
Abstract
Senescence was classically defined as an irreversible cell cycle arrest in G1 phase (G1 exit) triggered by eroded telomeres in aged primary cells. The molecular basis of this G1 arrest is thought to be due to a DNA damage response, resulting in accumulation of the cyclin dependent kinase (Cdk) inhibitors p21 and p16 that block the inactivating phosphorylation of the retinoblastoma tumor suppressor pRb, thereby preventing DNA replication. More than a decade ago, several studies showed that p21 also mediates permanent DNA damage-induced cell cycle arrest in G2 (G2 exit) by inhibiting mitotic Cdk complexes and pRb phosphorylation. The idea that the senescence program can also be launched after G2 arrest has gained support from several recent publications, including evidence for its existence in vivo.
Senescence, an Ultimate Anti-proliferative Barrier . . . and Beyond
Cellular senescence is a state of permanent cell cycle arrest characterized by distinct metabolic activity and dramatic changes in cell morphology.Citation1,2 Originally, its causes were proposed to be due to physiological erosion of telomeres that occurs upon extended cell proliferation (replicative senescence), as well as inappropriate oncogenic signaling (premature senescence). Senescence can be also induced by genotoxic agents, which like eroded telomeres or hyperactive oncogenes, trigger the DNA damage response (DDR) pathways and activate cell cycle checkpoints to halt proliferation. The ultimate DDR targets are cyclin dependent kinases (Cdk), key regulators of cell cycle progression. In addition to directly controlling the onset and progression of DNA replication and mitosis, Cdks phosphorylate the retinoblastoma (pRb) tumor suppressor and the related “pocket-proteins” p107 and p130, thus maintaining them in an inactive state. Continuous Cdk-mediated inactivation of pRb family proteins is a prerequisite for cell cycle progression, because active (hypo-phosphorylated) pocket-proteins drive cell cycle exit by sequestering E2F family transcription factors and repressing the genes required for DNA replication and cell division.Citation3 Deregulation of pRb and of the associated pathways plays a central role in tumorigenesis.Citation4 The action of Cdk is closely intertwined with that of another tumor suppressor, p53 (TP53), which is a major target of the PI3K-related protein kinases (PI3K) ATM and ATR and that plays a pivotal role in DDRCitation5 (). Following activation by panoply of stress signals, p53 induces the expression of the Cdk inhibitor p21Waf1/Cip1 (p21), an essential mediator of senescence (see below). More recently, mTOR (mechanistic target of rapamycin), another member of the PI3K family that controls cell growth in response to nutritional cues, has also been implicated in senescence, thus linking cellular growth with the p53 and DDR pathways.Citation6-9
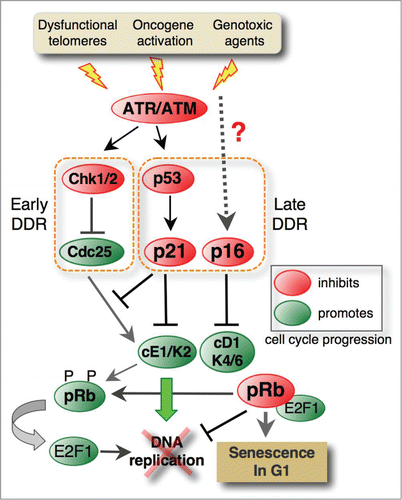
Figure 1. Molecular pathways involved in irreversible G1 arrest proceeding senescence. Dysfunctional telomeres, hyperactive oncogenes and various genotoxic agents stimulate the ATM/ATR-mediated DNA damage response (DDR) pathway. The ATM/ATR kinases induce the early response by activating Chk1/2 checkpoint kinases that, in turn, by inhibiting Cdc25 phosphatases, prevent activation of cyclin E1-Cdk2, which is a key regulator of G1/S transition. ATM also phosphorylates the p53 tumor suppressor that, by inducing the Cdk inhibitor p21Waf1/Cip1 (p21), plays a central role in the G1 exit program (late response). In addition to cyclin E1-Cdk2 (cE1-K2) complexes, p21 also inhibits cyclin D1-Cdk4/6 (cD1-K4/6) complexes that phosphorylate and inactivate pRb family pocket proteins. In turn, active pRb inhibits the E2F1-dependent expression of genes controlling G1/S progression, thus irreversibly blocking the cell cycle entry. Senescence is also associated with p16Ink4A (p16) upregulation, but the pathways leading to its induction are not entirely elucidated. This Cdk inhibitor specifically targets Cdk4/6 and prevents their association with D-type cyclins. While p16 does not intervene in G1 arrest (p16 is upregulated after induction of p21 and G1 arrest), it plays a key role in senescence maintenance.
Senescence is increasingly considered to be an essential early line of defense against tumor development by preventing proliferation of cells with damaged DNA,Citation10-12 Indeed, the markers of activated DDR are found in many pre-cancerous lesions, and mutations compromising DNA damage checkpoint (for example, defects in ATM/p53 pathway) are suggested to be a prerequisite for tumor progression.Citation13,14 Moreover, senescent cells are observed in pre-cancerous lesions in mice and in humans, but not in the subsequent malignant stages. This is consistent with the hypothesis that senescence restricts tumor progression during the early stages of tumorigenesis, whereas progression to malignancy would involve evading senescence.Citation1 However, senescence has also been implicated both in promoting carcinogenesisCitation15 and in age-related pathologiesCitation16,17 through its autocrine and paracrine effects, known as the senescence-associated secretory phenotype (SASP) originally described by the Campisi group.Citation18,19 SASP factors can act cell autonomously by reinforcing cell-cycle arrest and non-cell autonomously by influencing cells in the surrounding environment and promoting immune surveillance, leading to the elimination of senescent cells.Citation20,12 While in some cases, SASP factors secreted by pre-malignant senescent cells lead to their clearance by the immune system, resulting in tumor regression,Citation17 in other cases, pro-inflammatory SASP molecules might support carcinogenesis by promoting epithelial-mesenchyme transition and invasion, tumor vascularization and abnormal cell morphology (reviewed in ref.Citation2). Therefore, senescent cells are not merely non-dividing bystanders but, due to their active metabolism (reviewed in refCitation8), they can elicit pro-and/or anti-tumorigenic responses, depending on the microenvironment.Citation12 Finally, recent work has shown that senescence contributes also to tissue remodeling during embryonic development,Citation21,22 thus highlighting its importance in non-pathological situations. In conjunction with its role in preventing fibrosis,Citation23,24 these observations also raise the question as to whether fundamentally different mechanistic pathways underlie these different physiological roles of senescence.
