
Abstract
Multiple Myeloma (MM) is a haematologic malignancy characterized by the accumulation of clonal plasma cells in the bone marrow. Over the last 10–15 y the introduction of the proteasome-inhibitor bortezomib has improved MM prognosis, however relapse due to bortezomib-resistance is inevitable and the disease, at present, remains incurable. To model bortezomib-resistant MM we generated bortezomib-resistant MM cell lines (n = 4 ) and utilised primary malignant plasma cells from patients relapsing after bortezomib treatment (n = 6 ). We identified enhanced Bruton's tyrosine kinase (BTK) activity in bortezomib-resistant MM cells and found that inhibition of BTK, either pharmacologically with ibrutinib (0.5 μM) or via lenti-viral miRNA-targeted BTK interference, re-sensitized previously bortezomib-resistant MM cells to further bortezomib therapy at a physiologically relevant concentration (5 nM). Further analysis of pro-survival signaling revealed a role for the NF-κB p65 subunit in MM bortezomib-resistance, thus a combination of BTK and NF-κB p65 inhibition, either pharmacologically or via further lenti-viral miRNA NF-κB p65 interference, also restored sensitivity to bortezomib, significantly reducing cell viability (37.5 ± 6 .9 %, ANOVA P ≤ 0 .001). Accordingly, we propose the clinical evaluation of a bortezomib/ibrutinib combination therapy, including in patients resistant to single-agent bortezomib.
Abbreviations
| MM – | = | multiple myeloma |
| PI – | = | proteasome inhibitor |
| NF-κB – | = | nuclear factor-kappa B |
| BMSC – | = | bone marrow stromal cells |
| BTK – | = | Bruton's tyrosine kinase |
Introduction
Multiple Myeloma (MM) is characterized by the accumulation of clonal plasma cells in the bone marrow. The American Cancer Society estimates that there will be approximately 24,000 new cases diagnosed and 11,000 deaths from MM in 2014 in the USCitation1 Clinical manifestations of the disease occur as a consequence of the tumor bulk; including anaemia and fatigue, immune paresis leading to infection, renal failure, and osteolytic bone breakdown by activated osteoclasts, resulting in painful lytic bone destruction and hypercalcaemiaCitation2 An effective MM therapeutic succeeds, therefore, by 'de-bulking' the MM tumor mass, thus reducing the associated symptoms and subsequently maintaining remission.
The proteasome inhibitor (PtdIns) bortezomib, approved for the treatment of MM by the FDA in 2003,Citation3 is one such de-bulking agent that has greatly contributed to improved outcomes observed in MMCitation4 Despite this advance, however, relapse following bortezomib therapy remains inevitable due to the emergence of bortezomib-resistant plasma cell sub-clonesCitation5 For example; recent 'whole genome' sequencing studies of MM patients confirmed a high level of genetic heterogeneity; occurring both between separate patients, and within patient samples before and after therapy,Citation6 indicating the presence of a variety of genetically distinct plasma cell 'sub-clones'Citation7,8 Different sub-clones further indicate the presence of numerous minor tumor initiating cell populations with complex and divergent evolutionary histories.Citation9 The potential quiescence of these MM sub-clones, coupled with their diversity, can contribute to enhanced tumorigenicity and an intrinsic resistance to therapy.Citation10 Therefore; the eradication of the majority bortezomib-sensitive sub-clones may ultimately promote the growth of pre-existing minority bortezomib-resistant sub-clones, limiting the efficacy of single-agent bortezomib. This finding is further supported by the clinical observation that approximately half of initially bortezomib-sensitive MM patients are no longer able to respond to bortezomib once relapsed.Citation11 This sub-clonal bortezomib-resistance has been attributed to a range of mechanisms; including enhanced growth factor expression,Citation12 mutated proteasome subunits,Citation13 deregulated plasma cell maturation markers,Citation14 and nuclear factor-kappa B (NF-κB) pathway 'addiction'Citation15 [for a more in-depth exploration of this topic see Murray et al. 2014Citation16]. Furthermore, pro-survival NF-κB signaling pathway members were also found to have a broader than anticipated profile in MM whole genome sequencing data.Citation6 These findings are consistent with our previous studies into the role of NF-κB signaling in haematological malignancies,Citation17-19 suggesting that a greater understanding of the NF-κB signaling network in bortezomib-resistant MM may be central to achieving therapeutic advances in this disease.
In its primary mode of action bortezomib successfully inhibits 'inducible' NF-κB expression in MM cells, such as the expression stimulated by MM-bone marrow stromal cell (BMSC) interaction,Citation20 via its function as an inhibitor of the 20s proteasome β5 subunit.Citation21 Conversely, however, bortezomib also enhances 'constitutive' levels of NF-κB through activation of IKKβ, ultimately leading to NF-κB nuclear translocation and the transcription of multiple NF-κB-induced genes, including Bruton's tyrosine kinase (BTK).Citation22 BTK, a non-receptor tyrosine kinase, is now known to be of key importance to a number of haematological malignancies, including MM,Citation23 chronic lymphocytic leukemia (CLL)Citation24 and acute myeloid leukemia (AML).Citation25 The potential feedback mechanism between NF-κB and BTK signaling, whereby BTK also lies upstream of several NF-κB inducible signaling pathways,Citation26,27 provides a rationale for investigation of combined NF-κB and BTK inhibition in MM.
Previously we and others have shown ex vivo efficacy of BTK inhibition in MM.Citation17,23 Specifically, we have shown that the irreversible BTK inhibitor ibrutinib can enhance the action of bortezomib via repression of the NF-κB survival pathway in primary tissue, e.g., bone marrow-derived MM cells from treatment-naïve patients.Citation17 Despite these findings, however, early phase II clinical trial data for ibrutinib monotherapy in MM have so far proved disappointing,Citation28 while the study of ibrutinib efficacy in patients with relapsed of refractory MM is currently recruiting (ClinicalTrials.gov Identifier:NCT01478581).
Taken together these data highlight the need to develop novel therapeutic strategies that can overcome bortezomib-resistance,Citation16 while still de-bulking the tumor and protecting the patient from related organ and tissue impairment.
