
Abstract
The mammalian ortholog of Drosophila ecdysoneless (Ecd) gene product regulates Rb-E2F interaction and is required for cell cycle progression. Ecd is overexpressed in breast cancer and its overexpression predicts shorter survival in patients with ErbB2-positive tumors. Here, we demonstrate Ecd knock down (KD) in human mammary epithelial cells (hMECs) induces growth arrest, similar to the impact of Ecd Knock out (KO) in mouse embryonic fibroblasts. Furthermore, whole-genome mRNA expression analysis of control vs. Ecd KD in hMECs demonstrated that several of the top 40 genes that were down-regulated were E2F target genes. To address the role of Ecd in mammary oncogenesis, we overexpressed Ecd and/or mutant H-Ras in hTERT-immortalized hMECs. Cell cycle analyses revealed hMECs overexpressing Ecd+Ras showed incomplete arrest in G1 phase upon growth factor deprivation, and more rapid cell cycle progression in growth factor-containing medium. Analyses of cell migration, invasion, acinar structures in 3-D Matrigel and anchorage-independent growth demonstrated that Ecd+Ras-overexpressing cells exhibit substantially more dramatic transformed phenotype as compared to cells expressing vector, Ras or Ecd. Under conditions of nutrient deprivation, Ecd+Ras-overexpressing hMECs exhibited better survival, with substantial upregulation of the autophagy marker LC3 both at the mRNA and protein levels. Significantly, while hMECs expressing Ecd or mutant Ras alone did not form tumors in NOD/SCID mice, Ecd+Ras-overexpressing hMECs formed tumors, clearly demonstrating oncogenic cooperation between Ecd and mutant Ras. Collectively, we demonstrate an important co-oncogenic role of Ecd in the progression of mammary oncogenesis through promoting cell survival.
Abbreviations
| Ecd | = | Ecdysoneless |
| hMEC | = | Human Mammary Epithelial Cells |
| MEFs | = | Mouse Embryonic Fibroblasts |
| Rb | = | Retinoblastoma |
| PCR | = | Polymerase Chain Reaction |
| KD | = | Knockdown |
| KO | = | Knockout |
Introduction
Precise regulation of cell cycle machinery is required for normal development and adult homeostasis. Deregulation of the cell cycle machinery promotes uncontrolled proliferation, which in turn promotes genomic instability towards eventual cancer formation.Citation1 Notably, deregulated cell cycle components have emerged as key biomarkers and therapeutic targets in cancer.Citation2 Thus, a better understanding of cell cycle machinery and its aberrations in cancer are of fundamental importance in understanding physiology as well as pathology.
We have previously identified Ecdysoneless (Ecd), an ortholog of Drosophila Ecdysoneless, as a p53 interactorCitation3 and a novel cell cycle regulator.Citation4 Previously, we have shown that Ecd is required for embryogenesis, and its conditional deletion in Ecdflox/flox mouse embryonic fibroblasts (MEFs) halts cells in G1 phase of cell cycle.Citation4 Mechanistically, Ecd directly binds to Retinoblastoma (Rb) protein on its pocket domain, and appears to be necessary for the release of Rb from E2F. Inactivation of Rb rescued MEFs from proliferative block imposed by Ecd deletion, demonstrates that Ecd functions as a positive regulator of cell cycle progression by specifically regulating the Rb-E2F interaction and E2F-dependent cell cycle progression.Citation4
Our recent studies showed that Ecd is overexpressed in breast cancer, with a strong positive correlation between Ecd overexpression and higher histological grade, mitotic index and Nottingham Prognostic Index score. Ecd overexpression was positively associated with HER2/ErbB2 overexpression, a known marker of poor prognosis in breast cancer. Significantly, increased Ecd expression showed strong positive association with shorter breast cancer specific survival and disease-free survival in HER2/ErbB2 overexpressing patients.Citation5 Similarly, Ecd was also overexpressed in pancreatic cancer and its overexpression correlates with tumor progression.Citation6
Given the key role of Ecd in cell cycle progression as revealed by genetic studies, and an association between its overexpression and tumor progression, current studies were designed to investigate if Ecd is a positive regulator of cell cycle in hMECs. Furthermore, we modeled the breast cancer-associated Ecd overexpression in hMECs, either by itself or together with mutant H-Ras, a downstream signaling molecule of ErbB2-mediated signaling. Our studies reveal that Ecd is essential for cell cycle progression in hMECs; its overexpression cooperates with mutant Ras to promote full oncogenic transformation of hMECs. Furthermore, Ecd+Ras-overexpressing hMECs exhibit better survival in nutrient deprivation by upregulation of autophagy. Taken together, the present study shows Ecd as an essential regulator of cell cycle progression in hMECs and co-driver of mammary oncogenesis in conjunction with mutant Ras.