Senescence as an Irreversible G1 Arrest
Originally, cellular senescence was defined as an irreversible arrest in the G1 phase of the cell cycle (reviewed in refs.Citation25,26). It is initiated by inactivation of cyclin D1-Cdk4/6 and cyclin E1-Cdk2 complexes that positively regulate G1/S phase progression.Citation27,28 The best-characterized role of cyclin D1-Cdk4/6 complexes is in triggering pRb phosphorylation,Citation29,30 while cyclin E1-Cdk2 complexes control the onset of DNA replication by directly phosphorylating a number of replication proteins (reviewed in ref.Citation31). Like in proliferating cells exposed to DNA damage,Citation32,33 the ATM/p53/p21 pathway triggered by telomere erosion or activated oncogenes plays a central role in senescence-associated G1 arrest (). Indeed, overly short telomeres elicit the same response as DNA breaks, leading to activation of the DDR signaling cascade and blocking the G1/S transition.Citation34-36 On the other hand, inappropriate oncogene activation causes a proliferative burst and rounds of error-prone DNA synthesis. The DNA damage accumulated during such deregulated DNA synthesis also activates p53 via the ATM and ATR damage signaling pathways, leading to p21 induction and permanent cell cycle arrest.Citation37,38
In addition to directly preventing DNA replication by inhibiting Cdk2, p21 also blocks the inactivating phosphorylation of pRb by Cdk2 and Cdk4/6 (), which is a key mechanism in the senescence program.Citation39 Indeed, absence of pRb phosphorylation,Citation40 together with the presence of G1 cyclins (cyclins D1 and E1)Citation27 and the lack of mitotic cyclins (cyclin B1),Citation41 is an early-recognized hallmark of senescence. Very soon after its discovery, p16Ink4a (p16), another pRb regulator that specifically inhibits the cyclin D1-associated kinases Cdk4 and Cdk6, was also implicated in senescenceCitation28,42-44 (). Together with β-galactosidase staining,Citation45 p16 is increasingly used as a senescence biomarker.Citation16,46,47 However, despite sharing common targets, the 2 Cdk inhibitors have distinct functions and biological roles. Unlike p21, p16 is not required for cell cycle arrest associated with replicative senescence, and its late induction, which occurs only after p21-mediated cell cycle arrest, was suggested to stabilize rather than induce the senescent state.Citation28,42,48-50 In agreement with p16's role as a guardian of senescence, mutations, silencing or loss of CDKN2A, the gene encoding p16, facilitate senescence bypass and it is frequently associated with carcinogenesis (reviewed in.Citation51,52 Although this feature earned p16 the rank of tumor suppressor, the results showing high-level p16 expression in number of cancers (reviewed in ref.Citation52) and in tumor-infiltrating immune cellsCitation47 suggests a more nuanced view regarding its role in senescence and cancer development. Moreover, while p53 directly controls p21 induction, at least during DDR, the mechanisms controlling p16 up-regulation in senescence seem to be p53-independent and have not been completely elucidated (reviewed in ref.Citation51)
Senescence in G2 – an Old Concept Awaiting Wider Recognition
The idea that senescence could also be induced during a prolonged G2 arrest emerged after the discovery that p21 is also involved in the G2/M checkpoint regulation.Citation53-55 According to the prevailing model,Citation56 DNA damage-induced G2 arrest is triggered by the ATM/ATR-activated checkpoint kinases Chk1/2 that inhibit Cdc25 phosphatases, which are activators of the key mitotic regulator, cyclin B1-Cdk1 (). Based on data obtained using HCT-116 colorectal cancer cell line, it was initially proposed that p21 could play a role in the “maintenance” of DNA damage-induced G2 arrest. Indeed, after transiently arresting in G2, HCT-116 cells in which p21 was knocked out eventually underwent aberrant mitoses, leading to polyploidy or cell death.Citation53,55 In this work, however, the mechanism whereby p21 sustains G2 arrest was not elucidated. The uncertainty regarding the role of p21 in the G2/M checkpoint was further supported by the findings that in p53-deficient cells arrested in G2, cyclin B1-Cdk1 complexes are inactiveCitation57 and that even after its overexpression in U2OS osteosarcoma cancer cells, p21 only weakly associated with cyclin B1-Cdk1).Citation58 Based on these observations, p21 was not considered to be a crucial component of the G2 arrest mechanism.Citation56
Figure 2. DNA damage response pathways involved in the G2 exit program leading to senescence. Pro-senescence stimuli activate the ATM/ATR DNA damage-signaling pathway, leading to stable G2 arrest that entails mitotic bypass (see also Fig. 3) and permanent cell cycle arrest in a tetraploid G1 phase. The early response involves Chk1/2-mediated inhibition of Cdc25 phosphatases, which promote mitosis by activating cyclin B1-Cdk1 complexes (cB1/K1). In p53-deficient cells, this pathway transiently blocks G2/M progression, but the p53/p21 pathway is required to stabilize G2 arrest. In addition to inhibiting cyclin B1-Cdk1 complexes, p21 mediates premature activation of APC/C-Cdh1, leading to degradation of cyclin B1 and other mitotic regulators. Like in G1 arrest, p21 also activates pRb by blocking Cdk-mediated pRb phosphorylation. Active pRb inhibits the expression of genes that control G2/M progression, leading to irreversible G2 arrest of the cell cycle. It is proposed that p53 might induce senescence independently of p21Citation93 through mechanisms that are not fully clear. Like in G1 arrest, p16 stabilizes senescence in G2 presumably by targeting pRb kinases. However, the pathways involved have not been elucidated.