MM clonal development and selection is impacted by the timing, order, and combinations of therapies received; however, all patients treated with bortezomib therapy are destined to relapse and become bortezomib-resistant. Here, we utilize in vitro modeling to demonstrate BTK pro-survival pathway activity in bortezomib-resistant MM. These data provide justification for further assessment of greater patient numbers in the clinic, which will establish whether ibrutinib therapy can be used to overcome bortezomib-resistance in MM in practice.
Results
Generation and characterization of bortezomib-resistant MM cells
To determine the importance of the BTK pro-survival pathway in bortezomib-resistance we first generated bortezomib-resistant cells in vitro. We cultured MM-derived B lymphocytic cell lines (H929, LP-1, RPMI-8226, and U266) in the continuous presence of bortezomib, increasing bortezomib concentration in stepwise increments up to 10 nM. Corresponding bortezomib-naïve MM cell lines were used as a control. Bortezomib-resistance in the cell lines was confirmed by a significant increase in IC50 log[bortezomib] () and enhanced cell viability following exposure to physiologically relevantCitation29 levels of bortezomib [5 nM ()] when compared to control 'bortezomib-naïve' MM cells. In addition, we explored relative chymotrypsin-like proteasome activity in bortezomib-naïve and bortezomib-resistant MM cell lines and observed significantly higher levels of proteasome activity in bortezomib-resistant MM cells (), further confirming their inability to respond to bortezomib's proteasome-inhibitor function.
Figure 1. Characterization of bortezomib-resistant MM cell lines, and primary relapsed MM patient samples. (A) The IC50 Log[bortezomib] (nM) of bortezomib-naïve and bortezomib-resistant MM cell lines was analyzed by luminescent cell viability assay. (B) Relative cell viability (5 nM/48 h) and (C) chymotrypsin-like proteasome activity in bortezomib-naïve and bortezomib-resistant MM cell lines in response to (5 nM/4 h) bortezomib. (D) Relative cell viability of primary naïve and primary relapsed MM patient samples in response to bortezomib (5 nM/24 h). Statistical significance between treatments was calculated by Student's t test; * indicates P ≤ 0.05. Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.05.
![Figure 1. Characterization of bortezomib-resistant MM cell lines, and primary relapsed MM patient samples. (A) The IC50 Log[bortezomib] (nM) of bortezomib-naïve and bortezomib-resistant MM cell lines was analyzed by luminescent cell viability assay. (B) Relative cell viability (5 nM/48 h) and (C) chymotrypsin-like proteasome activity in bortezomib-naïve and bortezomib-resistant MM cell lines in response to (5 nM/4 h) bortezomib. (D) Relative cell viability of primary naïve and primary relapsed MM patient samples in response to bortezomib (5 nM/24 h). Statistical significance between treatments was calculated by Student's t test; * indicates P ≤ 0.05. Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.05.](/cms/asset/2f7f835c-59ff-4b63-ab44-a8698e61f480/kccy_a_998067_f0001_b.gif)
We also analyzed primary human treatment-naïve MM patient samples (primary naïve; n=5 ) and primary human MM samples from patients that had initially responded to bortezomib but subsequently relapsed (primary relapsed; n=6 ). All primary naïve patient samples analyzed (n=5 /5) showed significantly reduced cell viability in response to bortezomib in vitro, while the majority (n=4 /6) of primary relapsed samples showed no significant reduction in viability. The remaining (n=2 /6) primary relapsed samples showed significantly decreased cell viability in response to bortezomib, in-keeping with the clinical observation that approximately half of patients retreated with bortezomib will respond again Citation30,31 ().Citation30,31
Basal BTK activity is enhanced in bortezomib-resistant MM cell lines and is resistant to inhibition with bortezomib
We have previously shown that the BTK pathway is active in MM.Citation17 Furthermore, others have shown that bortezomib can reduce the expression of BTK mRNA and protein via an NF-κB p65-dependent mechanism.Citation22 We therefore examined whether BTK expression and activity in bortezomib-resistant MM cell lines reflects a mechanistic change in the BTK pro-survival signaling pathway in response to bortezomib treatment. We first examined basal levels of BTK activity in bortezomib-naïve and bortezomib-resistant U266 MM cells by immunocytochemistry. Utilizing antibodies against total BTK (BTK) and Y223-phosphorylated BTK (pBTK) we observed higher levels of pBTK in bortezomib-resistant cells [n ≥ 80] compared to bortezomib-naïve cells, suggesting a greater level of BTK activity in the resistant cells (; left panel). Further quantitative analysis of the immunocytochemistry confirmed significantly enhanced basal pBTK levels in bortezomib-resistant MM cell lines (; right panel [n ≥ 20 ]). We next examined what effect this enhanced level of basal BTK activity has on bortezomib-resistant MM cell response to further bortezomib exposure. Following bortezomib treatment (4 h), we observed a significant decrease in relative BTK mRNA in the bortezomib-naïve but not bortezomib-resistant MM cell lines (), suggesting BTK expression is not inhibited by bortezomib treatment in the bortezomib resistant cells.
Figure 2. Enhanced BTK activity in bortezomib-resistant MM cell lines is resistant to inhibition with bortezomib. (A) Representative immunocytochemistry of basal levels of total and phosphorylated BTK (pBTK) in bortezomib-naïve and bortezomib-resistant U266 MM cells with DAPI nuclear staining [left panel] and subsequent quantification of basal pBTK/total BTK (%) staining intensity [right panel]. Box and whisker indicates the mean ± SEM (n ≥ 80). (B) qRT-PCR analysis of BTK mRNA following bortezomib (5 nM/4 h) exposure in bortezomib-naïve and bortezomib-resistant U266 MM cells relative to GAPDH. Values indicate the mean ± SEM from 3 independent experiments. (C) Representation of wild type pGL4.BTK promoter-luciferase vector [including κB binding sites] transfected into bortezomib-naïve and bortezomib-resistant U266 MM cells. (D) Luciferase activity of pGL4.BTK promoter-luciferase vectors in bortezomib-naïve and bortezomib-resistant U266 MM cells following bortezomib (5 nM/24 h) exposure, normalized by co-transfection with pRL-TK Renilla Luciferase Reporter Vectors. Values indicate the mean ± SEM from 3 independent experiments. Statistical significance between treatments was calculated by Student's t test; * indicates P ≤ 0.05. Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.01.
![Figure 2. Enhanced BTK activity in bortezomib-resistant MM cell lines is resistant to inhibition with bortezomib. (A) Representative immunocytochemistry of basal levels of total and phosphorylated BTK (pBTK) in bortezomib-naïve and bortezomib-resistant U266 MM cells with DAPI nuclear staining [left panel] and subsequent quantification of basal pBTK/total BTK (%) staining intensity [right panel]. Box and whisker indicates the mean ± SEM (n ≥ 80). (B) qRT-PCR analysis of BTK mRNA following bortezomib (5 nM/4 h) exposure in bortezomib-naïve and bortezomib-resistant U266 MM cells relative to GAPDH. Values indicate the mean ± SEM from 3 independent experiments. (C) Representation of wild type pGL4.BTK promoter-luciferase vector [including κB binding sites] transfected into bortezomib-naïve and bortezomib-resistant U266 MM cells. (D) Luciferase activity of pGL4.BTK promoter-luciferase vectors in bortezomib-naïve and bortezomib-resistant U266 MM cells following bortezomib (5 nM/24 h) exposure, normalized by co-transfection with pRL-TK Renilla Luciferase Reporter Vectors. Values indicate the mean ± SEM from 3 independent experiments. Statistical significance between treatments was calculated by Student's t test; * indicates P ≤ 0.05. Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.01.](/cms/asset/f410d49a-dfc3-41ec-9b5a-7f580c1a3ae0/kccy_a_998067_f0002_c.gif)
To further investigate this difference in BTK mRNA expression and activity in bortezomib-resistant MM cells we examined activity of the BTK promoter, utilizing a chemi-luminescent BTK promoter-luciferase reporter construct, specifically containing 2 NF-κB transcription factor binding sites [κB1 and κB2]; pGL4.BTK (). Following 24 h bortezomib treatment bortezomib-naïve U266 MM cells transfected with pGL4.BTK showed significantly reduced BTK promoter activity (). Conversely, there was no change in BTK promoter activity when bortezomib-resistant U266 MM cells were transfected with pGL4.BTK, and the level of BTK promoter activity in these cells remained significantly higher than in bortezomib-naïve cells exposed to bortezomib. These results demonstrate that the ability of bortezomib to repress BTK promoter activity is lost in bortezomib-resistant MM cells.
Pharmacological BTK inhibition with ibrutinib restores sensitivity to bortezomib in bortezomib-resistant MM cells
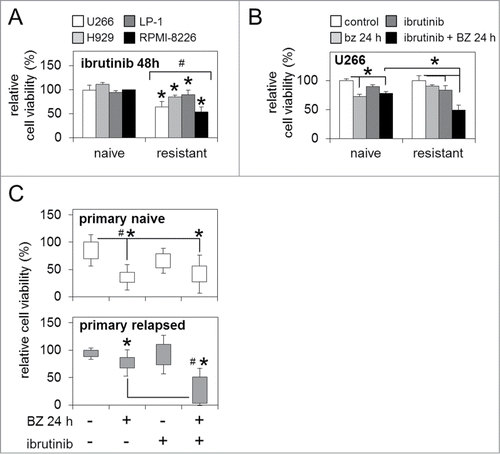
As BTK is emerging as an important regulator of downstream survival pathways in MM,Citation17,23 and further to our observations of enhanced BTK activity and expression in bortezomib-resistant MM cells (), we investigated the functional effect of combined pharmacological BTK inhibition and bortezomib treatment in bortezomib-naïve and bortezomib-resistant MM cell lines and primary human MM samples. Ibrutinib 'pulse' treatment [0.5 μM/1 h] alone significantly reduced cell viability in bortezomib-resistant MM cells lines after 48 h (). When combined with bortezomib exposure [5 nM/24 h], the ibrutinib 'pulse' also significantly reduced cell viability in bortezomib-resistant U266 MM cells below levels observed in bortezomib-resistant cells receiving bortezomib alone, and bortezomib-naïve cells receiving the same combination therapy (). Importantly, ibrutinib 'pulse' treatment followed by bortezomib exposure also dramatically reduced cell viability in primary relapsed MM samples [n=6 ], likewise below levels observed in primary relapsed samples receiving bortezomib alone, and primary naïve MM samples [n=5 ] also receiving the combination therapy ().
Figure 3. BTK inhibition enhances sensitivity to bortezomib in bortezomib-naïve and bortezomib-resistant MM cells. (A) Relative cell viability of bortezomib-naïve and bortezomib-resistant MM cell lines 48 h post-ibrutinib 'pulse' treatment. (B) Relative cell viability of bortezomib-naïve and bortezomib-resistant MM cells post-ibrutinib 'pulse' treatment in combination with bortezomib (5 nM/24 h). (C) Relative cell viability of primary naïve and primary relapsed MM patient samples post in vitro ibrutinib 'pulse' treatment in combination with bortezomib (5 nM/24 h). Statistical significance between treatments was calculated by Student's t test; * indicates p ≤ 0.05. Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.01.
BTK inhibition via lenti-viral miRNA targeting also restores sensitivity to bortezomib in bortezomib-resistant MM cells
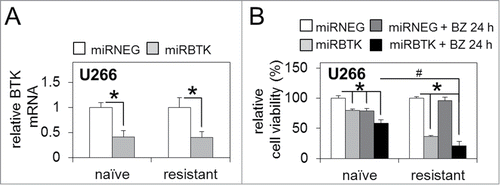
Although originally developed as an inhibitor of BTK, ibrutinib has more recently been shown to have 'off-target' effects, inhibiting multiple members of the TEC kinase family, including interleukin-2-inducible T-cell kinase (ITK)Citation32 Therefore, to ensure our results were not due to potential off-target inhibitor activity, we evaluated bortezomib-resistant and bortezomib-naïve U266 MM cell line response to lenti-viral-mediated BTK repression. We generated artificial and exogenous miRNA sequences specifically targeting the BTK transcript [miRBTK] and utilised lenti-viral infection to achieve semi-stable BTK mRNA knockdown [as described previouslyCitation25] in both bortezomib-naïve and bortezomib-resistant U266 MM cells (). Furthermore, the introduction of miRBTK in conjunction with bortezomib treatment (5 nM/24 h) confirmed that bortezomib-resistant U266 MM cells experienced significantly reduced cell viability compared to bortezomib-naïve cells also infected with miRBTK and exposed to bortezomib, and compared to bortezomib-resistant cells infected with a non-targeting control miRNA [miRNEG] (). This strongly suggests that the results observed following ibrutinib and bortezomib combination therapy in bortezomib-resistant cells are due to ibrutinib-driven irreversible BTK inhibition, rather than any off target ibrutinib effects.