Results
Depletion of Ecd leads to a proliferation defect in hMECs
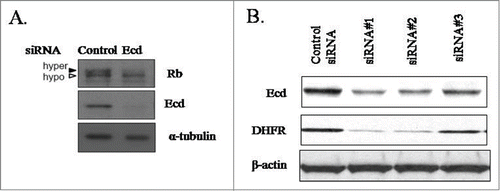
Given our previous findings that Ecd KO mice were embryonic lethal and that cre-mediated conditional deletion of Ecd in Ecdfl/fl MEFs led to cell cycle arrest in G1, we engineered siRNAs for Ecd KD in hMECs.Citation3 Ecd siRNA expression but not the scrambled siRNA expression led to a reduction in the level of hyper-phosphorylated Rb (pRb) () suggestive of growth arrest in G1. Consistently, a downregulation of DHFR, a well-known E2F target gene that is induced during G1 to S progression was observed (). To gain insights into mechanisms of growth arrest of hMECs upon Ecd KD, we performed genome-wide mRNA expression profiling of 76N cells expressing either a scrambled control siRNA or Ecd siRNA using the Affymetrix Human Genome U133 Plus2.0 chip. These analyses revealed that several of the top 40 genes that were downregulated upon Ecd KD were E2F target genes (Table S1). These results support a critical cell cycle regulatory role of Ecd in hMECs.
Figure 1. Ecd knockdown decreases the levels of hyperphosphorylated Rb and E2F target DHFR. Lysates of cells expressing control or Ecd siRNA were blotted with anti-Rb, anti-Ecd or anti-α-tubulin (used as a loading control) in (A) or anti-Ecd, anti-DHFR and β-actin (loading control) in (B). In (B) 3 distinct siRNAs were used for Ecd.
Ecd overexpression synergizes with mutant H-Ras to promote cell cycle progression
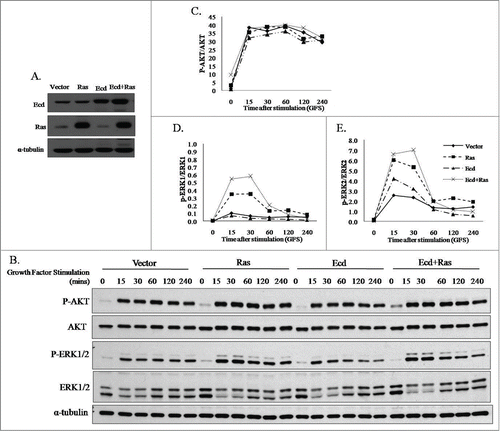
Evidence presented above for a role of Ecd in regulating cell cycle progression in hMECs, together with our recent findings of Ecd overexpression in breast cancer progression where Ecd overexpression correlated with poorer prognosis in ErbB2-positive patients, we assessed if Ecd overexpression drives cell cycle progression during mammary oncogenesis. Ras activation provides a major branch of signaling to link ErbB2 oncogene to cell cycle progression, and mutant H-Ras is very commonly used in cooperative oncogenesis protocols to transform hMECs.10,30 Therefore, we used retroviral infection to stably overexpress Ecd and/or H-RasQ61L in hTERT-immortalized but non-tumorigenic hMEC line 76N.TERT that is strictly dependent on exogenous growth factors for cell cycle progression and proliferation.Citation7,8 Western blot analysis of cell lysates confirmed the expression of overexpressed Ecd and/or Ras in respective single or double transductants (). Next, we assessed the impact of Ecd and/or Ras overexpression on the levels of known downstream effectors of Ras pathways. For this purpose, cells were cultured for 72 hours in growth factor deprivation conditions (DFCI-3) and then supplemented with complete medium DFCI-1.Citation8 Prior to shifting the cultures into growth factor-containing medium p-AKT and p-ERK signals were relatively low and comparable between the control and various transductants (0 time points- ). A significant increase was observed in the levels of p-AKT and p-ERK upon growth factor stimulation in all cases while the total levels for AKT and ERK levels remained comparable. Densitometry analysis was performed using ImageJ software and the values were graphically presented as ratio of phospho vs. total protein levels for AKT, ERK1 and ERK2 (). Notably, higher levels of p-ERK1/2 were observed after growth factor stimulation (GFS) with complete medium in cells overexpressing Ecd+Ras as compared to other transductants. These results suggest that Ecd cooperates with Ras to stimulate the downstream effector p-ERK, a known physiological regulator of Ras-mediated cell proliferation.
