migration
The high proliferation and migration of cells are critical for any cancer to become metastatic. Of different factors, oxygen and metabolites’ availability play a significant role in cancer cells proliferation and migration. Also, high reactive oxygen species (ROS) are the most common factor in all cancers, promoting tumor development and progression through the activation of survival (PI3K/Akt) and proliferative (MAPK/ERK) pathways.Citation30 For instance, the increased oxygen consumption in invasive melanoma cells upregulates cellular glycolysis and activates the mitochondrial ROS mediated focal adhesion kinase (FAK).Citation31 Further, integrin-mediated FAK signaling that support the migration of invasive pancreatic cancer cells.Citation32 In ROS response, cancer cells increase the expression of cellular antioxidants such as NADPH, nuclear factor erythroid 2–related factor2 (NRF2), superoxide dismutase (SOD), and glutathione, facilitating the proliferative and migratory mechanism of cancer cells.Citation30
The low cellular oxygen level also induces an oxidative environment, which transforms cancer cells to metastatic cells through up-regulation of proteins related to cell proliferation, migration, redox homeostasis, and metabolic reprogramming. Under a hypoxic environment, cancer cells’ either migrate toward the oxygen-rich organs or increase the expression of HIF-1/2. HIF-1 ubiquitously promotes vasculogenesis and Wnt/β-catenin mediated cell proliferation,Citation33 while HIF-2 shows contrasting proliferative effects in the lungs, kidneys, cardiac, pancreas, and intestinal, hepatocytes, and endothelial cells.Citation34 During the lung cancer cell migration toward oxygen-rich secondary sites, HIF upregulates cellular NADPH and suppresses E-cadherin expression.Citation35 Similarly, upregulated glutamate dehydrogenase-1 (GDH-1) and glutathione peroxidase (GPx) expression favor the adaptation of proliferating lung and breast cancer cells to oxidative stress.Citation36
The altered metabolism fulfills proliferative cells’ metabolic needs and acts as an inducer for cancer cell migration. In general, healthy brain cells oxidize glucose; however, the GBM cells and brain colonizing breast, melanoma, non-small cell lung carcinoma (NSCLC), renal, and endometrial cancer cells increase the oxidation of acetate.Citation37 Acetate adaptation generates acetyl-CoA, a substrate for the lipogenesis and TCA cycle, favoring the cancer cell’s proliferation and survival,Citation38 and maintaining the malignancy features in prostate cancer.Citation39 Significantly, LDH-A mediated aerobic glycolysis has been reported in cancer cells such as pancreatic, colorectal, breast, and NSCLC cells.Citation40 At the developing cancer site, LDH-A regulates MMP9, cathepsin B, PLAU,Citation41 Tropomyosin (e.g., TM4, Tm5NM1, and Tm5NM5), and actin filament rearrangement during cancer cell multiplication, migration, and invasion.Citation42 Additionally, LDH-A also favors the expression of metastasis-associated protein 1 (MTA1)Citation43 and proliferative marker (e.g., Ki-67) in migratory breast cancer cells.Citation44 In contrast, the inhibition of LDH-A has been reported to arrest neuroblastoma,Citation45 pediatric osteosarcoma,Citation46 and prostate cancer.Citation41 In liver colonizing breast cancer cells, high lactate also increases glutamine oxidation and suppresses OXPHOs.Citation47 Furthermore, pyruvate dehydrogenase kinase 1 (PDK1) diverts tumor metabolism toward glycolysis under hypoxia, supporting glioma cell migration, autophagy, apoptosis resistance, and redox homeostasis.Citation48 However, PDK inhibition upregulates the TCA cycle α-ketoglutarate and arrests metastasis of renal cell carcinoma (RCC).Citation49
Under metabolic and oxygen scarcity, cancer cells maintain proliferation through shifting mitochondrial oxidation toward aerobic glycolysis. Therefore, to sustain the Warburg effect, colon cancer cells suppress the oxidation of mitochondrial pyruvate and upregulate the expression of embryonic pyruvate kinase (PKM2)Citation50 and mitochondrial ACAT-1.Citation51 Subsequently, overexpression of glycolysis enzymes such as Phosphofructokinase-1 (PFK-1),Citation52 phosphoglycerate mutase (PGAM), hexokinase, and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) helps to reduce metabolic stress in the growing TME.Citation53 However, the elevated Warburg effect results in high lactate production, inducing a shortage of fatty acid, acetyl-CoA, and TCA intermediates. Therefore, to maintain metabolites homeostasis, cancer cells divert glycolysis intermediates to the PPP, TCA, and amino acid synthesisCitation54 (). Additionally, glutamine favors anabolic growthCitation55 and maintains acetate and fatty acids in RCC.Citation56 However, proliferating cancer cells increase the anaplerosis reactions to substitute limiting metabolites (e.g., TCA intermediates, NADPH, acetyl-CoA) with available metabolites (e.g., glucose, glutamine, α-ketoglutarate, fatty acid). The glucose-mediated anaplerosis reaction gives citric acid cycle intermediates such as oxaloacetate, pyruvate, and acetyl-CoA to proliferate GBM cells; however, glutamine acts under glucose scarcity as a gluconeogenic precursor.Citation2 Additionally, certain non-essential amino acids, such as glutamine and asparagine in cancer cells, favor cancer prognosis, while their shortage induces apoptosis of cancer cells.Citation57
Figure 3. Healthy cells use a sequential regulated metabolism (glycolysis-TCA-oxidative phosphorylation and fatty acid oxidation) for energy generation and anabolic biosynthesis. But in cancer cells according to available nutrient and unfavorable surrounding they divert their sequential metabolism toward the altered metabolism. Under the oxygen limiting conditions cancer cells increase the cytosolic metabolism and reduce the mitochondrial oxidation
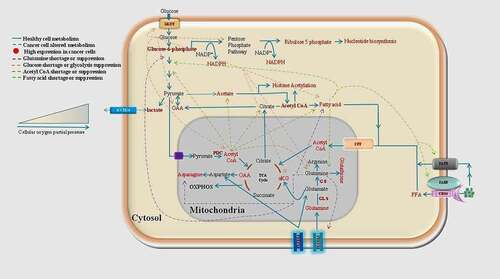
In general, cancer cells use carbon units of fatty acids for cell membrane expansion and ATP generation; however, aggressive cancer cells such as melanoma, breast, prostate, and ovarian divert them to synthesize structural and signaling lipid molecules,Citation58 and increases the expression of FAO acetyl-CoA acetyltransferase-1 (ACAT1).Citation51 Also, acetyl-CoA synthetase (ACS) regulates the fatty acid synthesis and oxidation in lung cancer cells.Citation59 In contrast, adipocyte FAO promotes PI3K/AKT mediated ovarian and breast cancer cell proliferation and migration.Citation60,Citation61 In addition, oleic and linoleic acid also activates the PI3K/AKT pathway to sustain the breast cancer cell migration,Citation62 and in gastric cancer cells, oleic acid-mediated PI3K/AKT activation increase the invasion of cancer cells to neighboring organs.Citation63
Due to the high metabolism in the TME, elevated glucose, lactate, fatty acid, amino acid, and ion transporters increase metabolites transport into the cancer cells. For instance, the upregulated expression of MCT and SLC1A3 in proliferating GBM cells is essential to maintain the high influx of lactate and aspartate.Citation64 The overexpression of MCT1/4 in breast cancer cells maintains high aerobic glycolysis.Citation65 Furthermore, overexpressed MCT1/4 are also associated with disease progression and reduced survival of clear cell carcinoma patients.Citation66 In contrast, MCT’s inhibition has been reported to suppress proliferation of breast and lung cancer cells and shift aerobic glycolysis to OXPHOS.Citation11 Similarly, MCT1/4 inhibition makes colorectal cancer cells sensitive to 5-fluorouracil and reduces tumor growth.Citation67
Surprisingly, metabolite reprogramming for energy generation also helps the cancer cells adaptation to changing surrounding environment. For instance, the altered metabolism in cancer cells suppresses the inhibitory effect of anti-cancer drugs. In squamous carcinoma cells, mTOR inhibitors suppress aerobic glycolysis. However, cancer cells shift their metabolism toward glutaminolysis to maintain a high proliferation rate in lung cancer cellsCitation68 or increase the mTOR expression to maintain glutamine synthetase (GS) expression in proliferating liver cancer cells.Citation69 Furthermore, in NSCLC cells mTOR mediated asparagine maintains protein synthesis during nutrient deficiency.Citation70 Additionally, glioma, breast, kidney, and pulmonary cancer cells excrete glutamine and uptake cysteine to synthesize glutathione to mimic mTOR inhibitory drug-induced oxidative stress.Citation71
Epithelial to mesenchymal transformation and cancer stem-like cell maintenance
Before the commencement of Stephen Paget’s Seed and soil theory, the researchers believed that only embryonic and adult tissue-stem cells retain the plasticity (ability to transform) and self-renewal phenotype (ability to divide). Subsequently, in 1889, Stephen Paget explained the EMT feature of cancer cells linked to the stemness feature in the malignant TME. Unlike embryonic stem cells, the cancer cells do not possess the true stemness; however, their plasticity and phenotypic transformation endow them with cancer stem-like cells. In the TME, some breast cancer cells maintain tumor growth owing to their self-renewal ability and plasticity.Citation72 PI3K/AKT mediated SMAD and Nanog expression regulates the self-renewal process of cells.Citation73 Of note, due to the plasticity, cancer cells show EMT during metastasis. In the TME, the EMT maintains the differential phenotype for dissemination and colonization of cancer cells, while the stem cell phenotype supports the polarity and outgrowth of cancer cells at the secondary site.Citation74,Citation75 For instance, in breast cancer, an elevated asparagine favors breast cancer cells’ colonization into the lung, liver, and brain,Citation76 while L-Proline maintains the migratory mesenchymal phenotype of stem cells.Citation77 Similarly, isocitrate dehydrogenase maintains EMT in colorectal cancer cells.Citation78 Thus, stem-like features maintain the self-renewal and high plasticity in cancer cells, leading to EMT-mediated secondary tumor outgrowth.