Figure 4. BTK inhibition via lenti-viral miRNA targeting enhances sensitivity to bortezomib in bortezomib-naïve and bortezomib-resistant MM cells. (A) qRT-PCR analysis of basal BTK mRNA expression in bortezomib-naïve and bortezomib-resistant MM U266 cells infected with lenti-viral miRNA constructs targeting BTK (miRBTK) transcription relative to GAPDH. (B) Relative cell viability of bortezomib-naïve and bortezomib-resistant MM U266 cells infected with lenti-viral miRBTK before and after bortezomib treatment (5 nM/24 h). Values indicate the mean ± SEM from 3 independent experiments. Statistical significance between treatments was calculated by Student's t test; * indicates P ≤ 0.05. Statistical significance between cohorts was calculated by ANOVA; # indicates p ≤ 0.01.
Bortezomib-resistance is driven by enhanced NF-κB p65 activity and can be reversed by BTK inhibition
We have previously shown that BTK inhibition reduces NF-κB p65 activity in bortezomib-naïve MM cells and primary naïve patient samples.Citation17 To determine the role of NF-κB p65 in the regulation of BTK activity in bortezomib-resistant MM cells we initially examined basal levels of sub-cellular NF-κB p65 protein distribution in bortezomib-naïve and bortezomib-resistant U266 MM cells by immunocytochemistry. Utilizing an antibody against the NF-κB p65 subunit we observed higher levels p65 in the nucleus of bortezomib-resistant cells compared to bortezomib-naïve cells [n ≥ 20 ], suggesting a greater level of NF-κB p65 transcription factor binding, and therefore NF-κB signaling in the bortezomib-resistant cells (; inset). Further analysis of nuclear vs. cytoplasmic NF-κB p65 revealed higher basal levels of nuclear localization in bortezomib-resistant compared to bortezomib-naïve U266 MM cells; both by quantitative analysis ( A) and western immunoblot (). Additionally, analysis of sub-cellular NF-κB p65 localization in response to ibrutinib 'pulse' treatment combined with bortezomib exposure [5 nM/4 h] revealed significant repression of NF-κB p65 nuclear localization in bortezomib-resistant and bortezomib-naïve U266 MM cells compared to those treated with bortezomib alone (). Importantly, bortezomib-resistant cells showed no significant reduction in NF-κB p65 nuclear localization in response to bortezomib treatment alone compared to bortezomib-naïve cells, potentially suggesting a level of constitutive NF-κB p65 activity in these cells.
Figure 5. Bortezomib-resistance is driven by enhanced NF-κB p65 activity and can be reversed by BTK inhibition. (A) Quantification of staining intensity of nuclear p65/total NF-κB p65 (%) in bortezomib-naïve and bortezomib-resistant U266 MM cells. Box and whisker indicates the mean ± SEM (n ≥ 20). Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.01. Inset shows example immunocytochemistry for NF-κB p65. Dotted line indicates nucleus as determined by DAPI staining. (B) Western immunoblot of bortezomib-naïve [N] and bortezomib-resistant [R] U266 nuclear [Nuc.] and cytoplasmic [Cyt.] cell fractions. GAPDH and Histone 3 [H3] show equal loading for cyt. and nuc. fractions, respectively. (C) Quantification of staining intensity of nuclear NF-κB p65/total NF-κB p65 (%) in bortezomib-naïve and bortezomib-resistant U266 MM cells. Box and whisker indicates the mean ± SEM (n ≥ 20). Statistical significance between cohorts was calculated by ANOVA; # indicates p ≤ 0.05. (D) qRT-PCR analysis of basal NF-κB p65 mRNA expression in bortezomib-naïve and bortezomib-resistant MM U266 cells infected with lenti-viral miRNA constructs targeting NF-κB p65 (miRp65) transcription relative to GAPDH. (E) Relative cell viability of bortezomib-naïve and bortezomib-resistant MM U266 cells infected with lenti-viral miRp65 24 h before and after ibrutinib 'pulse' treatment. Values indicate the mean ± SEM from 3 independent experiments. Statistical significance between treatments was calculated by Student's t test; * indicates P ≤ 0.05. Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.01.
![Figure 5. Bortezomib-resistance is driven by enhanced NF-κB p65 activity and can be reversed by BTK inhibition. (A) Quantification of staining intensity of nuclear p65/total NF-κB p65 (%) in bortezomib-naïve and bortezomib-resistant U266 MM cells. Box and whisker indicates the mean ± SEM (n ≥ 20). Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.01. Inset shows example immunocytochemistry for NF-κB p65. Dotted line indicates nucleus as determined by DAPI staining. (B) Western immunoblot of bortezomib-naïve [N] and bortezomib-resistant [R] U266 nuclear [Nuc.] and cytoplasmic [Cyt.] cell fractions. GAPDH and Histone 3 [H3] show equal loading for cyt. and nuc. fractions, respectively. (C) Quantification of staining intensity of nuclear NF-κB p65/total NF-κB p65 (%) in bortezomib-naïve and bortezomib-resistant U266 MM cells. Box and whisker indicates the mean ± SEM (n ≥ 20). Statistical significance between cohorts was calculated by ANOVA; # indicates p ≤ 0.05. (D) qRT-PCR analysis of basal NF-κB p65 mRNA expression in bortezomib-naïve and bortezomib-resistant MM U266 cells infected with lenti-viral miRNA constructs targeting NF-κB p65 (miRp65) transcription relative to GAPDH. (E) Relative cell viability of bortezomib-naïve and bortezomib-resistant MM U266 cells infected with lenti-viral miRp65 24 h before and after ibrutinib 'pulse' treatment. Values indicate the mean ± SEM from 3 independent experiments. Statistical significance between treatments was calculated by Student's t test; * indicates P ≤ 0.05. Statistical significance between cohorts was calculated by ANOVA; # indicates P ≤ 0.01.](/cms/asset/6d6ddcc8-f276-4df1-bfe4-8eec8fb2dc13/kccy_a_998067_f0005_b.gif)
To confirm the functional relevance of enhanced basal nuclear NF-κB p65 localization in bortezomib-resistant MM cells, and to confirm that any response to bortezomib observed was specifically due to its ability to repress inducible NF-κB p65 activity, we generated artificial and exogenous miRNA sequences specifically targeting NF-κB p65 (miRp65) and utilised lenti-viral infection to achieve semi-stable p65 mRNA knockdown in bortezomib-naïve and bortezomib-resistant U266 MM cells (). Introduction of miRp65, in combination with ibrutinib 'pulse' treatment, significantly reduced cell viability in bortezomib-resistant U266 MM cells compared to bortezomib-naïve cells exposed to the same treatment, and compared to bortezomib-resistant cells infected with a non-targeting control miRNEG or ibrutinib 'pulse' treatment alone ().
Discussion
The proteasome inhibitor bortezomib is licensed to treat newly diagnosed and relapsed MM in the clinic. Single agent bortezomib has a response rate of approximately 30% but, when used in combination with chemotherapy and/or corticosteroids, response rates range from approximately 60% to over 90% depending on the regimen.Citation16 Despite this relative success, however, clinical relapse following bortezomib therapy presently remains inevitable and resistance to further bortezomib treatment is common,Citation30,31 not only as a consequence of, but also further driving the selection and emergence of drug-resistant clones.Citation7,8 An improved understanding of the mechanisms underlying bortezomib-resistance is, therefore, vital for the progressive development of novel pharmacologic strategies to overcome the clinical phenomenon of bortezomib-resistance. In this study we have explored bortezomib-resistance in MM in vitro, utilizing bortezomib-resistant MM cell lines generated in the laboratory, and primary patient samples from both treatment naïve patients and patients that have relapsed following bortezomib therapy. Despite the limited sample size, this data provides an important foundation for future in vivo assessment of ibrutinib therapy in bortezomib-relapsed MM in a larger number of patients in the clinic.
We and others have previously demonstrated activity of the BTK survival pathway in treatment-naïve MM cells.Citation17,33 Furthermore, bortezomib has been reported to inhibit total BTK expression in a treatment-naive B lymphocyte cell line.Citation22 Here, utilizing in vitro models of post-bortezomib relapsed MM, we describe a role for BTK activity in bortezomib-resistance, demonstrating higher basal levels of active pBTK protein in bortezomib-resistant MM cell lines and a failure of these cells to respond to bortezomib in terms of BTK mRNA and BTK promoter activity repression. In particular, the higher basal BTK activity and altered BTK promoter activity in bortezomib-resistant cells would be consistent with process of clonal selection, driven by constant exposure to bortezomib.Citation8 This identifies BTK as a candidate therapeutic target in bortezomib-resistant MM. In addition, this may also be relevant to bortezomib-naïve MM, in which potentially bortezomib-resistant sub-clones as yet form a minority population within of the tumor ‘bulk’.
The oral BTK inhibitor ibrutinib, currently licensed for use in CLL and MCL,Citation34,35 has shown promising clinical activity and a favorable side-effect profile in a range of B cell malignancies.Citation36-39 Ibrutinib has also been found to reduce BTK expression in MMCitation22 and enhance the cytotoxicity of bortezomib in bortezomib-naïve primary malignant plasma cells and MM cell lines.Citation17 By inhibiting BTK with ibrutinib 'pulse' treatment we were also able to restore a measurable cytotoxic response to bortezomib in bortezomib-resistant MM cell lines and primary relapsed MM cells. Importantly, although ibrutinib is known to target other kinases, we were able to reproduce these results with highly specific artificial BTK-targeting microRNA, miRBTK, introduced by lenti-virus. This suggests that it is BTK inhibition which is restoring bortezomib activity in bortezomib-resistant MM cells and not an off target effect of ibrutinib.
Multiple mechanisms of action of bortezomib have been described,Citation20,16 including the inhibition of BTK expression driven via repression of inducible NF-κB transcription factor translocation to the nucleus.Citation17,22, Citation40 We found enhanced nuclear localization of NF-kB p65 in the bortezomib-resistant cells. This is in-keeping with previous reports that bortezomib-resistance in MM is conveyed through the NF-κB signaling network. For example; constitutive expression of NF-κB is frequently seen in bortezomib refractory primary patient samples.Citation41 Furthermore, the acquisition of bortezomib-resistance can follow up-regulation of heat shock protein (HSP)90 and HSP27, which, in their action as ubiquitin chaperones, facilitate the activation of NF-κB in MM.Citation42,43 Active pBTK is also known to be essential for NF-κB activation and B cell survival.Citation44 Thus, there exists a positive auto-regulatory feedback loop that stimulates transcription of BTK via 2 functionally competent NF-κB p65 sites in the BTK promoter.Citation22 Here we show that bortezomib-resistance in MM is associated with higher basal nuclear NF-κB p65 in bortezomib-resistant MM cells, which, in turn, is associated with a greater reliance on pBTK for survival. These observations suggest that BTK inhibition by ibrutinib may be useful in the clinical treatment of bortezomib-resistant MM.
Taken together, these results show that bortezomib-resistance observed in the clinic occurs, at least in part, because bortezomib-resistant MM cells lose the capacity to be influenced by the inhibition of BTK-driven NF-κB p65, and the consequent NF-κB p65-driven auto-regulation of BTK. Accordingly, introduction of ibrutinib to the treatment regimens of bortezomib-resistant MM patients may lead to enhanced cell death of the bortezomib-resistant sub-clones through inhibition of this BTK/NF-κB p65 signaling axis. Here we provide a biologic and molecular rationale for the clinical evaluation of bortezomib and ibrutinib combination therapy in post-bortezomib relapsed MM, including patients that have previously been refractory to bortezomib.
Materials and Methods
Materials
All reagents were obtained from Sigma-Aldrich Co. (St. Louis, MO), unless stated otherwise. All MM/B-lymphoblast cell lines were obtained from the European Collection of Cell Cultures (ECCC).