Figure 2. Co-overexpression of Ecd and H-RasQ61L upregulates phospho-ERK1/2 levels upon growth factor stimulation. (A) Retrovirally transduced stable transductants of 76N.TERT were generated and analyzed for expression of Ecd and Ras using protein gel blotting, (B) The retroviral transductants expressing vector, Ras, Ecd and Ecd+Ras were deprived of growth factors for 72 hours in DFCI-3 medium, and switched to growth factor containing DFCI-1 medium. Growth factor stimulation (GFS) done for the indicated time (mins). Lysates were blotted for indicated proteins. Densitometry signals using ImageJ were plotted as a ratio of signals for phospho vs total proteins over time for AKT (C) and ERK1/ ERK2 (D and E).
To test our hypothesis that overexpressed Ecd may promote cell cycle progression in an oncogenic context, we used growth factor deprivation in DFCI-3 medium to synchronize various cell lines in G1 phase followed by synchronous release into cell cycle by switching to growth factor-enriched DFCI-1 medium, as described in Materials and Methods. The cell cycle profile of the transductants was analyzed using FACS analysis. Data was collected from 4 independent experiments for the % of cells in G1 after synchronization at 0 hour time point. Statistical analysis showed significant difference among the 4 groups in G1 0 h percentage (P < 0001, ). Further analysis with Tukey's pairwise comparison to control for multiple testing revealed that the mean G1 0 h percentage of Ecd+Ras group (Mean ± SD of Ecd+Ras: 56.6% ± 11.0%) was significantly less than that of Vector group (Mean ± SD of Vector: 88.6% ± 3.4%, P < .0001) and that of Ras group (Mean ± SD of Ras: 82.7% ± 3.0%, p = 0.0006) and that of Ecd group (Mean ± SD of Ecd: 88.9% ± 6.0%, P < 0 .0001). In contrast, there was no evidence of difference in the mean G1 0 h percentage between Vector group with Ecd group (p = 0.99) and Ras group (p = 0.61 ; Table S2).
Figure 3. Co-overexpression of Ecd and Ras in 76N.TERT cells impairs G1 cell cycle arrest and promotes rapid and enhanced cell cycle progression. Cells were growth factor deprived for 72 hours in DFCI-3, followed by release into cell cycle in complete medium (DFCI-1). (A) The cell cycle profiles at the indicated time points were analyzed after propidium iodide staining using FACS. (B) Percentage of cells in G1 phase at ‘0’ hour timepoint (prior to switch to DFCI-1). Mean +/− SD with p-values as shown for 4 experimental replicates (N = 4). (C) Cell lysates at the indicated time points of growth factor stimulation were analyzed by blotting for the indicated proteins. (D) Cells subjected to growth factor deprivation in DFCI-3 medium were counted at the indicated time points.