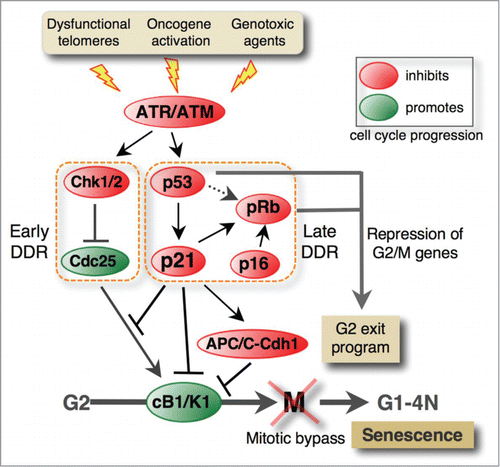
A breakthrough in understanding how p21 might exercise its G2 function(s) came from the study of normal human fibroblasts (NHF) exposed to ICRF-193, a topoisomerase II inhibitor that specifically causes G2 arrest. Prolonged exposure to this drug led to p53/p21-dependent irreversible cell cycle exit in G2 that was associated with down-regulation of both cyclin A and cyclin B1 and accumulation of non-phosphorylated pRb family pocket proteins.Citation59 G2-arrested fibroblasts, in which either p53 or pRb was compromised by viral oncogenes, failed to permanently exit the cell cycle and expressed mitotic regulators even after prolonged exposure to genotoxic agents. This suggested that p21 induces senescence upon G2 arrest by blocking pRb/p130 phosphorylation. ()Similar results were obtained in carcinoma cell lines that conditionally express p53 or following exposure to γ-irradiation or genotoxic drugsCitation60 but the notion of G2 cell cycle exit was not evoked. Moreover, in response to DNA damage, the expression of G2/M-specific genes is reduced in a p53- or pRb-dependent manner.Citation61,60,62 The discovery of G2/M targets of E2F1Citation63 and of the pocket protein-dependent transcriptional repression of mitotic regulators following stable G2 arrest Citation64 provided additional credibility to the G2 exit paradigm. Based on these findings, it was proposed that the “G2 exit program” might serve as a safeguard mechanism to prevent cell adaptation to the G2 checkpoint and consequently the passage into mitosis of cells with damaged DNA (;Citation59). Noteworthy, the “G2 exit program” could in principle apply to replicative senescence because telomeric attritions are expected to become apparent (and to be detected as DNA damage) at the end of the S phase (reviewed in ref.Citation36). Mechanistically, G2 exit is distinct from p53/p21-dependent tetraploid G1 arrest that is caused by adaptation to the spindle checkpoint (mitotic slippage) or by cytokinesis failure and that leads to cells with 2 G1 nucleiCitation65-67 (reviewed in ref.Citation68).
Although clearly essential for senescence,Citation39 blocking pRb phosphorylation is not the only role of p21 in the G2 exit program. In primary fibroblasts exposed to genotoxic drugs, in addition to inhibiting cyclin A-Cdk1/2 complexes,Citation59 p21 also associates with and sequesters inactive cyclin B1-Cdk1 complexes in the nucleus, thereby preventing mitotic entryCitation69 (). This mechanism was proposed to be paramount for cell cycle exit in G2 because, unlike transient inhibition of Cdk1 activation through the Chk1/2 pathway, p21 completely and irreversibly inhibits cyclin B1-Cdk1 complexes, thus rendering impossible their activation by CAK, PLK1 or Cdc25 phosphatases (). The pre-mitotic nuclear localization of cyclin B1 was rather unexpected because cyclin B1 nuclear entry is invariably associated with mitosis onset, whereas cytoplasmic cyclin B1 is thought to be a hallmark of G2 arrest.Citation70 Although also observed after ectopic over-expression of p21 or p53 in tumor cell linesCitation58,71, cyclin B1 nuclear localization associated with DNA damage-induced G2 arrest was not reported in other studies.Citation53,55 This is probably because most of these works were carried out in p53-proficient tumor cell lines, such as HCT-116 or U2OS cells, in which p21 induction in G2 is inefficient due to impairment of the ATM pathway.Citation72 A notable exception was the finding that p21/p27-dependent nuclear sequestration of cyclin B1-Cdk1 complexes also occurs in serum-deprived mouse embryonic fibroblasts (MEFs) lacking expression of all 3 retinoblastoma protein family members.Citation73 However, in this study, cyclin B1 nuclear localization was associated with reversible, rather than irreversible, cell cycle exit.
Originally, downregulation of mitotic regulators following G2 arrest was attributed to pRb-mediated suppression of their expression (). However, subsequent work in several laboratories provided evidence that this is also due to protein degradation triggered by anaphase-promoting complex/cyclosome (APC/C) and its co-activator Cdh1Citation74-76 (). This premature and p21-dependent activation of APC/C-Cdh1 in G2-arrested cells was thought to be due to down-regulation of the APC/C inhibitor Emi1,Citation74 although an implication of the Cdc14B phosphatase in this process was not formally excluded.Citation77
Despite the accumulating evidence, the G2 role of p21 and the concept of senescence in G2 failed to gain wider acceptance and are conspicuously missing in a number of influential reviews.Citation5,36,68,78
Senescence in G2 Comes of Age – New Insights
Several recent publications have confirmed these old observations and provided additional evidence supporting the existence of a G2 exit program and its role in senescence. First, 2 reports showed that replicative senescence could also be triggered in the G2 phase. The first paper demonstrated that the presence of a significant fraction of senescent fibroblasts with a 4N DNA content and strongly expressing cyclin D1 is not due to gradual accumulation of bi-nucleate or tetraploid G1 cells in aging cultures, but the consequence of cell cycle exit after G2 arrest.Citation79 Accumulation of cyclin D1 and E1 was also observed in G2-arrested cells following ectopic induction of p21.Citation80 More recently, it has been shown that in telomerase-negative cells eroded telomeres elicit DDR predominantly in G2, leading to p53/p21-dependent cell cycle arrest/exit with 4N DNA content.Citation81 Unlike functional telomeres that trigger a transient DNA damage response in G2 that is required for their protection,Citation82 telomeric DNA damage in aging cells persists at a subset of the shortest telomeres and fully activates DDR.Citation81 These observations corroborate the idea that telomeric attrition might preferentially activate the G2/M checkpoint.Citation36 However, one might wonder why not all senescent cells are stably arrested in G2. Although several explanations are plausible, the most likely is that the G2/M checkpoint is not as robust as the G1/S checkpoint, which relies on strong p21 induction (reviewed in ref.Citation78). As a consequence, some damaged cells can escape the G2/M checkpoint, progress into mitosis and arrest in the subsequent G1 phase by the more efficient G1/S checkpoint.