Cell culture
MM cell lines (H929, LP-1, RPMI-8228, and U266) were cultured in a humidified atmosphere at 37°C and 5% (v/v) CO2 in RPMI-1640 media +L-Glutamine (Gibco Life Technologies) supplemented with 10% (v/v) foetal bovine serum ([FBS]; Biosera). Bortezomib-resistant cells were cultured as above but supplemented with 20% (v/v) FBS. Bortezomib-resistance was selected for by dose escalation of a once-weekly bortezomib treatment, through 0.5, 1, 2.5 and 5 nM, until cells could tolerate 10 nM bortezomib (10–15 weeks; confirmed by luminescent cell viability assay). Each concentration was maintained for approximately 2–3 weeks to allow for proliferation of bortezomib-resistant sub-clones. Build-up of cell debris due to the high level of cell death was removed by centrifugation at 300 g for 5 minutes before gently removing the supernatant and re-suspending the pellet.
Primary MM cells were obtained under local ethical approval (LREC ref. 07/H0310/146) and were isolated from the bone marrow aspirates of MM patients, as described previously.Citation17 Primary MM cells from bortezomib-relapsed patients were not exposed to further bortezomib treatment until experimentation.
Ibrutinib 'pulse' treatment
To mimic in vivo pharmacokinetics of rapid adsorption and elimination of the irreversible BTK inhibitor ibrutinib,Citation45 we employed a method of 'pulse' exposure to physiologically relevant levels of ibrutinib in vitro, as described previously.Citation46 Briefly, cells were exposed to 500 nM ibrutinib for 1 h before washing, followed by any additional drug treatment or assay in fresh media.
Luminescent cell viability assay and chymotrypsin-like proteasome activity assay
One×105 MM cells were treated as indicated. Relative cell viability was assayed using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega), as per the manufacturer's instructions. Proteasome activity was measured using the Proteasome-Glo™ Chymotrypsin-Like Cell-Based Assay (Promega), as per the manufacturer's instructions. Luminescence was measured with the FLUOstar Omega Microplate Reader (BMGLabtech) and calculated as relative to untreated control samples.
Quantification of protein activity and subcellular localization by immunocytochemistry
Relative activity of phosphorylated versus total BTK (pBTK vs. BTK), and subcellular distribution of nuclear vs. cytoplasmic NF-κB p65 (nuc. vs. cyt. p65) were analyzed by immunocytochemistry. Briefly, 2 × 104 U266 MM cells were fixed onto microscope slides using the CytoSpin 4 Cytocentrifuge system (Thermo Fisher Scientific) and 4% (w/v) paraformaldehyde (PFA). Fixed cells were incubated with rabbit anti-human primary mAbs against pBTK (Tyr223) and goat anti-human primary mAbs against total BTK, or goat anti-human primary mAbs against NF-κB p65 (Cell Signaling Technology). Cells were then incubated with Alexa Fluor® goat anti-rabbit and rabbit anti-goat IgG (H+L) secondary antibodies (Molecular Probes Life Technologies), or rabbit anti-goat IgG alone, respectively. Nuclei were localized by 4′,6-diamidino-2-phenylindole (DAPI) staining. After mounting, cells were viewed using the Axio Imager.D2 microscope and Axiovision software (Carl Zeiss Microscopy).
Image analyses and quantification of pBTK vs. BTK, and nuc. vs. cyt. p65 staining intensity was performed using ImageJ 1.46 software (National Institute of Health) and plugins, as described previously.Citation25 Briefly, a threshold of staining intensity was applied and the area above the threshold (pixles2) was measured (n ≥ 80 [BTK] and n ≥ 20 [NF-κB] individual cells). Staining intensity (pixles2) was converted to a percentage as follows: pBTK/total BTK (%), or, nuclear/[cytoplasmic + nuclear NF-κB p65] (%).
Western Immunoblotting
Protein fractions of equal total protein concentration were extracted from U266 MM cells using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. SDS/PAGE and western immunoblot analyses were performed as described previously.Citation47 ECL detection using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) and subsequent quantification were performed with the ChemiDoc-It2 Imaging System (UVP).
RNA extraction and quantitative real time-PCR
Total RNA was extracted from 1 × 106 U266 MM cells using Total RNA Lysis Solution (Applied Biosystems-Life Technologies) according to the manufacturer's instructions. Reverse transcription of total RNA was performed using the GeneAmp® Gold RNA PCR Core Kit (Applied Biosystems-Life Technologies). Resultant cDNA was analyzed by quantitative real-time PCR (qRT-PCR) using LightCycler® 480 SYBR Green I Master mix (Roche) and qRT-PCR primers for GAPDH [forward 5′-ACCAGCCTCAAGATCATCAGC-3′ and reverse 5′-TGCTAAGCAGTTGGTGGTGC-3′], BTK [forward 5′-CACACAGGTGAACTCCAGAAAG-3' and reverse 5'-AGAGATACTGCCCATCGATCCAGA-3'] and NF-κB p65 [forward 5′-ACCGCTGCATCCACAGTT-3′ and reverse 5′-GGATGCGCTGACTGATAGC-3′] (Invitrogen-Life Technologies), on the LightCycler® 480 Real-Time PCR system (Roche), as previously described.Citation18 Gene expression was analyzed using the comparative cycle threshold algorithm (ΔΔCT); mRNA expression was standardised against GAPDH expression.
Lenti-virus construction and infection
Lenti-virus containing artificial exogenous microRNA (miRNA) sequences targeting human NF-κB p65 (miR-p65 [5′-TACGTTTCTCCTCAATCCGGT-3′]), BTK (miR-BTK [5′-TTCACTGGACTCTTCACCTCT-3′]) or a control/scrambled sequence (miR-NEG), and an EmGFP–pre-miRNA encoding fragment, were constructed and produced as described previously.Citation25 Briefly, artificial miRNA targets were identified and designed to interact with the relevant exon using Block-iT RNAi designer software (Invitrogen). This ensures high specificity of miR-p65 and miR-BTK, with no 3′UTR interaction, unlike endogenous miRNAs.
Five × 105 U266 MM cells were infected and transduced with each lenti-virus (MOI: 15), in serum-free medium with 8 μg/mL Polybrene™. Following infection and before experimentation, transduced cells were analyzed for target knock-down and efficiency by qRT-PCR for BTK and NF-κB p65, and flow cytometry to detect GFP using the Accuri-C6 flow cytometer (BD Biosciences).
Generation and transfection of the wild-type human BTK promoter-luciferase reporter construct
To generate the wild-type human BTK promoter-luciferase reporter construct (pGL4.BTK), the BTK promoter region, including 2 innate κB binding sites, was amplified from genomic DNA via PCR with specific forward [5′-TATCTCGAGGAAGAAAAGAGCCTGGGCA-3′] and reverse [5′-ATAAGATCTGTCTTTTTTTCTTCTCAGCAGCA-3′] primers. The amplified fragment was cloned into the Xho I/Bgl II site of the Promoterless Firefly Luciferase 'pGL4.11[luc2P]' Vector, according to the manufacturers' instructions (Promega).
U266 MM cells were co-transfected with a total of 1 μg DNA, composed of the pGL4.BTK promoter-luciferase reporter construct and a pRL-TK Renilla Luciferase Control Reporter Vector (Promega) using FuGENE® HD Transfection Reagent (Promega) and incubated for 24 h at 37°C and 5% (v/v) CO2 before any further treatment.
Chemi-luminescent luciferase reporter assay
Firefly and Renilla promoter-luciferase reporter activity was analyzed and quantified sequentially in a single sample with the Dual-Luciferase® Reporter Assay System (Promega), according to the manufacturer's instructions, and the EnVision 2103 Multilabel Plate Reader (Perkin Elmer).
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Funding
This work was funded by NIHR Flexibility and Sustainability Funding, the Big C Appeal, Worldwide Cancer Research, and the Humane Research Trust.
References
- American Cancer Society. Cancer Facts & Figures 2014. (2015). 1st ed. [ebook] Atlanta, Georgia: American Cancer Society, p.4. Available at: http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2014/ [Accessed 6 May 2014]
- Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Multiple myeloma. Lancet 2009; 374:324-39; PMID:19541364; http://dx.doi.org/10.1016/S0140-6736(09)60221-X
- Twombly R. First proteasome inhibitor approved for multiple myeloma. J Natl Cancer Inst 2003; 95:845; PMID:12813164; http://dx.doi.org/10.1093/jnci/95.12.845
- Palumbo A, Attal M, Roussel M. Shifts in the therapeutic paradigm for patients newly diagnosed with multiple myeloma: maintenance therapy and overall survival. Clin Cancer Res 2011; 17:1253-63; PMID:21411441; http://dx.doi.org/10.1158/1078-0432.CCR-10-1925
- Kapoor P, Ramakrishnan V, Rajkumar SV. Bortezomib combination therapy in multiple myeloma. Seminars Hematol 2012; 49:228-42; PMID:22726546; http://dx.doi.org/10.1053/j.seminhematol.2012.04.010
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet J-P, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471:467-72; PMID:21430775; http://dx.doi.org/10.1038/nature09837
- Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, Middha S, Asmann Y, Schmidt J, Braggio E, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012; 120:1060-6; PMID:22529291; http://dx.doi.org/10.1182/blood-2012-01-405977
- Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, Van Wier S, Blackburn PR, Baker AS, Dispenzieri A, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012; 120:1067-76; PMID:22498740; http://dx.doi.org/10.1182/blood-2012-01-405985
- Magrangeas F, Avet-Loiseau H, Gouraud W, Lode L, Decaux O, Godmer P, Garderet L, Voillat L, Facon T, Stoppa AM, et al. Minor clone provides a reservoir for relapse in multiple myeloma. Leukemia 2013; 27:473-81; PMID:22874878; http://dx.doi.org/10.1038/leu.2012.226
- Chen Z, Orlowski RZ, Wang M, Kwak L, McCarty N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood 2014; 123:2204-8; PMID:24425802; http://dx.doi.org/10.1182/blood-2013-07-517136
- Lonial S, Mitsiades CS, Richardson PG. Treatment options for relapsed and refractory multiple myeloma. Clin Cancer Res 2011; 17:1264-77; PMID:21411442; http://dx.doi.org/10.1158/1078-0432.CCR-10-1805
- Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, Davis RE, Lin P, Wang H, Madden TL, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 2012; 120:3260-70; PMID:22932796; http://dx.doi.org/10.1182/blood-2011-10-386789
- Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, et al. Molecular basis of bortezomib resistance: proteasome subunit {β}5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008; 112:2489-99; PMID:18565852; http://dx.doi.org/10.1182/blood-2007-08-104950
- Stessman HA, Mansoor A, Zhan F, Linden MA, Van Ness B, Baughn LB. Bortezomib resistance can be reversed by induced expression of plasma cell maturation markers in a mouse in vitro model of multiple myeloma. PLoS One 2013; 8:e77608; PMID:24204892; http://dx.doi.org/10.1371/journal.pone.0077608
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, et al. Frequent Engagement of the Classical and Alternative NF-κB Pathways by Diverse Genetic Abnormalities in Multiple Myeloma. Cancer Cell 2007; 12:115-30; PMID:17692804; http://dx.doi.org/10.1016/j.ccr.2007.07.004
- Murray MY, Auger MJ, Bowles KM. Overcoming bortezomib resistance in multiple myeloma. Biochem Soc Trans 2014; 42:4; http://dx.doi.org/10.1042/BST20140126
- Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-kappaB. Cell Signal 2013; 25:106-12; PMID:22975686; http://dx.doi.org/10.1016/j.cellsig.2012.09.008
- Murray MY, Rushworth SA, Zaitseva L, Bowles KM, MacEwan DJ. Attenuation of dexamethasone-induced cell death in multiple myeloma is mediated by miR-125b expression. Cell Cycle 2013; 12:2144-53; PMID:23759586; http://dx.doi.org/10.4161/cc.25251
- Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM, MacEwan DJ. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012; 120:5188-98; PMID:23077289; http://dx.doi.org/10.1182/blood-2012-04-422121
- Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann T, Anderson K. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood 1996; 87:1104-12; PMID:8562936
- Lu S, Wang J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomark Res 2013; 1(1):13; PMID:24252210; http://dx.doi.org/10.1186/2050-7771-1-13
- Yu L, Mohamed AJ, Simonson OE, Vargas L, Blomberg KE, Bjorkstrand B, Arteaga HJ, Nore BF, Smith CI. Proteasome-dependent autoregulation of Bruton tyrosine kinase (Btk) promoter via NF-kappaB. Blood 2008; 111:4617-26; PMID:18292289; http://dx.doi.org/10.1182/blood-2007-10-121137
- Tai YT, Chang BY, Kong SY, Fulciniti M, Yang G, Calle Y, Hu Y, Lin J, Zhao JJ, Cagnetta A, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood 2012; 120:1877-87; PMID:22689860; http://dx.doi.org/10.1182/blood-2011-12-396853
- O'Brien S, Furman R, Coutre S, Burger J, Blum K, Sharman J, Flinn I, Grant B, Heerema N, Johnson A, et al. The Bruton's tyrosine kinase inhibitor Ibrutinib is highly active and tolerable in relapsed or refractory (R/R) and treatment naïve (TN) CLL patients, updated results of a phase IB/II study. Haematologica 2012 97:256, abstr 0542.
- Rushworth SA, Murray MY, Zaitseva L, Bowles KM, MacEwan DJ. Identification of Bruton's tyrosine kinase as a therapeutic target in acute myeloid leukemia. Blood 2014; 123:1229-38; PMID:24307721; http://dx.doi.org/10.1182/blood-2013-06-511154
- Khan W. Regulation of B lymphocyte development and activation by Bruton's tyrosine kinase. Immunol Res 2001; 23:147-56; PMID:11444380; http://dx.doi.org/10.1385/IR:23:2-3:147
- Jefferies CA, O'Neill LA. Bruton's tyrosine kinase (Btk)-the critical tyrosine kinase in LPS signalling? Immunol Lett 2004; 92(1-2):15-22; PMID:15081522
- Pharmacyclics, Inc., (2012). Pharmacyclics Reports Fiscal 2013 First Quarter Financial Results and Multiple PCI-32765 Presentations at the 54th American Society of Hematology Annual Meeting. [online] Available at: http://ir.pharmacyclics.com/releasedetail.cfm?releaseid=718446 [Accessed 5 Mar. 2014]
- Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, et al. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol 2004; 22:2108-21; PMID:15169797; http://dx.doi.org/10.1200/JCO.2004.02.106
- Taverna C, Voegeli J, Trojan A, Olie RA, von Rohr A. Effective response with bortezomib retreatment in relapsed multiple myeloma–a multicentre retrospective survey in Switzerland. Swiss Med Weekly 2012; 142:w13562; PMID:22544478
- Petrucci MT, Giraldo P, Corradini P, Teixeira A, Dimopoulos MA, Blau IW, Drach J, Angermund R, Allietta N, Broer E, et al. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br J Haematol 2013; 160:649-59; PMID:23293914; http://dx.doi.org/10.1111/bjh.12198
- Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, Hessler JD, Liu TM, Chang BY, Larkin KM, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013; 122:2539-49; PMID:23886836; http://dx.doi.org/10.1182/blood-2013-06-507947
- Tai Y-T, Chang BY, Kong S-Y, Fulciniti M, Yang G, Calle Y, Hu Y, Lin J, Zhao J-J, Cagnetta A, et al. Bruton's tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood 2012; 120:1877-87; PMID:22689860; http://dx.doi.org/10.1182/blood-2011-12-396853.
- McDermott J, Jimeno A. Ibrutinib for the treatment of chronic lymphocytic leukemia and mantle cell lymphoma. Drugs of today (Bar) 2014; 50:291-300.
- Cameron F, Sanford M. Ibrutinib: first global approval. Drugs 2014; 74:263-71; PMID:24464309; http://dx.doi.org/10.1007/s40265-014-0178-8
- Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013; 369:32-42; PMID:23782158; http://dx.doi.org/10.1056/NEJMoa1215637
- Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, et al. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med 2013; 369:507-16; PMID:23782157; http://dx.doi.org/10.1056/NEJMoa1306220
- Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88-94; PMID:23045577; http://dx.doi.org/10.1200/JCO.2012.42.7906
- O'Brien SM, Furman RR, Coutre SE, Flinn I, Burger JA, Blum KA, Sharman JP, Jones JA, Wierda WG, Zhao W, Heerema NA, Johnson AJ, Tran A, Zhou C, Bilotti E, James DF, Byrd JC. “Leukemia.” [Abstract]. Independent evaluation of ibrutinib efficacy 3 years post-initiation of monotherapy in patients with chronic lymphocytic leukemia/small lymphocytic leukemia including deletion 17p disease. J Clin Oncol 32:5s, 2014 (suppl; abstr 7014) ed. Chicago: 2014 ASCO Annual Meeting. (Jun 2014).
- Dasmahapatra G, Patel H, Dent P, Fisher RI, Friedberg J, Grant S. The Bruton tyrosine kinase (BTK) inhibitor PCI-32765 synergistically increases proteasome inhibitor activity in diffuse large-B cell lymphoma (DLBCL) and mantle cell lymphoma (MCL) cells sensitive or resistant to bortezomib. Br J Haematol 2013; 161:43-56; PMID:23360303; http://dx.doi.org/10.1111/bjh.12206
- Markovina S, Callander NS, O'Connor SL, Kim J, Werndli JE, Raschko M, Leith CP, Kahl BS, Kim K, Miyamoto S. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res 2008; 6:1356-64; PMID:18708367; http://dx.doi.org/10.1158/1541-7786.MCR-08-0108
- Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Treon SP, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A 2002; 99:14374-9; PMID:12391322; http://dx.doi.org/10.1073/pnas.202445099
- Navas TA, Nguyen AN, Hideshima T, Reddy M, Ma JY, Haghnazari E, Henson M, Stebbins EG, Kerr I, O'Young G, et al. Inhibition of p38alpha MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating Hsp27, Bcl-X(L), Mcl-1 and p53 levels in vitro and inhibits tumor growth in vivo. Leukemia 2006; 20:1017-27; PMID:16617327; http://dx.doi.org/10.1038/sj.leu.2404200
- Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010; 463:88-92; PMID:20054396; http://dx.doi.org/10.1038/nature08638
- Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med 2013; 369:32-42; PMID:23782158; http://dx.doi.org/10.1056/NEJMoa1215637
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A 2010; 107:13075-80; http://dx.doi.org/10.1073/pnas.1004594107
- Rushworth SA, Bowles KM, Raninga P, MacEwan DJ. NF-kappaB-inhibited acute myeloid leukemia cells are rescued from apoptosis by heme oxygenase-1 induction. Cancer Res 2010; 70:2973-83; PMID:20332229; http://dx.doi.org/10.1158/0008-5472.CAN-09-3407