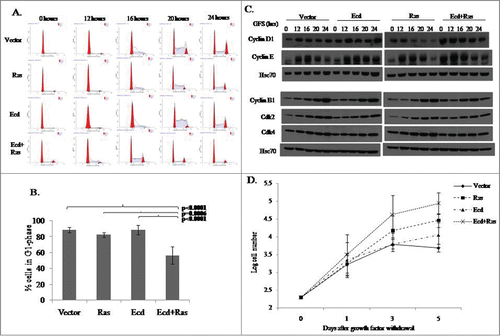
Western blot analyses of cell lysates prepared at various time points of cell cycle progression showed that Ecd+Ras-overexpressing cells had higher levels of G1 and G2 cyclins at time 0, indicative of deregulated cell cycle (). Taken together, these results support the idea that Ecd cooperates with Ras to promote more rapid cell cycle progression, and appears to further relax the requirement of exogenous growth factors for cell cycle progression.
Ecd plus Ras overexpressing hMECs exhibit enhanced survival under growth factor deficient conditions
Given the ability of Ecd+Ras-overexpressing cells to continue to enter the S-phase of cell cycle under growth factor deprivation conditions, we further assessed their proliferation under conditions of growth factor deprivation. We cultured various transductants in growth factor deprived medium DFCI-3, and counted cells as a direct indicator of cell proliferation at different times over a 5-day period. There was a statistically significant difference among the 4 groups in log cell count on day 3 and day 5 (p = 0.009 and p = 0.0006 respectively), but not on day 0 and day1 (p = 0.99 and p = 0.67 respectively) (; Table S3). There was a significant difference in log number of cells in Ecd+Ras group on day 3 as compared to Vector and Ecd alone (p = 0.01). Ras alone cells did not show a significant difference with Ecd+Ras group at this time point. The difference became more significant at day 5 where Ecd+Ras group showed a greater significance as compared to Vector or Ecd alone (p = 0.0002 and p = 0.009). Ras alone group showed a moderately significant difference as compared to Vector (p = 0.02) at this time point but no significant difference as compared to Ecd alone (Table S3). These results suggest that Ecd+Ras overexpression relaxes the requirements for growth factors for proliferation.
Ecd plus Ras overexpression promotes anchorage independent growth
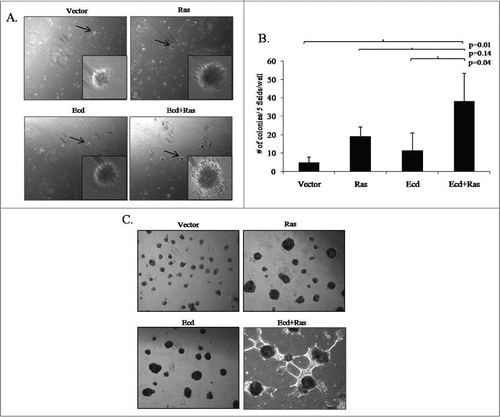
While normal epithelial cells require matrix adhesion to proliferate, transformed cells can survive and proliferate under anchorage-independent conditions.Citation9 We have previously shown that normal or immortal hMECs do not exhibit anchorage independence, while their oncogenically-transformed derivatives do.Citation10 Consistent with previous findings, vector control 76N.TERT cells showed few smaller colonies when plated on soft agar (), whereas enhanced colony numbers as well as sizes were seen with various transductants. Statistical analysis showed significant (p = 0.01) difference in the 4 groups for number of soft agar colonies. The mean colony number of Ecd+Ras group was significantly greater than that of Vector (p = 0.01), or Ecd (p = 0.04). There was no significant difference observed in colony numbers of Ras group as compared to Vector or Ecd groups (Table S2). These results clearly demonstrate co-oncogenic role of Ecd and Ras.