In agreement with this hypothesis, a recent report showed that, unlike genomic breaks, DNA damage signaling elicited by partial telomere deprotection is insufficient to fully activate the ATM-mediated G2/M checkpoint.Citation83 Instead, after completion of mitosis, DNA damage induces p53-dependent cell cycle exit in G1. Why do intermediate-state telomeres not activate the checkpoint and block G2/M progression? The authors suggested that the culprit is altered DDR and insufficient Chk2 activation. However, it is not clear how DNA damage can fully activate ATM and many of its targets (such as H2AX and 53BP1), but not Chk2. Moreover, the authors argued that their experimental model mimics replicative senescence, which is also associated with G1 cell cycle exit and weak Chk2 phosphorylation (based on the analysis of threonine 68 phosphorylation). A major caveat of this interpretation is that Chk2 does not seem to play an essential role in G2 arrestCitation84,85,72 and it is more likely that damaged cells progress into mitosis due to inefficient Chk1 activationCitation86 and/or lack of robust p21 induction. It should be noted that, at least in non-transformed cells, DNA damage-induced Chk2 phosphorylation at Thr68 is very transientCitation72 and hence not the best indicator of Chk2 activation (as opposed to SDS-PAGE shift). Furthermore, it is unclear to what extent this telomere-deprotection-DNA damage response actually contributes to replicative senescence because both Chk1 and Chk2 are activated by short telomeres,Citation34,87,81 whereas telomere erosion in aging fibroblasts activates mainly the G2/M checkpoint.Citation81 Another intriguing result is the absence of mitotic bypass and endoreplication in cells that lack p53 and that experience prolonged DNA damage signaling due to persistent loss of telomere protection,Citation83 which is in stark contrast with the results documented by the De Lange teamCitation88 (see below).
With respect to the expression of cell cycle regulators, the phenotype of cells arrested in G2 is similar to that of cells arrested in G1, notably, absence of G2/M cyclins and accumulation of G1 cyclins. What is molecular basis for that? A model originally proposed by the Lahav group suggests that cells stably blocked in G2 eventually undergo mitotic bypass - a passage from G2 to G1 without chromosome segregation - and enter a tetraploid G1 phase where they cannot replicate due to p21-mediated inhibition of cyclin E1-Cdk2.Citation89 The absence of mitotic regulators in these 4N cells could be explained by both their degradation and p53/pRb-mediated repression (). This model has been confirmed experimentally by several recent publications. Ye el al. showed that radiation-induced cellular senescence results from mitotic bypass of stably G2-arrested cells into a tetraploid G1 phase, where they accumulate cyclin D1.Citation90 Conversely, in p53/pRb-deficient MEFs, DNA damage generated by persistent telomere dysfunction induces prolonged G2 arrest that leads to genome reduplication and tetraploidy due to the absence of the p53/p21-dependent G1/S checkpoint (;Citation88). Tetraploidization is caused by mitotic bypass that leads to DNA synthesis in the ensuing 4N G1 phase, as shown elegantly using FUCCI imaging. This technique uses fluorescent probes engineered to label a G1-specific marker (CDT1) and a G2-specific marker (geminin) to follow cell cycle progression in live cells.Citation91 Subsequent work by the same team has shown that tetraploidization triggered by permanently damaged telomeres promotes tumorigenic transformation of mouse cells.Citation92
Figure 3. Mitotic bypass associated with p21 induction triggers senescence in tetraploid G1 cells. In p53-proficient cells, DNA damage-induced G2 arrest leads to p21-dependent nuclear sequestration of cyclin B1-Cdk1 (cB1) complexes, thus locking them in the inactive state (cf. Fig. 2). Subsequent APC/C-Cdh1-mediated degradation of cyclin B1 and other mitotic regulators precedes mitotic bypass (skip), leading to irreversible arrest (senescence) in a tetraploid (4N) G1 phase, characterized by accumulation of G1 cyclins (cyclin D1, cD1). In p53-deficient cells, in the case of persistent telomere dysfunction, DNA damage induces strong and sustained Chk1/2 activation leading to stable G2 arrest that, via mitotic bypass, leads to endoreplication, resulting in tetraploidy.Citation88 In the absence of sustained Chk1/2 activation, p53/p21-deficient cells exposed to genotoxic agents arrest only transiently in G2 and then progress into mitosis and eventually die.Citation59,93 Similarly, in aging p53/pRb-deficient cells, extensive telomere erosion induces crisis with massive cell death and tetraploidization through endoreplication or mitotic failure.Citation92
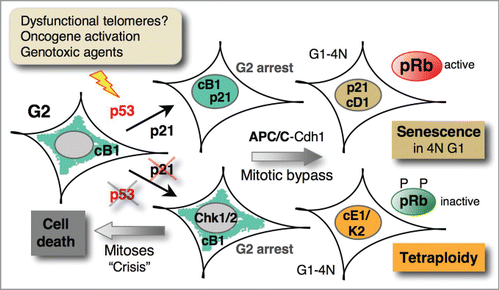