In cancer cells, TGF-β is a well-known molecule responsible for cancer cell transformation and regulates the expression of mesenchymal markers such as Snail, ZEB1,Citation79 SMA, and vimentin.Citation80 During cellular transformation, it maintains the fatty acid synthase (FAS) mediated anabolic synthesisCitation80 and shift the cellular metabolism toward aerobic glycolysis and amino acid metabolism, suppressing the expression of epithelial markers such as occludin and zonula occludens-1 (ZO-1).Citation81 At the time of EMT induction, increased glycolysis and mitochondrial oxidation maintain the high proliferation of epithelial cells. In contrast, upregulated amino acid synthesis and FAO maintain the mesenchymal phenotype of migratory breast cancer cells.Citation3 Wnt signaling positively regulates the EMT process in colorectal cancer cells by expressing epithelial markers followed by suppressing mesenchymal markers such as MMP2, MMP9, ZEB1, N-cadherin, and vimentin.Citation82
Fatty acids in the regulation of cancer stem-like cells phenotype
In cancer cells, cellular fatty acid content has a significant role in cancer initiation and cellular transformation, that divert the glycolytic intermediates toward the anabolic biosynthesis of fatty acids, thereby increasing lipid accumulation that acts as a good energy source and protects cancer cells from oxidative stress-induced cell death.Citation83 In colon cancer, elevated stearoyl-CoA desaturase-1 (SCD-1) increases the monounsaturated fatty acid (MUFA) mediated Notch and Wnt signaling, favoring the stem cell phenotypeCitation84 and also maintains the oxidative stress in Glioblastoma stem cells (GSC).Citation85 Giudetti et al. reported a differential fatty acid reprogramming in epithelial and mesenchymal breast cancer cells. The epithelial cells upregulate the accumulation of MUFAs and de novo lipogenesis, while mesenchymal cells suppress lipogenesis. However, the increase in the β-catenin mediated triacylglycerol synthesis and the accumulation of high cholesterol and polyunsaturated fatty acids (PUFA)Citation86 in breast cancer cells favor their colonization in the lungs.Citation87
Furthermore, inhibition of fatty acid synthesis arrests the expression of β-catenin mediated colorectal cancer cell metastasis cascade.Citation88 In breast cancer, lipogenesis inhibition increases oxidative stress, and acetyl-CoA mediated SMAD acetylation, including the expression of EMT markers in metastatic breast cancer cells.Citation89 Similarly, in colorectal cancer, a cascade of signaling begins due to the loss of acyl-CoA thioesterase-12 (ACOT-12), which induces the TWIST2 mediated EMT transformation epithelial marker E-cadherin suppression for the migration and invasion of cancer cells.Citation90
Glycolysis and acetyl-CoA production in cancer stem-like cells
The TNBC cells shift glycolysis toward acetyl-CoA synthesis by acetylating the H4 histone of stem cell genes SOX-2, KLF-4, OCT-4, and c-Myc to maintain stem cell behavior.Citation91 Thus to keep an adequate amount of acetyl-CoA, cancer cells increase glycolysis to maintain stem cell and self-renewal properties in GBM, ovarian, and pancreatic cancer cells.Citation92 For instance, glycolysis inhibition promotes the OXPHOS and lineage differentiation of stem cells.Citation93 The EMT induction results in increased glucose uptake and maintains the expression of mesenchymal markers such as Snail, SMAD, N-cadherin, and vimentin in pancreatic cancer cells.Citation94 Furthermore, Snail overexpression increases glucose and lactate uptake in transforming pancreatic cancer cells.Citation95 However, aerobic glycolysis mediated TME acidosis also increases the expression of stem cell markers such as Oct-4, OLIG2, and Nanog in glioma cells.Citation96
Metabolites and mitochondrial oxidation in the maintenance of cancer stem-like cells
The energy-generating mitochondria also regulate the epigenetic switches through acetyl-CoA during the somatic cell’s transformation.Citation97 The mitochondrial oxidation arrests the stem cell multiplication; however, its inhibition increases the proliferation of stem cellsCitation98 and EMT progressionCitation99 by suppression of E-cadherin and overexpression of fibronectin in colon and pancreatic cancer cells.Citation100 In general, cancer cells use glycolysis instead of mitochondrial oxidation; however, Curry et al. reported their occurrence in cancer cells to generate amino acids, fatty acids, and other metabolic intermediates.Citation101 For instance, the head and neck cancer cells use aerobic glycolysis and mitochondrial oxidation to maintain stem cell features. Besides metabolites, oxygen partial pressure (PO2) also regulates the stem cell phenotype in cancer cells. In the presence of oxygen, somatic cells use mitochondrial oxidation for energy generation; however, on the induction of stem cell phenotype, somatic cells shift their metabolism to aerobic glycolysis, or in other words, hypoxic condition favors the expression of stem cell markers.Citation97 Similarly, Curry et al. reported the proliferative marker (Ki-67) with the cellular energy demand and showed that OXPHOS maintains high proliferation in head and neck stem cells.Citation101 However, dysfunction of mitochondrial oxidation shifts cellular metabolism toward aerobic glycolysis, resulting in increased expression of stemness markers, such as ALDH, CD44, Nanog, and Notch in ovarian cancer cells.Citation102
Another critical factor, the HIF-1α, not only maintains the sufficient energy level in stem cells but also regulates the cell quiescence by Pdk2/Pdk4Citation103 and activates the Wnt/β-catenin for the maintenance of EMT mediated cellular proliferation and plasticity.Citation104 HIF-1α also induces the increased expression of EMT markers such as N-cadherin, Snail, β-catenin, vimentin,Citation105 ZEB1, and Slug.Citation106 However, lower cellular oxygen also reduces ROS production, favoring the self-renewal property of stem cells. To maintain self-renewal under high ROS, stem cells activate the PI3K/AKT pathwayCitation107 or increase the expression of cystine/glutamate transporter (SLC7A11) for the production of antioxidant molecules.Citation108 Additionally, high cellular ROS also induces the cellular transition to manage intracellular ROS levels and keep high glucose uptake followed by ERK-mediated NADPH synthesis.Citation109
Angiogenesis and lymphogenesis in the tumor microenvironment
Besides the role and importance of metabolites in tumor outgrowth and cancer cell migration, specific metabolites (e.g., fatty acid, glucose, and glutamine) also take part in the progression of vasculogenesis (e.g., angiogenesis and lymph-angiogenesis) and sustain the friendliness behavior with the TME. The initial investigation of increased vascularization in the TME marked them as a prognostic factor for tumor overgrowth and metastasis, supported by the constant supply of metabolites and oxygen. Also, during extravasation and invasion, it facilitates the migratory route to anticipating cancer cells. However, how cancer cells induce vasculogenesis in the tumor environment is still a matter of discussion. Cancer cells induce abnormal (leaky type) and normal (fully matured) vascularization in the TME. For instance, Wenes, et al. showed mTOR-mediated heterogenic TAMs and their association with glycolysis during vascular growth. Elevated macrophage glycolysis induces abnormal vascularization, i.e., leaky type blood vessels support cancer cells’ intravasation.Citation110 However, the low glycolysis rate induces blood vessel maturation by increasing the deposition of perivascular cells and tight vascular endothelial (VE) cadherin junctions between endothelial cells. However, glycolysis inhibition downregulates VE-cadherin protein and suppresses blood vessels’ maturation.Citation111 Besides macrophages, several other types of cells such as platelets, neutrophils, endothelial cells, and CAFs also secrete the angiogenic factors including angiogenin, VEGF, FGF, PDGF, IL-8, CXCL-8/12, MMPs, Osteopontin, angiotensin, erythropoietin, and angiopoietin-2 for the vascular development from the existing vascular vessels.Citation112,Citation113 Vasculogenic factors induce the proliferation and migration of endothelial cells, followed by colonization in the tube-like structures. For Instance, MMP-9 from the neutrophils digests the ECM, favoring the migration of endothelial cells and their colonization into the tube-like structures.Citation114
During angiogenesis and lymphogenesis, the altered metabolism in cancer cells plays a vital role in expressing the vasculogenic factors. Elevated aerobic glycolysis not only generates sufficient ATP molecules for proliferating endothelial cells, but high lactate in endothelial cells also maintains their proliferative nature and they increase the expression of LDH-A and MCT-1.Citation115 In the endothelial cells, lactate activates the NF-κβ/IL-8 mediated CXCL8 expression, which induces endothelial cells’ migration,Citation20 and PI3K/AKT mediated increased expression of VEGF-A for the new blood vessel development.Citation116 Lactate also polarizes the M2-type macrophages and HIF-1α expression for angiogenesis. Additionally, amino acids (e.g., glutamine and asparagine) and TCA intermediate (e.g., α-ketoglutarate) also regulate endothelial cell migration and vessel development.Citation117
Adipocytes from the surrounding tumor environment also support the proliferation and migration of endothelial cells.Citation118 For instance, 1-butyryl-glycerol from adipocytes induces the angiogenesis cascade,Citation119 and fatty acid Sphingosine-1-phosphate activates the ERK and p38 MAPK signaling for endothelial cell proliferation and their migration.Citation120 Besides, to facilitate the increased fatty acid transport in the endothelial cells, both VEGF-A and NOTCH increase the expression of FABP-4 mediated lipid transport. Other fatty acids (e.g., linoleic, and oleic acid) and cholesterol also induce vascular growth.Citation121 However, the scarcity of fatty acids directs the metabolic shift toward glycolysis. Together, the shortage of glucose and fatty acid reduces the α-ketoglutarate, which activates the HIF-1α mediated VEGF-A expression to increase the vasculogenesis to maintain fatty acid availability.Citation122 During vascular architecture development, glutamine metabolism regulates the proliferation and migratory behavior of endothelial cells.Citation123 Glutamine scarcity affects the endothelial cells’ proliferation, migration, causing disparity in TCA intermediates and fatty acid synthesis. Elevated glutamine (substituting α-ketoglutarate and asparagine) in the TME also induces the polarization of TAMs into M2-type macrophages. Subsequently, activated M2-type macrophages increase the vascular development and metastasis potential of cancer cells. During the vasculogenesis, macrophages from the TME sense the hypoxic condition and secrete the angiogenic factors in response to the hypoxia. Macrophages from the hypoxic cancer area secrete angiogenic factors such as VEGF,Citation124 PDGF, and FGF,Citation125 which increase the glycolysis in endothelial cells and stimulate blood vessel development.Citation110
The hypoxic TME acts as a critical inducer for the progression of vasculogenesis. In response to reduced PO2, HIF-1α increases aerobic glycolysis, favoring the VEGF-mediated angiogenesis.Citation126 However, lactate and succinate from TME also increase the expression of HIF-1α mediated VEGF secretion for the vascular growth around the growing tumor.Citation127 Under the hypoxic or oxidative environment, lactate maintains the oxidative environment and activates TAMs for angiogenesis.Citation128 For instance, the HIF-1α expression in NSCLC,Citation129 gall bladder carcinoma,Citation130 and breast cancer govern the VEGF expression and vascular density in the TME.Citation131 Both the isoforms of HIF (1/2) regulate vasculogenesis and induce the expression of pro-angiogenic factors under hypoxia.Citation132 The HIF-1α activates the β–catenin signaling pathway, which increases the proliferation and tumor development from endothelial cells. However, its suppression reduces the expression of angiogenic factors VEGF, FGF, and angiogenin in colorectal cancer patients.Citation133
In aggressive cancers such as melanoma, breast, hepatic, and lung cancers, the lymphogenesis process modulates the TME for cancer cell migration. The low fluidic shear, absence of pericytes, elastic discontinuous basement membrane, the weak cell-cell junction between lymph endothelial cells (LECs), and high expression of adhesion molecules in the lymphatic system favors the invasion and circulation of cancer cells through lymph vessels. Besides, high dietary fat and other available nutrients (e.g., glucose and glutamine) in the lymphatic fluid work as an excellent nutritional source for circulating cancer cells. The lymphatic system also circulates lymph fluid or exchanges lymph fluid with tissues to maintain ambient tissue fluid homeostasis and transport immune cells, dying cells, and cancer cells from tissue to lymph vessels supporting the extravasation.Citation134 For instance, in the marginal region of metastatic lung carcinoma, high VEGF C and D expression favor the lymph node invasion in lung cancer patients,Citation135 further inhibition of VEGFR2 not only suppresses the vasculogenesis but also arrest the metastasis of breast cancer.Citation136
Additionally, in hepatic cancer, CXCL-1 from cancer cells activates the F-actin in LECs and induces lymph vessel growth and lymph node metastasis.Citation137 Lymphangiogenic inducers from cancer cells, CAFs, endothelial cells, and TAMs also induce proliferation and migration of LECs. In the TME, lactate-induced macrophages secrete the VEGF-C to enhance LECs and vessel outgrowth.Citation138
During lymphatic vessel development, differential metabolism in the cancer environment also regulates LECs’ differentiation and migration. An altered metabolism leads to an increased PDGF-BB, VEGF-C, and D expression for lymph vessel outgrowth. Cancer cells also increase the glycolysis and FAO in the proliferating LECs. Elevated glycolysis maintains both the ATP and NADPH concentration, while FAO maintains the ATP, NADPH (through TCA cycle citrate), and acetyl-CoA in proliferating LECs. NADPH regulates the oxidative environment and acetyl-CoA used in the TCA cycle and fatty acid synthesis.Citation139,Citation140 Fatty acids from the TME also regulate the epigenetic switch of lymphogenesis. Acetyl-CoA from the FAO acetylates the histone (H3K9) and improves the PROX1 mediated VEGFR3 expression and acetyl-CoA synthesis. PROX1 increases the expression of β-oxidation regulating enzyme carnitine palmitoyltransferase1A (CPT1A), while inhibition of CPT1A leads to suppression of new lymphatic vessel development.Citation141 One critical study performed by Jung et al. on a high-fat diet in melanoma rat models reported the induction of lymphogenesis in cancer cells during metastasis. High fat induces pro lymphogenic markers M-CSF, VEGF-D, and VEGFR-3, followed by increased CCL19/21 in lymph nodes and CCR7 in tumors.Citation142
Immune evasion and resistance for anoikis
Anoikis, a form of programmed cell death, occurs in anchorage-dependent cells after detaching from the surrounding ECM. In general, ECM maintains morphological intactness; however, its breakdown activates the intrinsic and extrinsic apoptotic pathway in healthy cells while metastatic cancer cells show resistance to anoikis. Anchorage-independent growth or anoikis resistance, one of the cancer cells’ hallmarks, is acquired by the malignant cells for their migration and survival in the circulatory system.Citation143 Cancer cells also show resistance to the host immune system and escape from the host immune surveillance (immune evasion) during metastasis. Both immune evasion and resistance to anoikis adds to cancer cells’ survivor process and play a vital role in the detachment from the surrounding ECM. In cancer cells, anoikis resistance maintains cell viability, while immune evasion protects cancer cells from an ambush of the host immune system. During the onset of tumor prognosis, the host immune system recruits tumor-infiltrating lymphocytes (TILs), secrete cytotoxic agents (e.g., IFN-γ and granzyme) at the TME to suppress the tumor growth. However, glycolysis-mediated TME acidification neutralizes TILs’ action against growing tumors. Besides maintaining the immunosuppressive environment at the tumor site, cancer cells also recruit the regulatory T-cells (Treg), antigen-presenting cells (APCs), and myeloid-derived suppressor cells (MDSCs) in the TME for the host immune suppression.Citation144 Altered metabolism and hypoxic condition at the TME make it possible for the cancer cell to escape from the immune surveillance and resist anoikis. Furthermore, low T-cell infiltration or reduced number of available T-cells (CD4+ and CD8+) at the developing tumor site is associated with poor survival of cancer patients.Citation145
Although excess aerobic glycolysis in sarcoma increases the secretion of CTL4 and PD-1 and creates a shortage of free glucose in the TME thereby reducing the T-cell glycolysis and suppressing the IFN-γ expression from T-cells.Citation146 Inhibition of IFN-γ expression from CD8+ T cells reduces immune cells’ infiltration into the TME.Citation147 Similarly, in ovarian cancer, T-cells’ restricted glycolysis promotes T-cells’ apoptosis.Citation148 Even so, the presence of arginine reprograms the exhausted T-cells and increases their anti-tumor activity against developing melanoma. Arginine diverts cellular metabolism toward mitochondrial oxidation,Citation149 and metabolic shifting from glycolysis to OXPHOS or FAO favors the survival of CD8+ T cells.Citation150 Additionally, glycolysis inhibition suppresses MDSCs and increases the T cell response against developing TNBC.Citation151 However, Ho et al. proposed that Treg (TH1CD4+) cells play a critical role against developing tumor immune response by regulating both the activator and suppressive nature of T-cells. Reduced glycolysis in T-cells induces the suppressive nature of Treg cells, and glucose deprivation results in reduced expression of IFN-γ and CD40L from the TH1 cells.Citation144 Besides, glucose scarcity also induces ROS-mediated Treg cell apoptosis, and apoptotic Treg cells show a more suppressive effect than the viable Treg cells. Dying Treg cells express a high level of immunosuppressive molecules (e.g., PD-1, CD39, CD73, and CTLA-4), secrete many ATP, and indirectly induce immunosuppressive effects through adenosine signaling.Citation152