Figure 4. Co-overexpression of Ecd and Ras significantly enhances anchorage independent growth in soft agar and branching morphology in 3D matrigel culture. (A) Representative pictures of soft agar colonies taken at 4 × magnifications; with insets at 20×. (B) Bar diagram shows number of colonies per 5 fields per well in triplicates from 3 independent experiments (N = 3); in the indicated cell lines. Mean +/− SD with p-values are shown. (C) Representative 3D structures formed by various cell lines at Day12. Pictures are taken at 10× magnification.
Ecd plus Ras overexpressing cells exhibit aberrant branching in 3-dimensional (3D) matrigel culture
We have previously shown that hMECs exhibit polarized organization and limited proliferation with acinar morphology, when cultured on 3D matrices such as matrigel to mimic the in vivo tissue microenvironment.Citation11,12 We have previously established that immortal hMECs form acini with self-limiting proliferation on Matrigel, while their oncogenically-transformed derivatives show loss of polarity and hyperproliferation, resulting in irregular acini and/or aberrant branching structures.Citation11-13 As expected, the vector-expressing 76N.TERT cells formed regular acinar structures in 3D culture as observed under phase-contrast microscopy on day 12 (; Fig. S1). Notably, overexpression of Ecd or Ras alone promoted moderately irregular but larger sized acini, as compared to vector expressing cells. Notably, Ecd+Ras-overexpressing cells exhibited a dramatic increase in branched structures, similar to those reported with transformed hMECs and breast cancer cell lines.Citation13,14 These results suggest that the increased proliferative drive in Ecd+Ras-overexpressing hMECs is maintained under 3D culture conditions under which normal cells show self-limited growth.
Ecd plus Ras overexpression promotes migration and invasion of hMECs
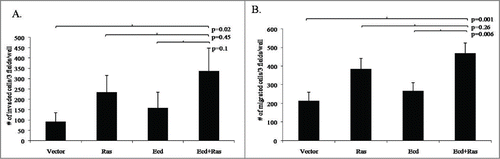
Given that cells expressing Ecd+Ras exhibited irregular branching morphology resembling that seen with oncogenically-transformed hMECs and breast cancer cell lines,Citation14 we assessed the impact of Ecd+Ras overexpression on the ability of cells to migrate and invade, using in vitro migration and invasion assays, as described in Materials and Methods. As anticipated from our previous studies,Citation13 control cells showed little migration toward the growth factor-enriched medium or invasion through matrix (Fig. S2). Statistical analysis showed significant difference among the 4 groups number of invaded and migrated cells (p = 0.03 and p = 0.001, respectively). Further analysis revealed that the mean number of cells invaded in Ecd+Ras group was significantly greater than that of Vector (p = 0.02 ). Ras or Ecd alone did not show a significant difference in invasion compared with Vector. On the other hand the mean number of cells migrated of Ecd+Ras group was significantly greater than that of Vector (p = 0.001) or Ecd (p = 0.006). Ras alone cells showed a significant difference in migration as compared to Vector (p = 0.01) but not in comparison with Ecd alone (p = 0.08 ; Table S2). These results further underscore the cooperative promotion of oncogenic traits by Ecd and Ras co-overexpression in hMECs.
Figure 5. Co-overexpression of Ecd and Ras enhances cell invasion and migration. The indicated cell lines were analyzed for invasion (A) and migration (B) as described in materials and methods. Histograms represent the number of cells per 3 fields per well in triplicates from 3 independent experiments (N = 3); that invaded or migrated after 24 hours. Mean +/− SD with p-values are shown.