The most recent evidence implicating the G2 exit program in senescence has come from 3 publications that also took advantage of FUCCI imaging system. The Nakanishi group showed that replicative senescence or exposure to various senescence-causing stimuli induces p53-dependent mitotic bypass (referred to as mitotic skipping), entailing irreversible cell cycle arrest in the 4N G1 state and accumulation of the senescent markers p16 and β-galactosidase.Citation93 Moreover, stable G2 arrest induced by ionizing radiation (IR) was associated with p53/p21-dependent premature APC/CCdh1 activation and degradation of mitotic cyclins, consistent with previous observations.Citation74-76 However, unlike p53-mediated mitotic bypass, APC/CCdh1 activation alone does not appear to be sufficient to induce senescence, that additionally requires repression of mitotic regulators by pRb family pocket proteins (). The finding that p16 is dispensable both for mitotic skipping and induction of senescence, but is required for its maintenance, is in full agreement with the models proposed earlier whereby p21 is a major mediator of irreversible cell cycle arrest.Citation28,49 Importantly, this work also provided evidence for the occurrence of 4N senescent cells in human melanocyte naevi, the first in vivo senescence model,Citation94 suggesting that the G2 exit program has a more general role in senescence. The results showing that mitotic bypass requires p53, as its absence leads to aberrant mitosis and cell death, corroborate the observations by the Karlseder team,Citation83 but they apparently contradict the finding that mitotic bypass in p53-deficient cells leads to endoreplication.Citation88 The most likely explanation is that the strong and persistent DNA damage signaling induced by POT1a/b double knock-downCitation88 causes sustained Chk1/2 activation that prevents Cdk1 activation (and mitosis), resulting in prolonged G2 arrest. This would enable the mitotic bypass and the onset of a new round of replication (). Conversely, transient Chk1/2 activation, induced partial TRF2 depletion Citation83 or acute DNA damage (by IR),Citation93 in the absence of (efficient) p21 induction can transiently delay, but not prevent mitosis.
In an accompanying paper, also using the FUCCI system and cell lines that express a cyclin B1 allele with a fluorescent tag, the Medema group investigated the role of p21-dependent cyclin B1 nuclear retention in senescence induction in G2-arrested cells upon IR.Citation95 They elegantly confirmed and extended previous observationsCitation58,69 and pinpointed this event as the first step in triggering the G2 exit program, resulting in permanent cell cycle withdrawal (). Surprisingly, and somewhat counter-intuitively, they showed that APC/CCdh1–mediated cyclin B1 degradation before cell cycle exit is actually dispensable for the latter. This result is in agreement with observations by the Nakanishi group showing that Cdh1 is not required for senescence onset.Citation93 Identical conclusions were reached in the recent paper by the Lindqvist team using a similar experimental approach.Citation96 In addition, by using fluorescent cyclin B1 and video-microscopy, they confirmed earlier observationsCitation72 that p21-dependent sequestration of cyclin B1 does not take place in U2OS cells. In this p53/pRb proficient cell line, p21 induction is impaired due to inefficient ATM response Citation72 and, probably, mutation of the Wip1 phosphatase that interferes with p53 activation.Citation97 It is not clear, however, how p21-mediated nuclear translocation of cyclin B1-Cdk1 complexes can induce senescence. Although this is the first detectable event, it is more likely that main role of p21 lies in its capacity to trigger pRb-mediated repression of cell cycle regulators (, for more information on p21 functions (see ref.Citation98). Indeed, as mentioned above, p21/p27-mediated cyclin B1 nuclear retention in mouse fibroblasts lacking pocket proteins does not induce senescence.Citation73 However, at the present time, the first crucial event in the irreversible journey to senescence has not been clearly identified yet.
A final comment should be made on the importance of p21 in inducing senescence upon G2 arrest. While the previously discussed worksCitation93,95 show that transient induction of p53 in G2 (but not in G1) is sufficient to induce mitotic bypass and senescence, this is not the case for p21.Citation93 Moreover, p21 depletion prevents senescence less efficiently than p53 depletion, prompting the authors to suggest that p53 might activate the pRb family independently of p21 (Fig. 5G in ref.Citation93). While this hypothesis cannot be entirely dismissed, a more likely explanation is that p16 might compensate for the absence of p21 in blocking pRb phosphorylation by cyclin D1-Cdk/6 (and ), thus justifying the proposed role of the p16-pRb pathway in senescence maintenance. In addition to a previous report,Citation48 this alternative possibility is also supported by data showing that cell cycle exit in p21-deficient cells is associated with p16 accumulation (Figure S3B in ref.Citation93). It is unclear, however, why in this experimental system, p16 induction was not observed in p53-deficient cells ( in ref.Citation93). Clearly, further studies are required to identify the missing components of the senescence program in G2.
In conclusion, a flurry of papers in the last 2 y has highlighted the relevance of the G2 exit program in senescence. Moreover, some data also suggest that the G2/M and not the G1/S checkpoint might be responsible for the onset of replicative senescence. The finding that the decision to enter quiescence also occurs at late cell cycle stages Citation99,100 might further change our understanding of the importance of the G2/M checkpoint in cell cycle exit.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank Drs. Etienne Schwob, Jacques Piette and Daniel Fisher for their encouragements to write this review and D.F. and J.P. for critically reading the manuscript. Finally, we thank Dr. Liliana Krasinska for help with the figures.
Funding
This work is supported by the grant E22013.LNCC/DF to Daniel Fisher's team (V.D.) and SIRIC N°095294 to Pierre Roux's team (V.G.).
Related Research Data
References
- Kuilman T, Michaloglou C, Mooi WJ Peeper DS. The essence of senescence. Genes Dev 2010; 24:2463-79; PMID:21078816; http://dx.doi.org/10.1101/gad.1971610
- Salama R, Sadaie M, Hoare M Narita M. Cellular senescence and its effector programs. Genes Dev 2014; 28:99-114; PMID:24449267; http://dx.doi.org/10.1101/gad.235184.113
- Burkhart DL Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 2008; 8:671-82; PMID:18650841; http://dx.doi.org/10.1038/nrc2399
- Polager S Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer 2009; 9:738-48; PMID:19776743; http://dx.doi.org/10.1038/nrc2718
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008; 9:759-69; PMID:18813293; http://dx.doi.org/10.1038/nrm2514
- Demidenko ZN, Korotchkina LG, Gudkov AV Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A 2010; 107:9660-4; PMID:20457898; http://dx.doi.org/10.1073/pnas.1002298107
- Kolesnichenko M, Hong L, Liao R, Vogt PK Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle 2012; 11:2391-401; PMID:22627671; http://dx.doi.org/10.4161/cc.20683
- Dulic V. Senescence regulation by mTOR. Methods Mol Biol 2013; 965:15-35; PMID:23296649; http://dx.doi.org/10.1007/978-1-62703-239-1_2
- Hasty P, Sharp ZD, Curiel TJ Campisi J. mTORC1 and p53: clash of the gods? Cell Cycle 2013; 12:20-5; PMID:23255104; http://dx.doi.org/10.4161/cc.22912
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633-7; PMID:17136093; http://dx.doi.org/10.1038/nature05268
- Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell 2009; 36:2-14; PMID:19818705; http://dx.doi.org/10.1016/j.molcel.2009.09.021
- Rodier F Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192:547-56; PMID:21321098; http://dx.doi.org/10.1083/jcb.201009094
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434:864-70; PMID:15829956; http://dx.doi.org/10.1038/nature03482
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr., Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434:907-13; PMID:15829965; http://dx.doi.org/10.1038/nature03485
- Collado M Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10:51-7; PMID:20029423; http://dx.doi.org/10.1038/nrc2772
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479:232-6; PMID:22048312; http://dx.doi.org/10.1038/nature10600
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011; 479:547-551; PMID:22080947; http://dx.doi.org/10.1038/nature10599
- Krtolica A, Parrinello S, Lockett S, Desprez PY Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A 2001; 98:12072-7; PMID:11593017; http://dx.doi.org/10.1073/pnas.211053698
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6:2853-68; PMID:19053174; http://dx.doi.org/10.1371/journal.pbio.0060301
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656-60; PMID:17251933; http://dx.doi.org/10.1038/nature05529
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M et al. Programmed cell senescence during mammalian embryonic development. Cell 2013; 155:1104-18; PMID:24238962; http://dx.doi.org/10.1016/j.cell.2013.10.019
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013; 155:1119-30; PMID:24238961; http://dx.doi.org/10.1016/j.cell.2013.10.041
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell 2008; 134:657-67; PMID:18724938; http://dx.doi.org/10.1016/j.cell.2008.06.049
- Jun JI Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 2010; 12:676-85; PMID:20526329; http://dx.doi.org/10.1038/ncb2070
- Stein GH Dulic V. Origins of G1 arrest in senescent human fibroblasts. Bioessays 1995; 17:537-43; PMID:7575495; http://dx.doi.org/10.1002/bies.950170610
- Smith JR Pereira-Smith OM. Replicative senescence: implications for in vivo aging and tumor suppression. Science 1996; 273:63-7; PMID:8658197; http://dx.doi.org/10.1126/science.273.5271.63
- Dulic V, Drullinger LF, Lees E, Reed SI Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci U S A 1993; 90:11034-8; PMID:8248208; http://dx.doi.org/10.1073/pnas.90.23.11034
- Stein GH, Drullinger LF, Soulard A Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 1999; 19:2109-17; PMID:10022898
- Harbour JW, Luo RX, Dei Santi A, Postigo AA Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999; 98:859-69; PMID:10499802; http://dx.doi.org/10.1016/S0092-8674(00)81519-6
- Sherr CJ Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004; 18:2699-711; PMID:15545627; http://dx.doi.org/10.1101/gad.1256504
- Hwang HC Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005; 24:2776-86; PMID:15838514; http://dx.doi.org/10.1038/sj.onc.1208613
- Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ Reed SI. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994; 76:1013-23; PMID:8137420; http://dx.doi.org/10.1016/0092-8674(94)90379-4
- Waldman T, Kinzler KW Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55:5187-90; PMID:7585571
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003; 426:194-8; PMID:14608368; http://dx.doi.org/10.1038/nature02118
- Gire V. Dysfunctional telomeres at senescence signal cell cycle arrest via Chk2. Cell Cycle 2004; 3:1217-20; PMID:15467458; http://dx.doi.org/10.4161/cc.3.10.1167
- d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 2008; 8:512-22; PMID:18574463; http://dx.doi.org/10.1038/nrc2440
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638-42; PMID:17136094; http://dx.doi.org/10.1038/nature05327
- Mallette FA, Gaumont-Leclerc MF Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 2007; 21:43-8; PMID:17210786; http://dx.doi.org/10.1101/gad.1487307
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M Lowe SW. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 2010; 17:376-87; PMID:20385362; http://dx.doi.org/10.1016/j.ccr.2010.01.023
- Stein GH, Beeson M Gordon L. Failure to phosphorylate the retinoblastoma gene product in senescent human fibroblasts. Science 1990; 249:666-9; PMID:2166342; http://dx.doi.org/10.1126/science.2166342
- Stein GH, Drullinger LF, Robetorye RS, Pereira-Smith OM Smith JR. Senescent cells fail to express cdc2, cycA, and cycB in response to mitogen stimulation. Proc Natl Acad Sci U S A 1991; 88:11012-6; PMID:1722313; http://dx.doi.org/10.1073/pnas.88.24.11012
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 1996; 93:13742-7; PMID:8943005; http://dx.doi.org/10.1073/pnas.93.24.13742
- Serrano M, Lin AW, McCurrach ME, Beach D Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593-602; PMID:9054499; http://dx.doi.org/10.1016/S0092-8674(00)81902-9
- Sandhu C, Peehl DM Slingerland J. p16INK4A mediates cyclin dependent kinase 4 and 6 inhibition in senescent prostatic epithelial cells. Cancer Res 2000; 60:2616-22; PMID:10825132
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O and et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363-7; PMID:7568133; http://dx.doi.org/10.1073/pnas.92.20.9363
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest 2004; 114:1299-307; PMID:15520862; http://dx.doi.org/10.1172/JCI22475
- Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH Sharpless NE. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013; 152:340-51; PMID:23332765; http://dx.doi.org/10.1016/j.cell.2012.12.010
- Wei W, Herbig U, Wei S, Dutriaux A Sedivy JM. Loss of retinoblastoma but not p16 function allows bypass of replicative senescence in human fibroblasts. EMBO Rep 2003; 4:1061-6; PMID:14566323; http://dx.doi.org/10.1038/sj.embor.7400001
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo J 2003; 22:4212-22; PMID:12912919; http://dx.doi.org/10.1093/emboj/cdg417
- Herbig U, Jobling WA, Chen BP, Chen DJ Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 2004; 14:501-13; PMID:15149599; http://dx.doi.org/10.1016/S1097-2765(04)00256-4
- Gil J Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7:667-77; PMID:16921403; http://dx.doi.org/10.1038/nrm1987
- Witkiewicz AK, Knudsen KE, Dicker AP Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 2011; 10:2497-503; PMID:21775818; http://dx.doi.org/10.4161/cc.10.15.16776
- Waldman T, Lengauer C, Kinzler KW Vogelstein B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 1996; 381:713-6; PMID:8649519; http://dx.doi.org/10.1038/381713a0
- Dulic V, Stein GH, Far DF Reed SI. Nuclear accumulation of p21Cip1 at the onset of mitosis: a role at the G2/M-phase transition. Mol Cell Biol 1998; 18:546-57; PMID:9418901
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497-501; PMID:9822382; http://dx.doi.org/10.1126/science.282.5393.1497
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15:2177-96; PMID:11544175; http://dx.doi.org/10.1101/gad.914401
- Levedakou EN, Kaufmann WK, Alcorta DA, Galloway DA Paules RS. p21CIP1 is not required for the early G2 checkpoint response to ionizing radiation. Cancer Res 1995; 55:2500-2; PMID:7780956
- Smits VA, Klompmaker R, Vallenius T, Rijksen G, Makela TP Medema RH. p21 inhibits Thr161 phosphorylation of Cdc2 to enforce the G2 DNA damage checkpoint. J Biol Chem 2000; 275:30638-43; PMID:10913154; http://dx.doi.org/10.1074/jbc.M005437200
- Baus F, Gire V, Fisher D, Piette J Dulic V. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. Embo J 2003; 22:3992-4002; PMID:12881433; http://dx.doi.org/10.1093/emboj/cdg387
- Flatt PM, Tang LJ, Scatena CD, Szak ST Pietenpol JA. p53 regulation of G(2) checkpoint is retinoblastoma protein dependent. Mol Cell Biol 2000; 20:4210-4223; PMID:10825186; http://dx.doi.org/10.1128/MCB.20.12.4210-4223.2000
- Passalaris TM, Benanti JA, Gewin L, Kiyono T Galloway DA. The G(2) checkpoint is maintained by redundant pathways. Mol Cell Biol 1999; 19:5872-5881; PMID:10454534
- Crawford DF Piwnica-Worms H. The G(2) DNA damage checkpoint delays expression of genes encoding mitotic regulators. J Biol Chem 2001; 276:37166-77; PMID:11483598; http://dx.doi.org/10.1074/jbc.M103414200
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA Dynlacht BD. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 2002; 16:245-256; PMID:11799067; http://dx.doi.org/10.1101/gad.949802
- Jackson MW, Agarwal MK, Yang J, Bruss P, Uchiumi T, Agarwal ML, Stark GR Taylor WR. p130/p107/p105Rb-dependent transcriptional repression during DNA-damage-induced cell-cycle exit at G2. J Cell Sci 2005; 118:1821-32; PMID:15827088; http://dx.doi.org/10.1242/jcs.02307
- Lanni JS Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol 1998; 18:1055-64; PMID:9448003
- Stewart ZA, Leach SD Pietenpol JA. p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol Cell Biol 1999; 19:205-15; PMID:9858545
- Andreassen PR, Lacroix FB, Lohez OD Margolis RL. Neither p21(WAF1) Nor 14-3-3sigma Prevents G(2) Progression to Mitotic Catastrophe in Human Colon Carcinoma Cells after DNA Damage, But p21(WAF1) Induces Stable G(1) Arrest in Resulting Tetraploid Cells. Cancer Res 2001; 61:7660-8; PMID:11606409
- Davoli T de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol 2011; 27:585-610; PMID:21801013; http://dx.doi.org/10.1146/annurev-cellbio-092910-154234
- Charrier-Savournin FB, Chateau MT, Gire V, Sedivy J, Piette J Dulic V. p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Mol Biol Cell 2004; 15:3965-76; PMID:15181148; http://dx.doi.org/10.1091/mbc.E03-12-0871
- Chan TA, Hermeking H, Lengauer C, Kinzler KW Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage ; see comments. Nature 1999; 401:616-620; PMID:10524633; http://dx.doi.org/10.1038/44188