In response to tumor acidic environment, Foxp3 from Treg cells’ shift glycolysis to OXPHOS and uplift their immune response against growing cancer.Citation153 In colon cancer cells, glutamine suppression arrests glycolysis, TCA, and PPP and increases CD8+ T cell immune activity. However, glutamine scarcity did not affect the CD8+ T cell activity; they maintain their metabolic plasticity through pyruvate carboxylase-mediated anaplerosis. Therefore, glutamine antagonists increase tumor infiltration of CD8+ T-cells. Additionally, glutamine targeting maintains anti-tumor memory of T-cells and increases their oxygen consumption, followed by a reduction in oxidative stress and a hypoxic environment. Increased oxygen consumption increases the acetate-mediated OXPHOS in CD8+ T-cells and shift glycolysis toward acetate-mediated OXPHOS.Citation154 However, increased glutamine metabolism in the TME also activates the HIF-1α mediated TAMs. Activated TAMs secrete IL-23 and increase the immunosuppressive IL-10 and TGF-β expression from Treg cells.Citation155 Hypoxia also induces the ER stress-mediated kill signal (e.g., calreticulin) expression on the cancer cells. However, in response to increased calreticulin, breast cancer cells also increase the expression of do not kill signals (e.g., CD47) to protect cancer cells from immune killer cells.Citation156
A hypoxic environment also modulates the host immune system and increases immunosuppressive and inflammatory mediators’ expression to hijack the host immune surveillance. In gastric cancer, HIF-1α mediated TGF-β increases the Treg deposition,Citation157 subsequently high CCL28 expression in the hypoxic TMECitation158 and CXCR4 expression on the Treg cells helps their deposition on the TME.Citation159 Although under a hypoxic environment, pheochromocytomas and paragangliomas cells also increase the PD-L1Citation160 and CD137 expression to suppress the host T-cell activation in colon cancer.Citation161 In RCC, HIF-1α induces GAL3ST1 mediated sulfonation of membrane fatty acids, protecting cancer cells from the attack of natural killer cells.Citation162 Additionally, HIF-2α expression reduces RCC patients’ survival rate, and it also increases the IL-10 and TGF-β secretion from mast cells to suppress CD8+ T-cells.Citation163 Similarly, Mayer et al. observed that in brain metastasis, hypoxic regions have low TC cell infiltration, and hypoxia suppression increases T-cells’ infiltration and reduces the growing tumor size. However, under hypoxia Treg cells, adenosine signaling also helps to develop an immunosuppressive environment.Citation164
Although during the initiation of anoikis, cancer cells reduce mitochondrial oxidation and shift their metabolism toward aerobic glycolysis. Increased mitochondrial oxidation leads to ROS-mediated anoikis. However, mitochondrial oxidation suppression shifts aerobic glycolysis intermediates toward synthesizing antioxidant molecules through fatty acidCitation165 and PPP.Citation166 Elevated FAO and fatty acid synthesis in anoikis-resistant cells suppress the cellular ROS and increase osteosarcoma cells’ lung colonization.Citation167 After the ECM detachment, cancer autophagy regulates the cellular metabolism and maintains the anoikis resistance in ovarian cancer cells.Citation168 For instance, Yoshino et al. observed that anchorage-independent cells rely on increased aerobic glycolysis and shift the glycolytic intermediates for the acetyl-CoA and TCA cycle.Citation169 However, instead of glycolysis, anoikis-resistant ovarian cancer cells increase their mitochondrial oxidation and shift their glutamine toward the TCA cycle intermediates synthesis.Citation170
In contrast, cancer cells do not always use glycolysis for ATP generation, but they also shift glycolysis or intermediates of glycolysis toward TCA and OXPHOS for the anabolic synthesis to maintain the viability of circulatory cancer cells. In cancer cells, AMPK act as a sensor and regulator of cellular energy expenditure. After detachment from the ECM, breast cancer cells conserve their energy to maintain essential cellular processes. During low cellular ATP, especially after the ECM detachment, AMPK suppresses the mTORC1 mediated unnecessary protein synthesis.Citation171 However, Bizjak et al. showed that low cellular glucose or glycolysis inhibition also activates the AMPK mediated anchorage-independent growth of breast cancer cells.Citation172
During ECM detachment, cellular integrin plays a vital role as an inducer of anoikis. After basement detachment, integrin/FAK induces apoptosis in detached cancer cells. Thus, to maintain anoikis resistance, breast cancer cells activate their survival pathways and suppress the expression of pro-apoptotic factors.Citation173 After the ECM detachment, elevated ROS activates the cancer cell’s survival pathway (EGFR/Src/ERK/AKT) and develops the anoikis resistance.Citation174 In epithelial and basal cell carcinoma, secreted angiopoietin-like protein 4 (ANGPTL-4) interacts with integrin and maintains the cellular antioxidant homeostasis followed by induction survival pathway (PI3K/ERK) and suppress pro-apoptotic factor (Bad) expression.Citation175 Additionally, Oleic acid also activates the ANGPTL-4 mediated anoikis resistance.Citation176 Similarly, under hypoxic conditions, ANGPTL-4 expression activates FAK-Src/PI3K/AKT/ERK survival pathway and suppresses Caspase-3, 8, 9, and PARP expression in gastric cancer cells.Citation177 However, in lung colonizing breast cancer cells, HIF-1α mediated PI3Ksignaling maintains anoikis resistanceCitation178 and inhibits the expression of anoikis proteins (Bim and Bmf).Citation179
Autophagy and cancer dormancy
Physiological stresses such as oxidative, metabolic, hypoxic, and therapeutic stress restrict tumor growth and start the pro-survival autophagy, followed by a reduced metabolic and proliferative activity. Autophagy is a natural and regulated cell mechanism to degrade and recycle the cellular component. In cancer cells, to continue the survival process, autophagy degrades and recycles the macromolecules (e.g., glucose, proline, α-ketoglutarate, glutamine, pyruvate, lactate, nucleotide, and fatty acids) for the generation of essential biomolecules. However, under the stress condition, autophagy-mediated low metabolic activity maintains the dormancy nature or quiescence (G0/G1) state of lung cancer cells.Citation180
Autophagy also maintains the viability of circulatory and colonizing cancer cells and protects them from anti-cancer drugs. For instance, in response to chemotherapeutics, the ROS-mediated autophagy activates the survival pathway to protect abdominal cancer cells,Citation181 while inhibiting autophagic metabolism restores drug sensitivity in cervical cancer cells.Citation182 In GBM cells, starvation conditions induce autophagy-mediated quiescence and therapeutic resistance.Citation183 It reduces cancer cells’ proliferative nature and suppresses proliferative markers (e.g., Ki-67 and PCNA). However, the stem cell-like feature of cancer cells maintains the differentiation state by expressing stemness markers in the dormant head and neck cancer cells.Citation184 In addition to metabolic starvation, low PO2 also increases the expression of dormancy markers (e.g., NR2F1, p27, and DEC2) and arrest breast cancer cells in the G0/G1-phase.Citation185 During the dormant condition, cancer cells increase the lactate, α-ketoglutarate, citrate, pyruvate, and FAO to maintain anabolic biosynthesis and suppress the AKT-mTOR/MAPK mediated metabolic activity. Additionally, dormant cancer cells also increase their antioxidant molecules to stop ROS-mediated oxidative stress.Citation186
In a limited nutrition environment, autophagy maintains energy homeostasis and anabolic biosynthesis. Therapeutic inhibition of aerobic glycolysis under low cellular glucose induces apoptosis in cancer cells, but colorectal cancer cells shift ATP generation toward autophagy to protect starved cells from apoptosis.Citation187 In the TME, glucose shortage activates the energy sensor AMPK, followed by ROS-mediated autophagosome formation. During the ROS-mediated autophagosome formation, pancreatic cancer cells suppress the NADPH and GPx-1 generation.Citation188 Cellular ROS helps in autophagy induction, but autophagy itself creates the oxidative environment. Thus, to balance ROS and autophagy, breast cancer cells also increase Nrf2 or glutathione expression for cancer cells’ sustained growth under the metabolic stress condition.Citation189 Furthermore, low cellular ATP also stimulates the AMPK mediated autophagyCitation190 and increases the membrane translocation of GLUT1 to increase the glucose uptake.Citation191 However, for autophagy induction, metabolites also maintain the H+ ions for autolysosome formation. The TME acidification by aerobic glycolysis and lactate-mediated NADPH oxidation generates the H+ ions for autophagosome acidification.Citation192 The TME acidosis even arrests cells in G0/G1-phase and maintains dormancy in cancer cells.Citation193
Cellular starvation directly regulates autophagosome development. For instance, suppression of cellular glycolysis makes alveolar rhabdomyosarcoma cells sensitive to autophagy inhibitors.Citation194 Thus, under the glucose starvation, AMPK activates ULK1 for autophagy initiation, while in the nutrient-rich condition, mTOR inhibits the ULK1 mediated autophagy induction.Citation195 Additionally, during amino acid starvation, autophagy also maintains the amino acid homeostasis and increases the free glutamine to synthesize non-essential amino acids (e.g., glutamate, proline, and arginine). However, sufficient free glutamine restores mTOR, directs protein synthesis, and inhibits autophagy.Citation196 For instance, to maintain the amino acid homeostasis, TNBC cells increase the localization of amino acid transporter (SLC38A2), while the shortage of glutamine results in proteasomal degradation of SLC38A2 and subsequently increases ROS-mediated autophagy.Citation197
Glutaminolysis inhibition shifts cancer metabolism toward fatty acid autophagy, while suppression of FAO and autophagy increases lung and breast cancer cell apoptosis.Citation198 Additionally, deleting the autophagy-related 7 (ATG7) gene in lung cancer cells makes them sensitive to starvation and induces mitochondrial dysfunction by increased fatty acid accumulation.Citation199 Thus, fatty acid autophagy plays a crucial role in maintaining mitochondrial function and TCA homeostasis. Amino acid shortage activates AMPK mediated lipid droplet generation and inhibits the mTOR pathway.Citation200 Additionally, TME adipocytes also secrete fatty acids and stimulate AMPK mediated autophagy in starved cancer cells.Citation201
Autophagy also supports cancer cell proliferation during hypoxic condition. For instance, the hypoxic condition increases the expression of autophagy-related proteins such as AMPK, PERK, BNIP3, REDD1, and PML.Citation202 However, during low oxygen levels, both HIF isoforms (HIF-1α and HIF-2α) increase the survival autophagyCitation203 by activating AKT/ERK1/2 and suppress the mTOR signaling.Citation204 The HIF-1α mediated AMPK activation induces autophagy to maintain cell viability.Citation205 Furthermore, the hypoxic condition increases ER stress and affects protein folding. PERK senses the protein misfolding in such stress conditions and activates the unfolded protein response (UPR) mediated autophagy to regulate the protein misfolding.Citation206 Joshi et al. observed that cancer cells increase the ROS in hypoxic conditions and suppress the AKT/mTORC1 pathway to increase the autophagy survival cascade.Citation207
In breast cancer cells, hypoxia increases autophagy and supports adapting in the low metabolic and oxygen environment. The hypoxic environment maintains both quiescence and stem cell state (CD44+) to regulate breast cancer cells’ proliferative nature.Citation208 To maintain the dormancy, HIFs suppress proliferative PI3K/AKT signaling and reduce the metabolic activityCitation209 by inhibiting mTOR-mediated protein expression. They also regulate the expression of EGFR/MEK/ERK-mediated stress factors and MAPK expression to maintain the dormant stage.Citation210 Elevated HIF-1α expression also increases protein phosphatase-2A to maintain low proliferation in cancer cells, uplift the cancer recurrence and reduce the disease-free survival of GBM patients.Citation211 Additionally, TME ROS regulates glucose and FAO under hypoxia to establish AMPK mediated dormancy.Citation212 Of note, hypoxia also maintains the dormancy condition under the presence of chemotherapeutics. In response to anti-estrogen drugs, HIF-1α increases the ERK1/2 activation and suppresses the ER-α expression, which further arrests proliferating breast cancer cells into the G0/G1-phase.Citation213
Drug resistance and cancer recurrence
Despite several therapeutic options for cancer management, cancer incidences are increasing every year. Indeed, many clinical studies reported drug resistance development followed by cancer recurrence and reduced disease-free survival of cancer patients. In the TME, both altered metabolism and PO2 modulate the cancer environment and reduce chemotherapeutics’ presence and effectiveness. Additionally, cancer cells’ autophagy and stemness properties create a resistant environment and help in cancer recurrence. Furthermore, escape from the host immune system also plays a critical role in cancer recurrence; for instance, patients with high blood glucose are more susceptible to prostate cancer recurrence;Citation214 breast and colorectal cancer.Citation215 Similarly, Walenta et al. also correlated high lactate with cervical cancer recurrence.Citation216
Lactate from aerobic glycolysis maintains cancer cell viability, creates TME acidification, and induces resistance for PI3K/mTOR inhibitors in breast cancer cells.Citation217 For instance, the high glycolysis rate is also associated with cisplatin resistance in head and neck carcinoma, and a lower rate of glycolysis in cancer cells favor cisplatin therapy.Citation218 However, metabolic inhibitors are often used for effective cancer management, including anti-cancer drugs, but metabolic rewiring also protects cancer cells from metabolic inhibitors. For instance, the mTOR inhibitory drugs are used to regulate cancer metabolism, and in response to the mTOR inhibitory environment, GBM cells increase glutaminolysis followed by α-ketoglutarate mediated TCA cycle anaplerosis for cellular homeostasis.Citation219 Furthermore, glutamine deprivation sensitizes the cancer cells for chemotherapeutic drugs and induces apoptosis,Citation220 but in response to glutamine shortage, liver cancer cells up-lift the aerobic glycolysis and activate serine mediated ERK to develop resistance against kinase inhibitors.Citation221 Excess glutamine oxidation also increases the drug resistance against gemcitabine in pancreatic cancer cells, and glutaminolysis inhibition increases cancer cells’ sensitivity to gemcitabine.Citation222 Elevated glutamine metabolism maintains the cellular glutathione to protect pancreatic cancer cells from drug effects,Citation223 and in cisplatin-resistant lung cancer cells, it also supports the ATP and TCA cycle intermediates synthesis.Citation224 Similarly, paclitaxel-mediated glycolysis and OXPHOS inhibition induce apoptosis in lung cancer cells, whereas FAO maintains resistance cancer cells’ viabilityCitation225 and NADH homeostasis for the suppression of drug-induced ROS mediated apoptosis.Citation226 Additionally, elevated fatty acid transporter (GPR120) maintains an adequate amount of fatty acid in breast cancer cells and increases ABC transporters’ expression to reduce drug accumulation in neoadjuvant chemotherapy-treated patients’ .Citation227
Although, therapeutic resistance also increases the stem cell phenotype and self-renewal property of cancer cells and favors cancer cell regeneration. The previous section discussed the FAO’s role in maintaining cancer stem cells and NADPH mediated redox homeostasis. Thus, ROS homeostasis not only maintains the chemoresistance phenotype in hepatocellular cancer cells but also supports cancer recurrence.Citation228 Similarly, another study showed that purine synthesis in glioma cells maintains cancer stemness and self-renewal properties.Citation229 In leukemia cells, the high aerobic glycolysis PKM2 maintains the imatinib resistance;Citation230 however, dexamethasone inhibits PKM2 and disrupts drug resistance. In response to PKM2 inhibition, peroxisome proliferator-activated receptor-α (PPAR-α) increases FAO and restores cellular metabolism in the drug environment.Citation231 In addition, elevated FAS in cancer cells protects cancer from DNA damaging drugs (e.g., mitoxantrone, doxorubicin, etoposide, cisplatin, and camptothecin). Nevertheless, it fails to protect from destructive microtubule drugs (e.g., paclitaxel and vinblastine). High FAS suppresses the drug-induced pro-apoptotic factors (e.g., TNF-α, ceramide, and caspase-8),Citation232 and FAS inhibition suppresses the PI3K/AKT signaling, followed by cancer cell apoptosis. Additionally, FAO and oxidative stress were also found in regenerated tumors and favor breast cancer recurrence.Citation233
The angiogenesis inhibitors are often used for tumor angiogenesis suppression and create a hypoxic environment at the developing tumor site. In such type of drug-induced hypoxic environment, HIF-mediated metabolic reprogramming maintains the viability of cancer cells. Hypoxia-mediated aerobic glycolysis protects cancer cells from nintedanib effectsCitation234 and increases the expression of metabolic transporter (e.g., GLUT1 and MCT1/4) in sunitinib treated RCC cells.Citation235 However, TME acidification under hypoxia reduces anti-cancer drugs’ effectiveness.Citation236 Similarly, in colon cancer patients, HIF-1α mediated PDK3 expression induces the development of drug resistance and recurrence of colon cancer cells.Citation237 For Instance, Li et al. reported the hypoxic environment’s involvement in the multi-drug-resistance development in the hepatoma cells against 5-FU, sorafenib, gemcitabine, cisplatin, adriamycin, and 6-thioguanine. Furthermore, the DNA damaging drug (5-FU) reduces the expression of the 5-FU target (thymidylate synthase) and protects cancer cells from DNA damage.Citation238 Hypoxia-mediated pro-apoptotic factor (e.g., Bid/Bax) expression could be a possible way of induced drug resistance in cancer cells.Citation239 In addition to metabolic reprogramming, regulators of metabolism are also differentially expressed in cancer cells and regulate different steps of metastasis cascade ().
Table 1. Cancer metabolite rewiring regulator and their role in cancer metastasis
Conclusion
Our understanding of tumor development and metastasis has significantly improved over recent years; however, the role of oxygen and metabolic availability affecting the metastatic cascade programming is still not explicit. As a typical feature of solid tumors, oxygen and metabolic nutrients together add to metastatic cells’ survival process. For instance, cancer cell generally develops their TME to maintain the continuous metabolites and oxygen availability. The elevated metabolites at the TME promote the proliferation of cancer cells, while their shortage induces metabolite reprogramming or cancer cell metastasis toward metabolites-rich organs. However, it is not clear how the metabolites variation regulates cancer metastasis. Furthermore, both normoxia and hypoxia also facilitate the metabolites rewiring for cancer cell survivability; however, it is unclear how hypoxia drives tumor progression and warrants substantial investigation pertinent to the mechanism and implication for diagnosis and treatment. Of note, the TME oxygen and metabolites rewiring also regulate the EMT feature of cancer cells that promote cancer cell self-renewal ability and plasticity. Additionally, cancer cells’ autophagy and stemness properties create a resistant environment and facilitate cancer recurrence. Furthermore, tumor vasculogenesis and host immune evasion play a critical role in cancer development and recurrence, adding to the global disease burden. Overall, we have highlighted the importance of the oxygen-dependent/independent processes and different metabolic factors associated with major metastatic pathways responsible for tumor development, invasion, and recurrence of cancer.
Ethical approval
No ethical approval necessary for this review.
Acknowledgments
This work was supported by the Intramural Research Program of Indian Institute of Technology Roorkee (IITR), Roorkee, India (FIG100642; to KA). VSB, K.G. iare thankful to CSIR (India) for providing a senior research fellowship. DK is thankful to CSIR-CDRI and CSIR-AcSIR for providing facilities and UGC, India, for providing a senior research fellowship.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Giri K, Mehta A, Ambatipudi K. In search of the altering salivary proteome in metastatic breast and ovarian cancers. FASEB BioAdvances. 2019;1(3):191–207. doi:https://doi.org/10.1096/fba.2018-00029.
- Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15(6):827–837. doi:https://doi.org/10.1016/j.cmet.2012.05.001.
- Halldorsson S, Rohatgi N, Magnusdottir M, Choudhary KS, Gudjonsson T, Knutsen E, Barkovskaya A, Hilmarsdottir B, Perander M, Mælandsmo GM, et al. Metabolic re-wiring of isogenic breast epithelial cell lines following epithelial to mesenchymal transition. Cancer Lett. 2017;396:117–129. doi:https://doi.org/10.1016/j.canlet.2017.03.019.
- Schild T, Low V, Blenis J, Gomes AP. Unique metabolic adaptations dictate distal organ-specific metastatic colonization. Cancer Cell. 2018;33(3):347–354. doi:https://doi.org/10.1016/j.ccell.2018.02.001.
- Devignes CS, Aslan Y, Brenot A, Devillers A, Schepers K, Fabre S, Chou J, Casbon AJ, Werb Z, Provot S. HIF signaling in osteoblast-lineage cells promotes systemic breast cancer growth and metastasis in mice. Proc Natl Acad Sci USA. 2018;115(5):992–1001. doi:https://doi.org/10.1073/pnas.1718009115.
- Hoang‐Minh LB, Siebzehnrubl FA, Yang C, Suzuki‐Hatano S, Dajac K, Loche T, Andrews N, Schmoll Massari M, Patel J, Amin K, et al. Infiltrative and drug‐resistant slow‐cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018;37(23):e98772. doi:https://doi.org/10.15252/embj.201798772.
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. doi:https://doi.org/10.1016/j.cmet.2006.02.002.
- Soukupova J, Malfettone A, Hyroššová P, Hernández-Alvarez MI, Peñuelas-Haro I, Bertran E, Junza A, Capellades J, Giannelli G, Yanes O, et al. Role of the transforming growth factor-β in regulating hepatocellular carcinoma oxidative metabolism. Sci Rep. 2017;7(1):1–15. doi:https://doi.org/10.1038/s41598-017-12837-y.
- Celià-Terrassa T, Kang Y. Metastatic niche functions and therapeutic opportunities. Nat Cell Biol. 2018;20(8):868–877. doi:https://doi.org/10.1038/s41556-018-0145-9.
- Benjamin D, Robay D, Hindupur SK, Pohlmann J, Colombi M, El-Shemerly MY, Maira SM, Moroni C, Lane HA, Hall MN. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018;25(11):3047–3058. doi:https://doi.org/10.1016/j.celrep.2018.11.043.
- Hong CS, Graham NA, Gu W, Espindola Camacho C, Mah V, Maresh EL, Alavi M, Bagryanova L, Krotee PAL, Gardner BK, et al. MCT1 modulates cancer cell pyruvate export and growth of tumors that co-express MCT1 and MCT4. Cell Rep. 2016;14(7):1590–1601. doi:https://doi.org/10.1016/j.celrep.2016.01.057.
- Fornetti J, Welm AL, Stewart SA. Understanding the bone in cancer metastasis. J Bone Miner Res. 2018;33(12):2099–2113. doi:https://doi.org/10.1002/jbmr.3618.
- Meyer KA, Neeley CK, Baker NA, Washabaugh AR, Flesher CG, Nelson BS, Frankel TL, Lumeng CN, Lyssiotis CA, Wynn ML, et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem Biophys Reports. 2016;7:144–149. doi:https://doi.org/10.1016/j.bbrep.2016.06.004.
- Grzegorczyk EA, Harasim-Symbor E, Lukaszuk B, Harasiuk D, Choromanska B, Mysliwiec P, Zendzian-Piotrowska M, Chabowski A. Lack of pronounced changes in the expression of fatty acid handling proteins in adipose tissue and plasma of morbidly obese humans. Nutr Diabetes. 2018;8(1):3. doi:https://doi.org/10.1038/s41387-017-0013-x.
- Hunter KW, Crawford NP, Alsarraj J. Mechanisms of metastasis. Breast Cancer Res. 2008;10(S1):1–10. doi:https://doi.org/10.1186/bcr1988.
- Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, Maxwell R, Stoll EA. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2017;19(1):43–54. doi:https://doi.org/10.1093/neuonc/now128.
- Weverwijk A, Koundouros N, Iravani M, Ashenden M, Gao Q, Poulogiannis G, Jungwirth U, and Isacke CM. Metabolic adaptability in metastatic breast cancer by AKR1B10-dependent balancing of glycolysis and fatty acid oxidation. Nat Commun. 2019;10:2698.
- Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, Yeh CC, Peng YJ, Kuo YY, Wen HT, et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. 2020;77:213–227. doi:https://doi.org/10.1016/j.molcel.2019.10.023.
- Kes MMG, Van den Bossche J, Griffioen AW, Huijbers EJM. Oncometabolites lactate and succinate drive pro-angiogenic macrophage response in tumors. Biochim Biophys Acta-Rev Cancer. 2020;1874(2):188427. doi:https://doi.org/10.1016/j.bbcan.2020.188427.
- Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550–2560. doi:https://doi.org/10.1158/0008-5472.CAN-10-2828.
- Lin S, Sun L, Lyu X, Ai X, Du D, Su N, Li H, Zhang L, Yu J, Yuan S. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: a positive metabolic feedback loop. Oncotarget. 2017;8(66):110426–110443. doi:https://doi.org/10.18632/oncotarget.22786.
- Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, Aranda Lopez P, Hahlbrock J, Muth S, Endo S, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. 2018;19(12):1319–1329. doi:https://doi.org/10.1038/s41590-018-0226-8.
- Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, Yang J, Pan J, Hu S, Zhang C, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. 2018;17:428–438.
- Stegen S, Laperre K, Eelen G, Rinaldi G, Fraisl P, Torrekens S, Van Looveren R, Loopmans S, Bultynck G, Vinckier S, et al. HIF-1α metabolically controls collagen synthesis and modification in chondrocytes. Nature. 2019;565(7740):511–515. doi:https://doi.org/10.1038/s41586-019-0874-3.
- Mah EJ, Lefebvre AEYT, McGahey GE, Yee AF, Digman MA. Collagen density modulates triple-negative breast cancer cell metabolism through adhesion-mediated contractility. Sci Rep. 2018;8(1):1–11. doi:https://doi.org/10.1038/s41598-018-35381-9.
- Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, Chow A, O’Connor STF, Li S, Chin AR, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17:183–194. doi:https://doi.org/10.1038/ncb3094.
- Sullivan WJ, Mullen PJ, Schmid EW, Flores A, Momcilovic M, Sharpley MS, Jelinek D, Whiteley AE, Maxwell MB, Wilde BR, et al. Extracellular matrix remodeling regulates glucose metabolism through TXNIP destabilization. Cell. 2018;175:117–132.
- Sai B, Dai Y, Fan S, Wang F, Wang L, Li Z, Tang J, Wang L, Zhang X, Zheng L, et al. Cancer-educated mesenchymal stem cells promote the survival of cancer cells at primary and distant metastatic sites via the expansion of bone marrow-derived-PMN-MDSCs. Cell Death Dis. 2019;10(12):1–13. doi:https://doi.org/10.1038/s41419-019-2149-1.
- Fujita S, Sumi M, Tatsukawa E, Nagano K, Katase N. Expressions of extracellular matrix‐remodeling factors in lymph nodes from oral cancer patients. Oral Dis. 2020;26:1424–1431. doi:https://doi.org/10.1111/odi.13419.
- Reczek CR, Chandel NS. ROS promotes cancer cell survival through calcium signaling. Cancer Cell. 2018;33(6):949–951. doi:https://doi.org/10.1016/j.ccell.2018.05.010.
- Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014;8(3):754–766. doi:https://doi.org/10.1016/j.celrep.2014.06.043.
- Costanza B, Rademaker G, Tiamiou A, De Tullio P, Leenders J, Blomme A, Bellier J, Bianchi E, Turtoi A, Delvenne P, et al. Transforming growth factor beta‐induced, an extracellular matrix interacting protein, enhances glycolysis and promotes pancreatic cancer cell migration. Int J Cancer. 2019;145(6):1570–1584. doi:https://doi.org/10.1002/ijc.32247.
- Qi C, Zhang J, Chen X, Wan J, Wang J, Zhang P, Liu Y, Morotti M, Zois CE, El-Ansari R, et al. Hypoxia stimulates neural stem cell proliferation by increasing HIF-1α expression and activating Wnt/β-catenin signaling. Cell Mol Biol. 2017;63(7):12–19. doi:https://doi.org/10.14715/cmb/2017.63.7.2.
- Zhao J, Du F, Shen G, Zheng F, Xu B. The role of hypoxia-inducible factor-2 in digestive system cancers. Cell Death Dis. 2015;6(1):e1600. doi:https://doi.org/10.1038/cddis.2014.565.
- Zhang Q, Wang S-Y, Nottke AC, Rocheleau JV, Piston DW, Goodman RH. Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc Natl Acad Sci. 2006;103(24):9029–9033.
- Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, Boggon TJ, Jin P, Yi H, Wright ER, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 2015;27(2):257–270. doi:https://doi.org/10.1016/j.ccell.2014.12.006.
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159(7):1603–1614. doi:https://doi.org/10.1016/j.cell.2014.11.025.
- Lakhter AJ, Hamilton J, Konger RL, Brustovetsky N, Broxmeyer HE, Naidu SR. Glucose-independent acetate metabolism promotes melanoma cell survival and tumor growth. J Biol Chem. 2016;291(42):21869–21879. doi:https://doi.org/10.1074/jbc.M115.712166.
- Björnson E, Mukhopadhyay B, Asplund A, Pristovsek N, Cinar R, Romeo S, Uhlen M, Kunos G, Nielsen J, Mardinoglu A. Stratification of hepatocellular carcinoma patients based on acetate utilization. Cell Rep. 2015;13(9):2014–2026. doi:https://doi.org/10.1016/j.celrep.2015.10.045.
- Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY, et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell. 2013;23(4):464–476. doi:https://doi.org/10.1016/j.ccr.2013.02.005.
- Xian ZY, Liu JM, Chen QK, Chen HZ, Ye CJ, Xue J, Yang HQ, Li JL, Liu XF, Kuang SJ. Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumor Biol. 2015;36(10):8093–8100. doi:https://doi.org/10.1007/s13277-015-3540-x.
- Arseneault R, Chien A, Newington JT, Rappon T, Harris R, Cumming RC. Attenuation of LDHA expression in cancer cells leads to redox-dependent alterations in cytoskeletal structure and cell migration. Cancer Lett. 2013;338(2):255–266. doi:https://doi.org/10.1016/j.canlet.2013.03.034.
- Guddeti RK, Bali P, Karyala P, Pakala SB. MTA1 coregulator regulates LDHA expression and function in breast cancer. Biochem Biophys Res Commun. 2019;520(1):54–59. doi:https://doi.org/10.1016/j.bbrc.2019.09.078.
- Huang X, Li X, Xie X, Ye F, Chen B, Song C, Tang H, Xie X. High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. Breast. 2016;30:39–46. doi:https://doi.org/10.1016/j.breast.2016.08.014.
- Rellinger EJ, Craig BT, Alvarez AL, Dusek HL, Kim KW, Qiao J, Chung DH. FX11 inhibits aerobic glycolysis and growth of neuroblastoma cells. Surgery. 2017;161(3):747–752. doi:https://doi.org/10.1016/j.surg.2016.09.009.
- Gao S, Tu D-N, Li H, Jiang J-X, Cao X, You J-B, Zhou X-Q. Pharmacological or genetic inhibition of LDHA reverses tumor progression of pediatric osteosarcoma. Biomed Pharmacother. 2016;81:388–393. doi:https://doi.org/10.1016/j.biopha.2016.04.029.
- Dupuy F, Tabariès S, Andrzejewski S, Dong Z, Blagih J, Annis MG, Omeroglu A, Gao D, Leung S, Amir E, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015;22(4):577–589. doi:https://doi.org/10.1016/j.cmet.2015.08.007.
- Chae YC, Vaira V, Caino MC, Tang HY, Seo JH, Kossenkov AV, Ottobrini L, Martelli C, Lucignani G, Bertolini I, et al. Mitochondrial Akt regulation of hypoxic tumor reprogramming. Cancer Cell. 2016;30(2):257–272. doi:https://doi.org/10.1016/j.ccell.2016.07.004.
- Kinnaird A, Dromparis P, Saleme B, Gurtu V, Watson K, Paulin R, Zervopoulos S, Stenson T, Sutendra G, Pink DB, et al. Metabolic modulation of clear-cell renal cell carcinoma with dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase. Eur Urol. 2016;69(4):734–744. doi:https://doi.org/10.1016/j.eururo.2015.09.014.
- Schell JC, Olson KA, Jiang L, Hawkins AJ, VanVranken JG, Xie J, Egnatchik RA, Earl EG, DeBerardinis RJ, Rutter J. A role for the mitochondrial pyruvate carrier as a repressor of the warburg effect and colon cancer cell growth. Mol Cell. 2014;56(3):400–413. doi:https://doi.org/10.1016/j.molcel.2014.09.026.
- Fan J, Lin R, Xia S, Chen D, Elf SE, Liu S, Pan Y, Xu H, Qian Z, Wang M, et al. Tetrameric acetyl-CoA acetyltransferase 1 is important for tumor growth. Mol Cell. 2016;64(5):859–874. doi:https://doi.org/10.1016/j.molcel.2016.10.014.
- Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337(6097):975–980. doi:https://doi.org/10.1126/science.1222278.
- Lincet H, Icard P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene. 2015;34(29):3751–3759. doi:https://doi.org/10.1038/onc.2014.320.
- De Berardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi:https://doi.org/10.1126/sciadv.1600200.
- Wang YP, Zhou W, Wang J, Huang X, Zuo Y, Wang TS, Gao X, Xu YY, Zou SW, Bin LY, et al. Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol Cell. 2016;64:673–687. doi:https://doi.org/10.1016/j.molcel.2016.09.028.
- Gameiro PA, Yang J, Metelo AM, Pérez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, López-Larrubia P, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17(3):372–385. doi:https://doi.org/10.1016/j.cmet.2013.02.002.
- Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36(10):1302–1315. doi:https://doi.org/10.15252/embj.201696151.
- Louie SM, Roberts LS, Mulvihill MM, Luo K, Nomura DK. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim Biophys Acta-Mol Cell Biol Lipids. 2013;1831(10):1566–1572. doi:https://doi.org/10.1016/j.bbalip.2013.07.008.
- Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S, Mitsche M, Yang C, Batten K, Huffman KE, Liu J, et al. Fatty acid oxidation mediated by acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 2016;16:1614–1628. doi:https://doi.org/10.1016/j.celrep.2016.07.009.
- Miranda F, Mannion D, Liu S, Zheng Y, Mangala LS, Redondo C, Herrero-Gonzalez S, Xu R, Taylor C, Chedom DF, et al. Salt-inducible kinase 2 couples ovarian cancer cell metabolism with survival at the adipocyte-rich metastatic niche. Cancer Cell. 2016;30(2):273–289. doi:https://doi.org/10.1016/j.ccell.2016.06.020.
- Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, Bhat VB, Bhowmik SK, Gupta V, Arora K, et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016;14(9):2154–2165. doi:https://doi.org/10.1016/j.celrep.2016.02.004.
- Serna-Marquez N, Diaz-Aragon R, Reyes-Uribe E, Cortes-Reynosa P, Salazar EP. Linoleic acid induces migration and invasion through FFAR4- and PI3K-/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Med Oncol. 2017;34(6):111. doi:https://doi.org/10.1007/s12032-017-0969-3.
- Xiang F, Wu K, Liu Y, Shi L, Wang D, Li G, Tao K, Wang G. Omental adipocytes enhance the invasiveness of gastric cancer cells by oleic acid-induced activation of the PI3K-Akt signaling pathway. Int J Biochem Cell Biol. 2017;84:14–21. doi:https://doi.org/10.1016/j.biocel.2016.12.002.
- Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, Papagiannakopoulos T, Molina H, Snuderl M, Lewis CA, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. 2018;20(7):775–781. doi:https://doi.org/10.1038/s41556-018-0118-z.
- Li Z, Wu Q, Sun S, Wu J, Li J, Zhang Y, Wang C, Yuan J, Sun S. Monocarboxylate transporters in breast cancer and adipose tissue are novel biomarkers and potential therapeutic targets. Biochem Biophys Res Commun. 2018;501(4):962–967. doi:https://doi.org/10.1016/j.bbrc.2018.05.091.
- Cao YW, Liu Y, Dong Z, Guo L, Kang EH, Wang YH, Zhang W, Niu HT. Monocarboxylate transporters MCT1 and MCT4 are independent prognostic biomarkers for the survival of patients with clear cell renal cell carcinoma and those receiving therapy targeting angiogenesis. Urol Oncol Semin Orig Investig. 2018;36:311.
- Amorim R, Pinheiro C, Miranda-Gonçalves V, Pereira H, Moyer MP, Preto A, Baltazar F. Monocarboxylate transport inhibition potentiates the cytotoxic effect of 5-fluorouracil in colorectal cancer cells. Cancer Lett. 2015;365(1):68–78. doi:https://doi.org/10.1016/j.canlet.2015.05.015.
- Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, Graeber TG, Parlati F, Demo S, Li R, et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell. 2018;33(5):905–921. doi:https://doi.org/10.1016/j.ccell.2018.04.002.
- Adebayo Michael AO, Ko S, Tao J, Moghe A, Yang H, Xu M, Russell JO, Pradhan-Sundd T, Liu S, Singh S, et al. Inhibiting glutamine-dependent mTORC1 activation ameliorates liver cancers driven by β-catenin mutations. Cell Metab. 2019;29(5):1135–1150. doi:https://doi.org/10.1016/j.cmet.2019.01.002.
- Gwinn DM, Lee AG, Briones-Martin-del-Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D, Sweet-Cordero EA. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell. 2018;33(1):91–107. doi:https://doi.org/10.1016/j.ccell.2017.12.003.
- Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, Ikegami S, Masui K, Gini B, Yang H, et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell. 2017;67(1):128–138. doi:https://doi.org/10.1016/j.molcel.2017.05.030.
- Zhang J, Liang Q, Lei Y, Yao M, Li L, Gao X, Feng J, Zhang Y, Gao H, Liu DX, et al. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res. 2012;72(17):4597–4608. doi:https://doi.org/10.1158/0008-5472.CAN-12-1045.
- Singh AM, Reynolds D, Cliff T, Ohtsuka S, Mattheyses AL, Sun Y, Menendez L, Kulik M, Dalton S. Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell. 2012;10(3):312–326. doi:https://doi.org/10.1016/j.stem.2012.01.014.
- Florian MC, Geiger H. Tissue-specific stem cells concise review: polarity in stem cells, disease, and aging. Stem Cells. 2010;28(9):1623–1629. doi:https://doi.org/10.1002/stem.481.
- Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer. 2011;12:23–38. doi:https://doi.org/10.1038/nrc3169.
- Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, Turgeon MO, Fish L, Erard N, Gable AL, et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature. 2018;554(7692):378–381. doi:https://doi.org/10.1038/nature25465.
- Comes S, Gagliardi M, Laprano N, Fico A, Cimmino A, Palamidessi A, De Cesare D, De Falco S, Angelini C, Scita G, et al. L-proline induces a mesenchymal-like invasive program in embryonic stem cells by remodeling H3K9 and H3K36 methylation. Stem Cell Reports. 2013;1(4):307–321. doi:https://doi.org/10.1016/j.stemcr.2013.09.001.
- Grassian AR, Lin F, Barrett R, Liu Y, Jiang W, Korpal M, Astley H, Gitterman D, Henley T, Howes R, et al. Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition (EMT). J Biol Chem. 2012;287(50):42180–42194. doi:https://doi.org/10.1074/jbc.M112.417832.
- Yalcin A, Solakoglu TH, Ozcan SC, Guzel S, Peker S, Celikler S, Balaban BD, Sevinc E, Gurpinar Y, Chesney JA. 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for transforming growth factor β1-enhanced invasion of Panc1 cells in vitro. Biochem Biophys Res Commun. 2017;484(3):687–693. doi:https://doi.org/10.1016/j.bbrc.2017.01.178.
- Jung M, Kang J, Hernandez DM, Yin X, Andrianifahanana M, Wang Y, Gonzalez‐Guerrico A, Limper AH, Leof EB. Fatty acid synthase is required for profibrotic TGF‐β signaling. FASEB J. 2018;32(7):3803–3815. doi:https://doi.org/10.1096/fj.201701187R.
- Huang L, Zhang C, Su L, Song Z. GSK3β attenuates TGF-β1 induced epithelial–mesenchymal transition and metabolic alterations in ARPE-19 cells. Biochem Biophys Res Commun. 2017;486(3):744–751. doi:https://doi.org/10.1016/j.bbrc.2017.03.113.
- Kishore C, Sundaram S, Karunagaran D. Vitamin K3 (menadione) suppresses epithelial-mesenchymal-transition and Wnt signaling pathway in human colorectal cancer cells. Chem Biol Interact. 2019;309:108725. doi:https://doi.org/10.1016/j.cbi.2019.108725.
- Jarc E, Kump A, Malavašič P, Eichmann TO, Zimmermann R, Petan T. Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim Biophys Acta-Mol Cell Biol Lipids. 2018;1863(3):247–265. doi:https://doi.org/10.1016/j.bbalip.2017.12.006.
- Choi SG, Yoo YJ, Kim H, Lee H, Chung H, Nam MH, Moon JY, Lee HS, Yoon S, Kim WY. Clinical and biochemical relevance of monounsaturated fatty acid metabolism targeting strategy for cancer stem cell elimination in colon cancer. Biochem Biophys Res Commun. 2019;519(1):100–105. doi:https://doi.org/10.1016/j.bbrc.2019.08.137.
- Pinkham K, Park DJ, Hashemiaghdam A, Kirov AB, Adam I, Rosiak K, Da Hora CC, Teng J, Cheah PS, Carvalho L, et al. Stearoyl CoA desaturase is essential for regulation of endoplasmic reticulum homeostasis and tumor growth in glioblastoma cancer stem cells. Stem Cell Reports. 2019;12(4):712–727. doi:https://doi.org/10.1016/j.stemcr.2019.02.012.
- Giudetti AM, De Domenico S, Ragusa A, Lunetti P, Gaballo A, Franck J, Simeone P, Nicolardi G, De Nuccio F, Santino A, et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim Biophys Acta-Mol Cell Biol Lipids. 2019;1864(3):344–357. doi:https://doi.org/10.1016/j.bbalip.2018.12.011.
- Wright HJ, Hou J, Xu B, Cortez M, Potma EO, Tromberg BJ, Razorenova OV. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc Natl Acad Sci USA. 2017;114(32):E6556–65. doi:https://doi.org/10.1073/pnas.1703791114.
- Wang Y, Guo D, He J, Song L, Chen H, Zhang Z, Tan N. Inhibition of fatty acid synthesis arrests colorectal neoplasm growth and metastasis: anti-cancer therapeutical effects of natural cyclopeptide RA-XII. Biochem Biophys Res Commun. 2019;512(4):819–824. doi:https://doi.org/10.1016/j.bbrc.2019.03.088.
- Rios Garcia M, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A, Christian S, Hess-Stumpp H, Augustin HG, Müller-Decker K, et al. Acetyl-CoA carboxylase 1-dependent protein acetylation controls breast cancer metastasis and recurrence. Cell Metab. 2017;26(6):842–855. doi:https://doi.org/10.1016/j.cmet.2017.09.018.
- Lu M, Zhu WW, Wang X, Tang JJ, Zhang KL, Yu GY, Shao WQ, Lin ZF, Wang SH, Lu L, et al. ACOT12-dependent alteration of acetyl-CoA drives hepatocellular carcinoma metastasis by epigenetic induction of epithelial-mesenchymal transition. Cell Metab. 2019;29(4):886–900. doi:https://doi.org/10.1016/j.cmet.2018.12.019.
- Yang D, Peng M, Hou Y, Qin Y, Wan X, Zhu P, Liu S, Yang L, Zeng H, Jin T, et al. Oxidized ATM promotes breast cancer stem cell enrichment through energy metabolism reprogram-mediated acetyl-CoA accumulation. Cell Death Dis. 2020;11:1–17.
- Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, Kitanaka C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget. 2015;6(2):651–661. doi:https://doi.org/10.18632/oncotarget.2892.
- Cliff TS, Wu T, Boward BR, Yin A, Yin H, Glushka JN, Prestegaard JH, Dalton S. MYC controls human pluripotent stem cell fate decisions through regulation of metabolic flux. Cell Stem Cell. 2017;21(4):502–516. doi:https://doi.org/10.1016/j.stem.2017.08.018.
- Liu M, Quek L-E, Sultani G, Turner N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. 2016;4(1):19. doi:https://doi.org/10.1186/s40170-016-0160-x.
- Liu M, Hancock SE, Sultani G, Wilkins BP, Ding E, Osborne B, Quek L-E, Turner N. Snail-overexpression induces epithelial-mesenchymal transition and metabolic reprogramming in human pancreatic ductal adenocarcinoma and non-tumorigenic ductal cells. J Clin Med. 2019;8(6):822. doi:https://doi.org/10.3390/jcm8060822.
- Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, McLendon R, Lindner D, Sloan A, Rich JN. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011;18(5):829–840. doi:https://doi.org/10.1038/cdd.2010.150.
- Panopoulos AD, Ruiz S, Yi F, Herrerías A, Batchelder EM, Belmonte JCI. Rapid and highly efficient generation of induced pluripotent stem cells from human umbilical vein endothelial cells. PLoS One. 2011;6(5):e19743. doi:https://doi.org/10.1371/journal.pone.0019743.
- Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, Flores A, Mohlman J, Sorensen LK, Earl CS, et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat Cell Biol. 2017;19(9):1027–1036. doi:https://doi.org/10.1038/ncb3593.
- Ohashi T, Eguchj H, Kawamoto K, Konno M, Asat A, Colvin H, Ueda Y, Takaoka H, Iwagami Y, Yamada D, et al. Mitochondrial pyruvate carrier modulates the epithelial-mesenchymal transition in cholangiocarcinoma. Oncol Rep. 2018;39:1276–1282.
- Takaoka Y, Konno M, Koseki J, Colvin H, Asai A, Tamari K, Satoh T, Mori M, Doki Y, Ogawa K, et al. Mitochondrial pyruvate carrier 1 expression controls cancer epithelial‐mesenchymal transition and radioresistance. Cancer Sci. 2019;110(4):1331–1339. doi:https://doi.org/10.1111/cas.13980.
- Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA, Anantharaman A, Butera A, Leiby B, Cognetti D, Sotgia F, Lisanti MP, et al. Cancer metabolism, stemness and tumor recurrence. Cell Cycle. 2013;12(9):1371–1384. doi:https://doi.org/10.4161/cc.24092.
- Li X, Han G, Li X, Kan Q, Fan Z, Li Y, Ji Y, Zhao J, Zhang M, Grigalavicius M, et al. Mitochondrial pyruvate carrier function determines cell stemness and metabolic reprogramming in cancer cells. Oncotarget. 2017;8(28):46363–46380. doi:https://doi.org/10.18632/oncotarget.18199.
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12(1):49–61. doi:https://doi.org/10.1016/j.stem.2012.10.011.
- Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang C, Xie S, Chen C, Hu L, Xu S, et al. Wnt/β-catenin signaling enhances hypoxia-induced epithelial–mesenchymal transition in hepatocellular carcinoma via crosstalk with HIF-1α signaling. Carcinogenesis. 2013;34(5):962–973. doi:https://doi.org/10.1093/carcin/bgt027.
- Xiong Y, Liu Y, Xiong W, Zhang L, Liu H, Du Y, Li N. Hypoxia-inducible factor 1α-induced epithelial-mesenchymal transition of endometrial epithelial cells may contribute to the development of endometriosis. Hum Reprod. 2016;31(6):1327–1338. doi:https://doi.org/10.1093/humrep/dew081.
- Liu J, Wu Q, Wang Y, Wei Y, Wu H, Duan L, Zhang Q, Wu Y. Ovol2 induces mesenchymal–epithelial transition via targeting ZEB1 in osteosarcoma. Onco Targets Ther. 2018;11:2963–2973. doi:https://doi.org/10.2147/OTT.S157119.
- Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Wu H, Kornblum HI. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. 2011;8(1):59–71. doi:https://doi.org/10.1016/j.stem.2010.11.028.
- Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Aboody KS. SLC7A11 overexpression in glioblastoma is associated with increased cancer stem cell-like properties. Stem Cells Dev. 2017;26(17):1236–1246. doi:https://doi.org/10.1089/scd.2017.0123.
- He T, Guan X, Wang S, Xiao T, Yang K, Xu X, Wang J, Zhao J. Resveratrol prevents high glucose-induced epithelial-mesenchymal transition in renal tubular epithelial cells by inhibiting NADPH oxidase/ROS/ERK pathway. Mol Cell Endocrinol. 2015;402:13–20. doi:https://doi.org/10.1016/j.mce.2014.12.010.
- Wenes M, Shang M, Di Matteo M, Goveia J, Martín-Pérez R, Serneels J, Prenen H, Ghesquière B, Carmeliet P, Mazzone M. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 2016;24(5):701–715. doi:https://doi.org/10.1016/j.cmet.2016.09.008.
- Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen LA, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell. 2016;30(6):968–985. doi:https://doi.org/10.1016/j.ccell.2016.10.006.
- Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, Biswas SK, Murdoch C, Plate KH, Reiss Y, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010;70(13):5270–5280. doi:https://doi.org/10.1158/0008-5472.CAN-10-0012.
- Kugeratski FG, Atkinson SJ, Neilson LJ, Lilla S, Knight JRP, Serneels J, Juin A, Ismail S, Bryant DM, Markert EK, et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci Signal. 2019;12(567):eaan8247. doi:https://doi.org/10.1126/scisignal.aan8247.
- Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci USA. 2007;104(51):20262–20267. doi:https://doi.org/10.1073/pnas.0706438104.
- Sonveaux P, Copetti T, De Saedeleer CJ, Végran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frérart F, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One. 2012;7(3):e33418. doi:https://doi.org/10.1371/journal.pone.0033418.
- Morland C, Andersson KA, Haugen ØP, Hadzic A, Kleppa L, Gille A, Rinholm JE, Palibrk V, Diget EH, Kennedy LH, et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat Commun. 2017;8(1):15557. doi:https://doi.org/10.1038/ncomms15557.
- Teuwen LA, Geldhof V, Carmeliet P. How glucose, glutamine and fatty acid metabolism shape blood and lymph vessel development. Dev Biol. 2019;447(1):90–102. doi:https://doi.org/10.1016/j.ydbio.2017.12.001.
- Vanetti C, Bifari F, Vicentini LM, Cattaneo MG, Bolego C. Fatty acids rather than hormones restore in vitro angiogenesis in human male and female endothelial cells cultured in charcoal-stripped serum. PLoS One. 2017;12(12):e0189528. doi:https://doi.org/10.1371/journal.pone.0189528.
- Dobson DE, Kambe A, Block E, Dion T, Lu H, Castellot JJ, Spiegelman BM. 1-Butyryl-Glycerol: a novel angiogenesis factor secreted by differentiating adipocytes. Cell. 1990;61(2):223–230. doi:https://doi.org/10.1016/0092-8674(90)90803-M.
- Kimura T, Watanabe T, Sato K, Kon J, Tomura H, Tamama K, Kuwabara A, Kanda T, Kobayashi I, Ohta H, et al. Sphingosine 1-phosphate stimulates proliferation and migration of human endothelial cells possibly through the lipid receptors, Edg-1 and Edg-3. Biochem J. 2000;348(1):71–76. doi:https://doi.org/10.1042/bj3480071.
- Samson FP, He W, Sripathi SR, Patrick AT, Madu J, Chung H, Frost MC, Jee D, Gutsaeva DR, Jahng WJ. Dual switch mechanism of erythropoietin as an antiapoptotic and pro-angiogenic determinant in the retina. ACS Omega. 2020;5(33):21113–21126. doi:https://doi.org/10.1021/acsomega.0c02763.
- Joyal JS, Sun Y, Gantner ML, Shao Z, Evans LP, Saba N, Fredrick T, Burnim S, Kim JS, Patel G, et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat Med. 2016;22(4):439–445. doi:https://doi.org/10.1038/nm.4059.
- Peyton KJ, Liu X, Yu Y, Yates B, Behnammanesh G, Durante W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem Pharmacol. 2018;156:204–214. doi:https://doi.org/10.1016/j.bcp.2018.08.032.
- Eubank TD, Roda JM, Liu H, O’Neil T, Marsh CB. Opposing roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood. 2011;117(1):323–332. doi:https://doi.org/10.1182/blood-2010-01-261792.
- Kuwabara K, Ogawa S, Matsumoto M, Koga S, Clauss M, Pinsky DJ, Lyn P, Leavy J, Witte L, Joseph-Silverstein J, et al. Hypoxia-mediated induction of acidic/basic fibroblast growth factor and platelet-derived growth factor in mononuclear phagocytes stimulates growth of hypoxic endothelial cells. Proc Natl Acad Sci USA. 1995;92(10):4606–4610. doi:https://doi.org/10.1073/pnas.92.10.4606.
- Curtarello M, Zulato E, Nardo G, Valtorta S, Guzzo G, Rossi E, Esposito G, Msaki A, Pastò A, Rasola A, et al. VEGF-targeted therapy stably modulates the glycolytic phenotype of tumor cells. Cancer Res. 2015;75(1):120–133. doi:https://doi.org/10.1158/0008-5472.CAN-13-2037.
- Li Y, Liu Y, Wang C, Xia WR, Zheng JY, Yang J, Liu B, Liu JQ, Liu LF. Succinate induces synovial angiogenesis in rheumatoid arthritis through metabolic remodeling and HIF-1α/VEGF axis. Free Radic Biol Med. 2018;126:1–14. doi:https://doi.org/10.1016/j.freeradbiomed.2018.07.009.
- Zabel DD, Feng JJ, Scheuenstuhl H, Hunt TK, Hussain MZ. Lactate stimulation of macrophage-derived angiogenic activity is associated with inhibition of poly(ADP-ribose) synthesis. Lab Investig. 1996;74:644–649.
- Giatromanolaki A, Koukourakis MI, Sivridis E, Turley H, Talks K, Pezzella F, Gatter KC, Harris AL. Relation of hypoxia inducible factor 1α and 2α in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br J Cancer. 2001;85(6):881–890. doi:https://doi.org/10.1054/bjoc.2001.2018.
- Giatromanolaki A, Sivridis E, Simopoulos C, Polychronidis A, Gatter KC, Harris AL, Koukourakis MI. Hypoxia inducible factors 1α and 2α are associated with VEGF expression and angiogenesis in gallbladder carcinomas. J Surg Oncol. 2006;94(3):242–247. doi:https://doi.org/10.1002/jso.20443.
- Helczynska K, Larsson AM, Mengelbier LH, Bridges E, Fredlund E, Borgquist S, Landberg G, Påhlman S, Jirström K. Hypoxia-inducible factor-2α correlates to distant recurrence and poor outcome in invasive breast cancer. Cancer Res. 2008;68(22):9212–9220. doi:https://doi.org/10.1158/0008-5472.CAN-08-1135.
- Hahne M, Schumann P, Mursell M, Strehl C, Hoff P, Buttgereit F, Gaber T. Unraveling the role of hypoxia-inducible factor (HIF)-1α and HIF-2α in the adaption process of human microvascular endothelial cells (HMEC-1) to hypoxia: redundant HIF-dependent regulation of macrophage migration inhibitory factor. Microvasc Res. 2018;116:34–44. doi:https://doi.org/10.1016/j.mvr.2017.09.004.
- Sui H, Zhao J, Zhou L, Wen H, Deng W, Li C, Ji Q, Liu X, Feng Y, Chai N, et al. Tanshinone IIA inhibits β-catenin/VEGF-mediated angiogenesis by targeting TGF-β1 in normoxic and HIF-1α in hypoxic microenvironments in human colorectal cancer. Cancer Lett. 2017;403:86–97. doi:https://doi.org/10.1016/j.canlet.2017.05.013.
- Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer. 2014;14(3):159–172. doi:https://doi.org/10.1038/nrc3677.
- Feng Y, Wang W, Hu J, Ma J, Zhang Y, Zhang J. Expression of VEGF-C and VEGF-D as significant markers for assessment of lymphangiogenesis and lymph node metastasis in non-small cell lung cancer. Anat Rec Adv Integr Anat Evol Biol. 2010;293(5):802–812. doi:https://doi.org/10.1002/ar.21096.
- Lee E, Koskimaki JE, Pandey NB, Popel AS. Inhibition of lymphangiogenesis and angiogenesis in breast tumor xenografts and lymph nodes by a peptide derived from transmembrane protein 45A. Neoplasia. 2013;15(2):112–124. doi:https://doi.org/10.1593/neo.121638.
- Xu J, Zhang C, He Y, Wu H, Wang Z, Song W, Li W, He W, Cai S, Zhan W. Lymphatic endothelial cell-secreted CXCL1 stimulates lymphangiogenesis and metastasis of gastric cancer. Int J Cancer. 2012;130(4):787–797. doi:https://doi.org/10.1002/ijc.26035.
- Zhang B, Wang J, Gao J, Guo Y, Chen X, Wang B, Gao J, Rao Z, Chen Z. Alternatively activated RAW264.7 macrophages enhance tumor lymphangiogenesis in mouse lung adenocarcinoma. J Cell Biochem. 2009;107(1):134–143. doi:https://doi.org/10.1002/jcb.22110.
- Ju H-Q, Lin J-F, Tian T, Xie D, Xu R-H. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduct Target Ther. 2020;5(1):1–12. doi:https://doi.org/10.1038/s41392-020-00326-0.
- Acevedo-Acevedo S, Millar DC, Simmons AD, Favreau P, Cobra PF, Skala M, Palecek SP. Metabolomics revealed the influence of breast cancer on lymphatic endothelial cell metabolism, metabolic crosstalk, and lymphangiogenic signaling in co-culture. Sci Rep. 2020;10(1):1–15. doi:https://doi.org/10.1038/s41598-020-76394-7.
- Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J, García-Caballero M, Missiaen R, Huang H, Brüning U, et al. The role of fatty acid β-oxidation in lymphangiogenesis. Nature. 2017;542(7639):49–54. doi:https://doi.org/10.1038/nature21028.
- Jung JI, Cho HJ, Jung YJ, Kwon S-H, Her S, Choi SS, Shin S-H, Lee KW, Park JHY. High-fat diet-induced obesity increases lymphangiogenesis and lymph node metastasis in the B16F10 melanoma allograft model: roles of adipocytes and M2-macrophages. Int J Cancer. 2015;136(2):258–270. doi:https://doi.org/10.1002/ijc.28983.
- Gupta P, Gupta N, Fofaria NM, Ranjan A, Srivastava SK. HER2-mediated GLI2 stabilization promotes anoikis resistance and metastasis of breast cancer cells. Cancer Lett. 2019;442:68–81. doi:https://doi.org/10.1016/j.canlet.2018.10.021.
- Ho PC, Bihuniak JD, MacIntyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor t cell responses. Cell. 2015;162(6):1217–1228. doi:https://doi.org/10.1016/j.cell.2015.08.012.
- Craig SG, Humphries MP, Alderdice M, Bingham V, Richman SD, Loughrey MB, Coleman HG, Viratham-Pulsawatdi A, McCombe K, Murray GI, et al. Immune status is prognostic for poor survival in colorectal cancer patients and is associated with tumour hypoxia. Br J Cancer. 2020;123(8):1280–1288. doi:https://doi.org/10.1038/s41416-020-0985-5.
- Chang C-H, Qiu J, Sullivan DO, Schreiber RD, Pearce EJ, Pearce EL. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. doi:https://doi.org/10.1016/j.cell.2015.08.016.
- Cham CM, Gajewski TF. Glucose availability regulates IFN-γ production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. 2005;174(8):4670–4677. doi:https://doi.org/10.4049/jimmunol.174.8.4670.
- Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, Wei S, Crespo J, Wan S, Vatan L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17(1):95–103. doi:https://doi.org/10.1038/ni.3313.
- Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meer F, Mann M, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3):829–842. doi:https://doi.org/10.1016/j.cell.2016.09.031.
- O’Sullivan D, vanderWindt GWJ, Huang SCC, Curtis JD, Chang CH, Buck MDL, Qiu J, Smith AM, Lam WY, DiPlato LM, et al. Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41(1):75–88. doi:https://doi.org/10.1016/j.immuni.2014.06.005.
- Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, Wei S, Zhao L, Vatan L, Wen B, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. 2018;28(1):87–103. doi:https://doi.org/10.1016/j.cmet.2018.04.022.
- Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, Zhao L, Vatan L, Shao I, Szeliga W, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. 2017;18(12):1332–1341. doi:https://doi.org/10.1038/ni.3868.
- Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, Kopinski PK, Wang L, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017;25(6):1282–1293. doi:https://doi.org/10.1016/j.cmet.2016.12.018.
- Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013–1021. doi:https://doi.org/10.1126/science.aav2588.
- Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, Zhou Q, Zeng H, Tong S, Wang T, et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol. 2019;75(5):752–763. doi:https://doi.org/10.1016/j.eururo.2018.09.030.
- Han YK, Park GY, Bae MJI, Kim JS, Jo WS, Lee CG. Hypoxia induces immunogenic cell death of cancer cells by enhancing the exposure of cell surface calreticulin in an endoplasmic reticulum stress-dependent manner. Oncol Lett. 2019;18:6269–6274.
- Deng B, Zhu J-M, Wang Y, Liu -T-T, Ding Y-B, Xiao W-M, Lu G-T, Bo P, Shen X-Z, Gangopadhyay N. Intratumor hypoxia promotes immune tolerance by inducing regulatory T cells via TGF-β1 in gastric cancer. PLoS One. 2013;8(5):e63777. doi:https://doi.org/10.1371/journal.pone.0063777.
- Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T reg cells. Nature. 2011;475(7355):226–230. doi:https://doi.org/10.1038/nature10169.
- Yan M, Jene N, Byrne D, Millar EKA, O’Toole SA, McNeil CM, Bates GJ, Harris AL, Banham AH, Sutherland RL, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res. 2011;13(2):1–10. doi:https://doi.org/10.1186/bcr2869.
- Pinato DJ, Black JR, Trousil S, Dina RE, Trivedi P, Mauri FA, Sharma R. Programmed cell death ligands expression in phaeochromocytomas and paragangliomas: relationship with the hypoxic response, immune evasion and malignant behavior. Oncoimmunology. 2017;6(11):e1358332. doi:https://doi.org/10.1080/2162402X.2017.1358332.
- Labiano S, Palazón A, Bolaños E, Azpilikueta A, Sánchez-Paulete AR, Morales-Kastresana A, Quetglas JI, Perez-Gracia JL, Gúrpide A, Rodriguez-Ruiz M, et al. Hypoxia-induced soluble CD137 in malignant cells blocks CD137L-costimulation as an immune escape mechanism. Oncoimmunology. 2016;5(1):e1062967. doi:https://doi.org/10.1080/2162402X.2015.1062967.
- Robinson CM, Poon BPK, Kano Y, Pluthero FG, Kahr WHA, Ohh M. A hypoxia-inducible HIF1-GAL3ST1-sulfatide axis enhances ccRCC immune evasion via increased tumor cell-platelet binding. Mol Cancer Res. 2019;17:2306–2317. doi:https://doi.org/10.1158/1541-7786.MCR-19-0461.
- Xiong Y, Liu L, Xia Y, Qi Y, Chen Y, Chen L, Zhang P, Kong Y, Qu Y, Wang Z, et al. Tumor infiltrating mast cells determine oncogenic HIF-2α-conferred immune evasion in clear cell renal cell carcinoma. Cancer Immunol Immunother. 2019;68(5):731–741. doi:https://doi.org/10.1007/s00262-019-02314-y.
- Mayer A, Haist M, Loquai C, Grabbe S, Rapp M, Roth W, Vaupel P, Schmidberger H. Role of hypoxia and the adenosine system in immune evasion and prognosis of patients with brain metastases of melanoma: a multiplex whole slide immunofluorescence study. Cancers (Basel). 2020;12(12):1–19. doi:https://doi.org/10.3390/cancers12123753.
- Kamarajugadda S, Stemboroski L, Cai Q, Simpson NE, Nayak S, Tan M, Lu J. Glucose oxidation modulates anoikis and tumor metastasis. Mol Cell Biol. 2012;32(10):1893–1907. doi:https://doi.org/10.1128/MCB.06248-11.
- Sousa B, Pereira J, Marques R, Grilo LF, Pereira SP, Sardão VA, Schmitt F, Oliveira PJ, Paredes J. P-cadherin induces anoikis-resistance of matrix-detached breast cancer cells by promoting pentose phosphate pathway and decreasing oxidative stress. Biochim Biophys Acta-Mol Basis Dis. 2020;1866(12):165964. doi:https://doi.org/10.1016/j.bbadis.2020.165964.
- Sun T, Zhong X, Song H, Liu J, Li J, Leung F, Lu WW, Liu ZL. Anoikis resistant mediated by FASN promoted growth and metastasis of osteosarcoma. Cell Death Dis. 2019;10(4):1–13. doi:https://doi.org/10.1038/s41419-019-1532-2.
- Cai Q, Yan L, Xu Y. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene. 2015;34(25):3315–3324. doi:https://doi.org/10.1038/onc.2014.264.
- Yoshino S, Hara T, Nakaoka HJ, Kanamori A, Murakami Y, Seiki M, Sakamoto T. The ERK signaling target RNF126 regulates anoikis resistance in cancer cells by changing the mitochondrial metabolic flux. Cell Discov. 2016;2(1):1–19. doi:https://doi.org/10.1038/celldisc.2016.19.
- Caneba CA, Bellance N, Yang L, Pabst L, Nagrath D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am J Physiol Metab. 2012;303(8):1036–1052.
- Ng TL, Leprivier G, Robertson MD, Chow C, Martin MJ, Laderoute KR, Davicioni E, Triche TJ, Sorensen PHB. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012;19(3):501–510. doi:https://doi.org/10.1038/cdd.2011.119.
- Bizjak M, Malavašič P, Dolinar K, Pohar J, Pirkmajer S, Pavlin M. Combined treatment with Metformin and 2-deoxy glucose induces detachment of viable MDA-MB-231 breast cancer cells in vitro. Sci Rep. 2017;7(1):1–14. doi:https://doi.org/10.1038/s41598-017-01801-5.
- Chen IH, Shih HC, Hsieh PW, Chang FR, Wu YC, Wu CC. HPW-RX40 restores anoikis sensitivity of human breast cancer cells by inhibiting integrin/FAK signaling. Toxicol Appl Pharmacol. 2015;289:330–340. doi:https://doi.org/10.1016/j.taap.2015.09.011.
- Giannoni E, Fiaschi T, Ramponi G, Chiarugi P. Redox regulation of anoikis resistance of metastatic prostate cancer cells: key role for Src and EGFR-mediated pro-survival signals. Oncogene. 2009;28(20):2074–2086. doi:https://doi.org/10.1038/onc.2009.77.
- Zhu P, Tan MJ, Huang RL, Tan CK, Chong HC, Pal M, Lam CRI, Boukamp P, Pan JY, Tan SH, et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2-: H2O2ratio and confers anoikis resistance to tumors. Cancer Cell. 2011;19(3):401–415. doi:https://doi.org/10.1016/j.ccr.2011.01.018.
- Shen CJ, Chan SH, Lee CT, Huang WC, Tsai JP, Chen BK. Oleic acid-induced ANGPTL4 enhances head and neck squamous cell carcinoma anoikis resistance and metastasis via up-regulation of fibronectin. Cancer Lett. 2017;386:110–122. doi:https://doi.org/10.1016/j.canlet.2016.11.012.
- Baba K, Kitajima Y, Miyake S, Nakamura J, Wakiyama K, Sato H, Okuyama K, Kitagawa H, Tanaka T, Hiraki M, et al. Hypoxia-induced ANGPTL4 sustains tumour growth and anoikis resistance through different mechanisms in scirrhous gastric cancer cell lines. Sci Rep. 2017;7(1):1–13. doi:https://doi.org/10.1038/s41598-017-11769-x.
- Maroni P, Bendinelli P, Matteucci E, Locatelli A, Nakamura T, Scita G, Desiderio MA. Osteolytic bone metastasis is hampered by impinging on the interplay among autophagy, anoikis and ossification. Cell Death Dis. 2014;5(1):e1005. doi:https://doi.org/10.1038/cddis.2013.465.
- Whelan KA, Caldwell SA, Shahriari KS, Jackson SRE, Franchetti LD, Johannes GJ, Reginato MJ, Mostov KE. Hypoxia suppression of Bim and Bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol Biol Cell. 2010;21(22):3829–3837. doi:https://doi.org/10.1091/mbc.e10-04-0353.
- Bhatt V, Khayati K, Hu ZS, Lee A, Kamran W, Su X, Guo JY. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient kras-driven lung tumorigenesis. Genes Dev. 2019;33:150–165. doi:https://doi.org/10.1101/gad.320481.118.
- Zhao Z, Han F, Yang S, Wu J, Zhan W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015;358(1):17–26. doi:https://doi.org/10.1016/j.canlet.2014.11.046.
- Peng X, Gong F, Chen Y, Jiang Y, Liu J, Yu M, Zhang S, Wang M, Xiao G, Liao H. Autophagy promotes paclitaxel resistance of cervical cancer cells: involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis. 2014;5(8):1367. doi:https://doi.org/10.1038/cddis.2014.297.
- Wang L, Shang Z, Zhou Y, Hu X, Chen Y, Fan Y, Wei X, Wu L, Liang Q, Zhang J, et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018;9:1–17.
- Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, Ekpin E, George A, Zheng Y, Lam HM, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat Commun. 2015;6(1):1–14. doi:https://doi.org/10.1038/ncomms7170.
- Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, Calvo V, Cheung JF, Bravo-Cordero JJ, Entenberg D, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. 2017;19(2):120–132. doi:https://doi.org/10.1038/ncb3465.
- Fox DB, Garcia NMG, McKinney BJ, Lupo R, Noteware LC, Newcomb R, Liu J, Locasale JW, Hirschey MD, Alvarez JV. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat Metab. 2020;2(4):318–334. doi:https://doi.org/10.1038/s42255-020-0191-z.
- Yao Z, Xie F, Li M, Liang Z, Xu W, Yang J, Liu C, Li H, Zhou H, Qu LH. Oridonin induces autophagy via inhibition of glucose metabolism in p53-mutated colorectal cancer cells. Cell Death Dis. 2017;8(2):2633. doi:https://doi.org/10.1038/cddis.2017.35.