Ecd plus Ras overexpression promotes tumor formation in vivo
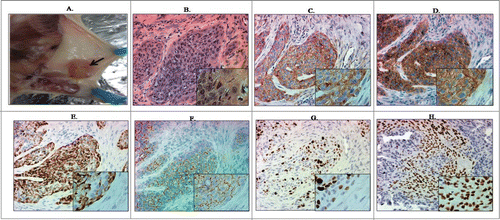
In vitro oncogenic changes observed in Ecd+Ras-overexpressing 76N.TERT cells suggested a full transformation of these cells. Therefore, we assessed the ability of various transductants to form tumors after mammary fat pad implantation in NOD/SCID mice. In keeping with modest increase in oncogenic traits in vitro, cells expressing vector, or Ras or Ecd alone did not form tumors over the full duration of observation, whereas 5 out of 5 mice implanted with cells co-overexpressing Ecd+Ras formed palpable tumors within 6–8 weeks after injection (). Hematoxylin and Eosin staining of histological sections of resected tumors showed epithelial tumor cells (adenocarcinoma) as well as mesenchymal (metaplastic) lesions (). Ecd and Ras expression was observed in the tumor tissue () confirming that the tumors were formed from the injected cells. Immunohistochemical analysis of these tumors showed significant expression of epithelial marker E-cadherin and mesenchymal marker vimentin (). Furthermore, these tumors stained strongly for Ki67, indicative of a high degree of proliferation ().Citation15 The cells within tumors also stained positive for p63, a marker for metaplastic carcinomas Citation16,17 (). Taken together, our results show a full oncogenic transformation of hTERT-immortalized hMECs upon Ecd and Ras co-overexpression.
Figure 6. 76N.TERT cells co-overexpressing Ecd and Ras form tumors upon mammary fat pad implantation in NOD/SCID mice. (A) Tumors were resected. Representative tumor in the mammary gland indicated by an arrow. (B) H & E staining of tumors formed by Ecd+Ras overexpressing cells resected at euthanasia. (C–H) Immunohistochemical analysis of tumor sections for Ecd (C), Ras (D), Vimentin (E), E-cadherin (F), Ki-67 (G), and p63 (H). Pictures were taken at 20X magnification (insets at 40×).
Ecd plus Ras overexpression induces upregulation of autophagy marker LC3
In order to mechanistically understand the increased survival of Ecd+Ras overexpressing hMECs under nutrient deprivation, we focused on the autophagy pathway, as it has been shown to promote cell survival under conditions of nutrient starvation, hypoxia and other stresses.Citation18 Given that recent studies have demonstrated that oncogenic H-Ras or K-Ras promotes basal autophagy as a mechanism to promote tumor cell survival,Citation19–22 we assessed if Ecd+Ras promote cell survival under nutrient deprivation by elevating autophagy markers.
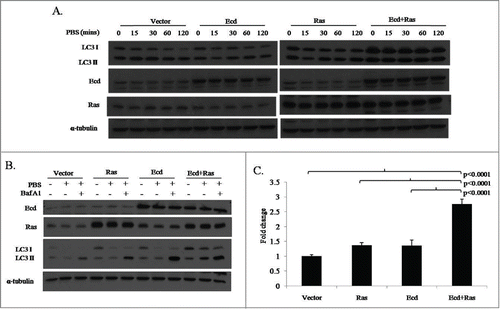
To directly assess the impact of Ecd and/or Ras overexpression on autophagy induction in hMECs, we exposed various transductants to a standard autophagy-induced nutrient deprivation protocol by culturing them in phosphate buffered saline.Citation23 As expected, Ras-overexpressing cells showed increased levels of LC3 at various time points. Notably, while Ecd overexpression by itself had no obvious impact on LC3 levels, Ecd+Ras-overexpressing cells exhibited much higher levels of LC3 I and LC3 II basally (0 time point), as well as throughout the course of autophagy induction ( and S3A). To confirm the induction of autophagy, we blocked autophagic flux using Bafilomycin A1.Citation24 A substantial accumulation of LC3 II was observed in all cell lines, indicating an active autophagic response (; Fig. S3B). In order to demonstrate if the observed upregulation is at transcript levels or protein levels, we performed quantitative PCR analysis for LC3 gene transcripts. This revealed upregulation of basal LC3 mRNA level in Ecd+Ras-overexpressing cells (); consistent with recent studies that upregulation of LC3 levels in autophagy involves increased transcription.Citation25,26 The fold change in expression of LC3, normalized to β-Actin and relative to the expression of vector, was calculated using Livak method. Statistical analysis showed significant difference among the 4 groups in fold change of LC3 (P < .0001). