
Methylation of DNA at CpG dinucleotides is dynamic at many genomic loci during normal development. The continuously differentiating haematopoietic system offers a well-characterized model to study these changes in the adult. During normal haematopoietic differentiation, DNA methylation levels have been shown to both increase and decrease as haematopoietic stem cell (HSC) lineage specification progresses.Citation1-3 Recent studies of DNA methylation in haematopoietic stem and progenitor (HSPC) populations have elucidated finely tuned methylation changes taking place during differentiation, as well as identified some of the key regulators and drivers of this epigenetic process.Citation1-4 Major questions remain, however, regarding the relationship between DNA methylation status, including location in the genome (intron, exon, promoter, intergenic, etc.), and the resultant effect on overall gene expression.
Following up on their recent generation and analysis of DNA methylomes in murine HSCs and 4 closely related multipotent progenitor populations (MPPs) using tagmentation-based whole genome bisulfite sequencing (TWGBS)Citation1 Lipka et al. further categorize the molecular landscape changes in HSPCs during the very early stages of HSC differentiation.Citation5 They found a number of epigenetic changes that were inversely correlated with gene expression during HSPC differentiation (). During the HSC to MPP1 transition, for example, genes known to be important for normal HSC function, such as Gata2,Citation6 exhibited increased DNA methylation and decreased gene expression. Across the genome, they identified nearly 16 thousand differentially methylated regions (DMRs), distinguishing several distinct clusters of methylation dynamics.Citation5
Figure 1. HSC differentiation into MPPs characterized by dynamic DNA methylation changes and subsequent gene expression changes. DMRs observed comparing populations separate into discrete clusters, either gaining (top left graph; clusters 1–4) or losing (bottom left graph; clusters 6–9) methylation during differentiation. These changes exhibited inverse correlation with gene expression, particularly in TFs with roles in haematopoietic regulation (e.g., Mecom & Gfi1).
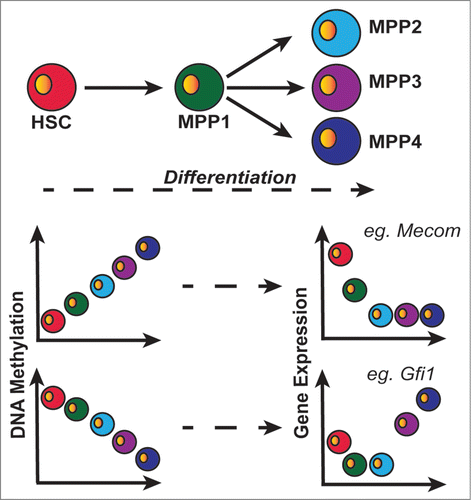
Importantly, the TWGBS approach enabled two-to-three times as many DMRs to be identified as in previous reports of haematopoietic differentiation and better correlation with gene expression changes.Citation2,3 Previous studies of DNA methylation during HSPC differentiation used reduced representation bisulfite sequencing (RRBS) or comprehensive high-throughput array-based relative methylation (CHARM) arrays, and a general correlation between DNA methylation levels and gene expression was not observed.Citation2,3 In the current study, the majority of DMRs (85%) were exclusively identified by TWGBS, and were found within intragenic regions (57%), which were more likely to be missed by other assays due to an overrepresentation of CpG-rich genomic regions.Citation1 Lipka et al. also identify a group of 264 “core-enriched” genes, which exhibited an inverse relationship between DNA methylation and gene expression during the HSC to MPP1 transition, but their functional significance remains to be determined.Citation5
The newly identified DMRs were further analyzed for overlap with regulatory regions associated with haematopoietic -specific transcription factors (TFs); each differentiation stage produced examples of DMRs associated with differentially expressed haematopoietic -specific TFs. The authors propose some of these stage-specific DMRs represent novel cis-acting regulatory regions of TFs important for haematopoietic differentiation, but this will require experimental validation. In addition, probing the DMRs for the presence of canonical TF binding motifs revealed potential involvement of about 30 TFs. The binding site enrichments for specific TF groups were, in turn, limited to specific DMR clusters, possibly implicating a more distinctive, active role for DNA methylation in regulating TF binding in early hematopoiesis.Citation5
The regulation of, and downstream effects of, DNA methylation changes in the haematopoietic system are crucial to elucidate because a main driver of DNA methylation in HSCs, DNA methyltransferase 3a (DNMT3A), both directly regulates DNA methylation levels in HSCs,Citation4 and is recurrently mutated in hematologic malignancies, including acute myeloid leukemia.Citation7 The connection between regulation of DNA methylation in HSCs and development of cancer is still largely unknown, but understanding the epigenetic mechanisms governing differentiation, and the downstream cellular impacts, could provide a great deal of insight.
Overall, this study provides important insights into DNA methylation dynamics of early HSC differentiation, and a roadmap for experimental exploration of potential downstream effects of epigenetic aberrations, such as DNMT3A mutations, in hematologic malignancy and disease. Many important questions remain for the future. For example, the functional relevance of the 9 DMR clusters, particularly given the enrichment of specific TF binding motifs, is intriguing. As the authors suggest, genome-wide mapping of protein binding via chromatin-Immunoprecipitation sequencing (ChIP-Seq) will be required to address this question, as well as determining the effect of DNA methylation dynamics on TF binding.Citation5 It would additionally be useful to determine the genome-wide binding of proteins involved in DNA methylation and demethylation to elucidate how these subtle, but significant, DNA methylation changes may be occurring during HSC differentiation.
References
- Cabezas-Wallscheid N, et al. Cell Stem Cell 2014; 15:507-22; PMID:25158935; http://dx.doi.org/10.1016/j.stem.2014.07.005
- Ji H, et al. Nature 2011; 467:338-42; PMID:20720541; http://dx.doi.org/10.1038/nature09367
- Bock CB, et al. Mol Cell 2012; 47:633-47; PMID:22841485; http://dx.doi.org/10.1016/j.molcel.2012.06.019
- Jeong M, et al. Nat Genet 2013; 46:17-23; PMID:24270360; http://dx.doi.org/10.1038/ng.2836
- Lipka DB, et al. Cell Cycle 2014; In Press.
- Ling KW, et al. JEM 2004; 200:871-82; PMID:15466621; http://dx.doi.org/10.1084/jem.20031556
- Ley TJ, et al. N Engl J Med 2010; 363:2424-33; PMID:21067377; http://dx.doi.org/10.1056/NEJMoa1005143