- Meng Q, Shi S, Liang C, Liang D, Hua J, Zhang B, Xu J, Yu X. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/snail signaling. Oncogene. 2018;37(44):5843–5857. doi:https://doi.org/10.1038/s41388-018-0392-z.
- Walker A, Singh A, Tully E, Woo J, Le A, Nguyen T, Biswal S, Sharma D, Gabrielson E. Nrf2 signaling and autophagy are complementary in protecting breast cancer cells during glucose deprivation. Free Radic Biol Med. 2018;120:407–413. doi:https://doi.org/10.1016/j.freeradbiomed.2018.04.009.
- Xi H, Barredo JC, Merchan JR, Lampidis TJ. Endoplasmic reticulum stress induced by 2-deoxyglucose but not glucose starvation activates AMPK through CaMKKβ leading to autophagy. Biochem Pharmacol. 2013;85(10):1463–1477. doi:https://doi.org/10.1016/j.bcp.2013.02.037.
- Roy S, Leidal AM, Ye J, Ronen SM, Debnath J. Autophagy-dependent shuttling of TBC1D5 controls plasma membrane translocation of GLUT1 and glucose uptake. Mol Cell. 2017;67(1):84–95. doi:https://doi.org/10.1016/j.molcel.2017.05.020.
- Brisson L, Bański P, Sboarina M, Dethier C, Danhier P, Fontenille MJ, Van Hée VF, Vazeille T, Tardy M, Falces J, et al. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell. 2016;30(3):418–431. doi:https://doi.org/10.1016/j.ccell.2016.08.005.
- Peppicelli S, Andreucci E, Ruzzolini J, Laurenzana A, Margheri F, Fibbi G, Del RM, Bianchini F, Calorini L. The acidic microenvironment as a possible niche of dormant tumor cells. Cell Mol Life Sci. 2017;74(15):2761–2771. doi:https://doi.org/10.1007/s00018-017-2496-y.
- Ramírez-Peinado S, León-Annicchiarico CL, Galindo-Moreno J, Iurlaro R, Caro-Maldonado A, Prehn JHM, Ryan KM, Muñoz-Pinedo C. Glucose-starved cells do not engage in prosurvival autophagy. J Biol Chem. 2013;288(42):30387–30398. doi:https://doi.org/10.1074/jbc.M113.490581.
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:https://doi.org/10.1038/ncb2152.
- Tan HWS, Sim AYL, Long YC. Glutamine metabolism regulates autophagy-dependent mTORC1 reactivation during amino acid starvation. Nat Commun. 2017;8(1):1–10. doi:https://doi.org/10.1038/s41467-017-00369-y.
- Morotti M, Zois CE, El-Ansari R, Craze ML, Rakha EA, Fan SJ, Valli A, Haider S, Goberdhan DCI, Green AR, et al. Increased expression of glutamine transporter SNAT2/SLC38A2 promotes glutamine dependence and oxidative stress resistance, and is associated with worse prognosis in triple-negative breast cancer. Br J Cancer. 2021;124(2):494–505. doi:https://doi.org/10.1038/s41416-020-01113-y.
- Halama A, Kulinski M, Dib SS, Zaghlool SB, Siveen KS, Iskandarani A, Zierer J, Prabhu KS, Satheesh NJ, Bhagwat AM, et al. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 2018;430:133–147. doi:https://doi.org/10.1016/j.canlet.2018.05.017.
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447–1461. doi:https://doi.org/10.1101/gad.219642.113.
- Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, Olzmann JA. DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev Cell. 2017;42(1):9–21. doi:https://doi.org/10.1016/j.devcel.2017.06.003.
- Wen YA, Xing X, Harris JW, Zaytseva YY, Mitov MI, Napier DL, Weiss HL, Mark Evers B, Gao T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017;8(2):2593. doi:https://doi.org/10.1038/cddis.2017.21.
- Jawhari S, Ratinaud MH, Verdier M. Glioblastoma, hypoxia and autophagy: a survival-prone “ménage-à-trois”. Cell Death Dis. 2016;7(10):2434. doi:https://doi.org/10.1038/cddis.2016.318.
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–2581. doi:https://doi.org/10.1128/MCB.00166-09.
- Gao L, Wang Z, Lu D, Huang J, Liu J, Hong L. Paeonol induces cytoprotective autophagy via blocking the Akt/mTOR pathway in ovarian cancer cells. Cell Death Dis. 2019;10(8):1–13. doi:https://doi.org/10.1038/s41419-019-1849-x.
- Tam SY, Wu VWC, Law HKW. Dynamics of oxygen level-driven regulators in modulating autophagy in colorectal cancer cells. Biochem Biophys Res Commun. 2019;517(2):193–200. doi:https://doi.org/10.1016/j.bbrc.2019.07.043.
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29(31):4424–4435. doi:https://doi.org/10.1038/onc.2010.191.
- Joshi S, Kumar S, Ponnusamy MP, Batra SK. Hypoxia-induced oxidative stress promotes MUC4 degradation via autophagy to enhance pancreatic cancer cells survival. Oncogene. 2016;35(45):5882–5892. doi:https://doi.org/10.1038/onc.2016.119.
- Carcereri de Prati A, Butturini E, Rigo A, Oppici E, Rossin M, Boriero D, Mariotto S. Metastatic breast cancer cells enter into dormant state and express cancer stem cells phenotype under chronic hypoxia. J Cell Biochem. 2017;118(10):3237–3248. doi:https://doi.org/10.1002/jcb.25972.
- Endo H, Okuyama H, Ohue M, Inoue M, Singh PK. Dormancy of cancer cells with suppression of AKT activity contributes to survival in chronic hypoxia. PLoS One. 2014;9(6):e98858. doi:https://doi.org/10.1371/journal.pone.0098858.
- Endo H, Okami J, Okuyama H, Nishizawa Y, Imamura F, Inoue M. The induction of MIG6 under hypoxic conditions is critical for dormancy in primary cultured lung cancer cells with activating EGFR mutations. Oncogene. 2017;36(20):2824–2834. doi:https://doi.org/10.1038/onc.2016.431.
- Hofstetter CP, Burkhardt J-K, Shin BJ, Gü Rsel DB, Mubita L. Protein phosphatase 2A mediates dormancy of glioblastoma multiforme-derived tumor stem-like cells during hypoxia. PLoS One. 2012;7(1):30059. doi:https://doi.org/10.1371/journal.pone.0030059.
- Chen C, Mahar R, Merritt ME, Denlinger DL, and Hahn DA. ROS and hypoxia signaling regulate periodic metabolic arousal during insect dormancy to coordinate glucose, amino acid, and lipid metabolism. Proc Natl Acad Sci USA. 2021;118(1) :e2017603118.
- Kronblad Å, Hedenfalk I, Nilsson E, Påhlman S, Landberg G. ERK1/2 inhibition increases antiestrogen treatment efficacy by interfering with hypoxia-induced downregulation of ERα: a combination therapy potentially targeting hypoxic and dormant tumor cells. Oncogene. 2005;24:6835–6841.
- Wright JL, Plymate SR, Porter MP, Gore JL, Lin DW, Hu E, Zeliadt SB. Hyperglycemia and prostate cancer recurrence in men treated for localized prostate cancer. Prostate Cancer Prostatic Dis. 2013;16:204–208.
- Barba M, Sperati F, Stranges S, Carlomagno C, Nasti G, Iaffaioli V, Caolo G, Mottolese M, Botti G, Terrenato I, et al. Fasting glucose and treatment outcome in breast and colorectal cancer patients treated with targeted agents: results from a historic cohort. Ann Oncol. 2012;23:1838–1845. doi:https://doi.org/10.1093/annonc/mdr540.
- Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, Mueller-Klieser W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000;60(4):916–921.
- Park S, Chang CY, Safi R, Liu X, Baldi R, Jasper JS, Anderson GR, Liu T, Rathmell JC, Dewhirst MW, et al. ERRα-regulated lactate metabolism contributes to resistance to targeted therapies in breast cancer. Cell Rep. 2016;15:323–335. doi:https://doi.org/10.1016/j.celrep.2016.03.026.
- Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016;381(1):96–103. doi:https://doi.org/10.1016/j.canlet.2016.07.035.
- Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, Kyotani K, Mizowaki T, Imahori T, Ejima Y, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest. 2015;125(4):1591–1602. doi:https://doi.org/10.1172/JCI78239.
- Valter K, Chen L, Kruspig B, Maximchik P, Cui H, Zhivotovsky B, Gogvadze V. Contrasting effects of glutamine deprivation on apoptosis induced by conventionally used anticancer drugs. Biochim Biophys Acta-Mol Cell Res. 2017;1864(3):498–506. doi:https://doi.org/10.1016/j.bbamcr.2016.12.016.
- Nwosu ZC, Piorońska W, Battello N, Zimmer AD, Dewidar B, Han M, Pereira S, Blagojevic B, Castven D, Charlestin V, et al. Severe metabolic alterations in liver cancer lead to ERK pathway activation and drug resistance. EBioMedicine. 2020;54:102699. doi:https://doi.org/10.1016/j.ebiom.2020.102699.
- Chen R, Lai LA, Sullivan Y, Wong M, Wang L, Riddell J, Jung L, Pillarisetty VG, Brentnall TA, Pan S. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci Rep. 2017;7:1–14.
- Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, Kim JK, Heo Y, Lee HS, Lee MY, et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 2020;31(2):267–283. doi:https://doi.org/10.1016/j.cmet.2019.11.020.
- Jiang ZF, Wang M, Xu JL, Ning YJ. Hypoxia promotes mitochondrial glutamine metabolism through HIF1α-GDH pathway in human lung cancer cells. Biochem Biophys Res Commun. 2017;483:32–38. doi:https://doi.org/10.1016/j.bbrc.2017.01.015.
- Li J, Zhao S, Zhou X, Zhang T, Zhao L, Miao P, Song S, Sun X, Liu J, Zhao X, et al. Inhibition of lipolysis by mercaptoacetate and etomoxir specifically sensitize drug-resistant lung adenocarcinoma cell to paclitaxel. PLoS One. 2013;8(9):e74623. doi:https://doi.org/10.1371/journal.pone.0074623.
- Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27(1):136–150. doi:https://doi.org/10.1016/j.cmet.2017.11.001.
- Wang X, He S, Gu Y, Wang Q, Chu X, Jin M, Xu L, Wu Q, Zhou Q, Wang B, et al. Fatty acid receptor GPR120 promotes breast cancer chemoresistance by upregulating ABC transporters expression and fatty acid synthesis. EBioMedicine. 2019;40:251–262. doi:https://doi.org/10.1016/j.ebiom.2018.12.037.
- Ko E, Seo HW, Jung G. Telomere length and reactive oxygen species levels are positively associated with a high risk of mortality and recurrence in hepatocellular carcinoma. Hepatology. 2018;67(4):1378–1391. doi:https://doi.org/10.1002/hep.29604.
- Wang X, Yang K, Xie Q, Wu Q, Mack SC, Shi Y, Kim LJY, Prager BC, Flavahan WA, Liu X, et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat Neurosci. 2017;20:661–673. doi:https://doi.org/10.1038/nn.4537.
- Tong L, Xu N, Zhou X, Huang J, Wan-er W, Chen C, Liang L, Liu Q, Xiaoli L. PKM2 Mediates chronic myeloid leukemia imatinib resistance by regulating glycolysis energy metabolism. Blood. 2018;132(Supplement 1):1724. doi:https://doi.org/10.1182/blood-2018-99-113960.
- Tung S, Shi Y, Wong K, Zhu F, Gorczynski R, Laister RC, Minden M, Blechert AK, Genzel Y, Reichl U, et al. PPARα and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia. Blood. 2013;122(6):969–980. doi:https://doi.org/10.1182/blood-2013-03-489468.
- Liu H, Wu X, Dong Z, Luo Z, Zhao Z, Xu Y, Zhang JT. Fatty acid synthase causes drug resistance by inhibiting TNF-α and ceramide production. J Lipid Res. 2013;54(3):776–785. doi:https://doi.org/10.1194/jlr.M033811.
- Herrera ACS, Victorino VJ, Campos FC, Verenitach BD, Lemos LT, Aranome AMF, Oliveira SR, Cecchini AL, Simão ANC, Abdelhay E, et al. Impact of tumor removal on the systemic oxidative profile of patients with breast cancer discloses lipid peroxidation at diagnosis as a putative marker of disease recurrence. Clin Breast Cancer. 2014;14(6):451–459. doi:https://doi.org/10.1016/j.clbc.2014.05.002.
- Pisarsky L, Bill R, Fagiani E, Dimeloe S, Goosen RW, Hagmann J, Hess C, Christofori G. Targeting metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep. 2016;15:1161–1174. doi:https://doi.org/10.1016/j.celrep.2016.04.028.
- Jiménez-Valerio G, Martínez-Lozano M, Bassani N, Vidal A, Ochoa-de-olza M, Suárez C, García-del-Muro X, Carles J, Viñals F, Graupera M, et al. Resistance to antiangiogenic therapies by metabolic symbiosis in renal cell carcinoma PDX models and patients. Cell Rep. 2016;15(6):1134–1143. doi:https://doi.org/10.1016/j.celrep.2016.04.015.
- Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283(42):28106–28114. doi:https://doi.org/10.1074/jbc.M803508200.
- Lu CW, Lin SC, Chien CW, Lin SC, Lee CT, Lin BW, Lee JC, Tsai SJ. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am J Pathol. 2011;179:1405–1414. doi:https://doi.org/10.1016/j.ajpath.2011.05.050.
- Li JQ, Wu X, Gan L, Yang XL, Miao ZH. Hypoxia induces universal but differential drug resistance and impairs anticancer mechanisms of 5-fluorouracil in hepatoma cells. Acta Pharmacol Sin. 2017;38(12):1642–1654. doi:https://doi.org/10.1038/aps.2017.79.
- Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C, Miller C, Demonacos C, Stratford IJ, Dive C. Hypoxia-mediated down-regulation of Bid and bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol. 2004;24:2875–2889. doi:https://doi.org/10.1128/MCB.24.7.2875-2889.2004.