- Winters ZE, Ongkeko WM, Harris AL Norbury CJ. p53 regulates Cdc2 independently of inhibitory phosphorylation to reinforce radiation-induced G2 arrest in human cells. Oncogene 1998; 17:673-84; PMID:9715268; http://dx.doi.org/10.1038/sj.onc.1201991
- Lossaint G, Besnard E, Fisher D, Piette J Dulic V. Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATM/p53/p21 pathway is functional. Oncogene 2011; 30:4261-74; PMID:21532626; http://dx.doi.org/10.1038/onc.2011.135
- Foijer F, Wolthuis RM, Doodeman V, Medema RH te Riele H. Mitogen requirement for cell cycle progression in the absence of pocket protein activity. Cancer Cell 2005; 8:455-66; PMID:16338659; http://dx.doi.org/10.1016/j.ccr.2005.10.021
- Lee J, Kim JA, Barbier V, Fotedar A Fotedar R. DNA damage triggers p21WAF1-dependent Emi1 down-regulation that maintains G2 arrest. Mol Biol Cell 2009; 20:1891-902; PMID:19211842; http://dx.doi.org/10.1091/mbc.E08-08-0818
- Gillis LD, Leidal AM, Hill R Lee PW. p21Cip1/WAF1 mediates cyclin B1 degradation in response to DNA damage. Cell Cycle 2009; 8:253-6; PMID:19158493; http://dx.doi.org/10.4161/cc.8.2.7550
- Wiebusch L Hagemeier C. p53- and p21-dependent premature APC/C-Cdh1 activation in G2 is part of the long-term response to genotoxic stress. Oncogene 2010; 29:3477-89; PMID:20383190; http://dx.doi.org/10.1038/onc.2010.99
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008; 134:256-67; PMID:18662541; http://dx.doi.org/10.1016/j.cell.2008.05.043
- Lobrich M Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 2007; 7:861-9; PMID:17943134; http://dx.doi.org/10.1038/nrc2248
- Mao Z, Ke Z, Gorbunova V Seluanov A. Replicatively senescent cells are arrested in G1 and G2 phases. Aging (Albany NY) 2012; 4:431-5; PMID:22745179
- Leontieva OV, Lenzo F, Demidenko ZN Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle 2012; 11:4642-9; PMID:23187803; http://dx.doi.org/10.4161/cc.22937
- Jullien L, Mestre M, Roux P Gire V. Eroded human telomeres are more prone to remain uncapped and to trigger a G2 checkpoint response. Nucleic Acids Res 2013; 41:900-11; PMID:23193277
- Verdun RE, Crabbe L, Haggblom C Karlseder J. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell 2005; 20:551-61; PMID:16307919; http://dx.doi.org/10.1016/j.molcel.2005.09.024
- Cesare AJ, Hayashi MT, Crabbe L Karlseder J. The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol Cell 2013; 51:141-55; PMID:23850488; http://dx.doi.org/10.1016/j.molcel.2013.06.006
- Ahn J, Urist M Prives C. Questioning the role of checkpoint kinase 2 in the p53 DNA damage response. J Biol Chem 2003; 278:20480-9; PMID:12654916; http://dx.doi.org/10.1074/jbc.M213185200
- Jallepalli PV, Lengauer C, Vogelstein B Bunz F. The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem 2003; 278:20475-9; PMID:12654917; http://dx.doi.org/10.1074/jbc.M213159200
- Gong Y de Lange T. A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol Cell 2010; 40:377-87; PMID:21070964; http://dx.doi.org/10.1016/j.molcel.2010.10.016
- Gire V, Roux P, Wynford-Thomas D, Brondello JM Dulic V. DNA damage checkpoint kinase Chk2 triggers replicative senescence. Embo J 2004; 23:2554-63; PMID:15192702; http://dx.doi.org/10.1038/sj.emboj.7600259
- Davoli T, Denchi EL de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010; 141:81-93; PMID:20371347; http://dx.doi.org/10.1016/j.cell.2010.01.031
- Toettcher JE, Loewer A, Ostheimer GJ, Yaffe MB, Tidor B Lahav G. Distinct mechanisms act in concert to mediate cell cycle arrest. Proc Natl Acad Sci U S A 2009; 106:785-90; PMID:19139404; http://dx.doi.org/10.1073/pnas.0806196106
- Ye C, Zhang X, Wan J, Chang L, Hu W, Bing Z, Zhang S, Li J, He J, Wang J, et al. Radiation-induced cellular senescence results from a slippage of long-term G2 arrested cells into G1 phase. Cell Cycle 2013; 12:1424-32; PMID:23574719; http://dx.doi.org/10.4161/cc.24528
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H Miyawaki A. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008; 132:487-98; PMID:18267078; http://dx.doi.org/10.1016/j.cell.2007.12.033
- Davoli T de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 2012; 21:765-76; PMID:22698402; http://dx.doi.org/10.1016/j.ccr.2012.03.044
- Johmura Y, Shimada M, Misaki T, Naiki-Ito A, Miyoshi H, Motoyama N, Ohtani N, Hara E, Nakamura M, Morita A, et al. Necessary and Sufficient Role for a Mitosis Skip in Senescence Induction. Mol Cell 2014; 55:73-84; PMID:24910096
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436:720-4; PMID:16079850; http://dx.doi.org/10.1038/nature03890
- Krenning L, Feringa FM, Shaltiel IA, van den Berg J Medema RH. Transient Activation of p53 in G2 Phase Is Sufficient to Induce Senescence. Mol Cell 2014; 55:59-72; PMID:24910099
- Müllers E, Cascales HS, Jaiswal H, Saurin AT Lindqvist A. Nuclear translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle 2014; 13:2733-2743; PMID:25486360; http://dx.doi.org/10.4161/15384101.2015.945831
- Kleiblova P, Shaltiel IA, Benada J, Sevcik J, Pechackova S, Pohlreich P, Voest EE, Dundr P, Bartek J, Kleibl Z, et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol 2013; 201:511-21; PMID:23649806; http://dx.doi.org/10.1083/jcb.201210031
- Abbas T Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009; 9:400-14; PMID:19440234; http://dx.doi.org/10.1038/nrc2657
- Chassot AA, Lossaint G, Turchi L, Meneguzzi G, Fisher D, Ponzio G Dulic V. Confluence-induced cell cycle exit involves pre-mitotic CDK inhibition by p27(Kip1) and cyclin D1 downregulation. Cell Cycle 2008; 7:2038-46; PMID:18604165; http://dx.doi.org/10.4161/cc.7.13.6233
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013; 155:369-83; PMID:24075009; http://dx.doi.org/10.1016/j.cell.2013.08.062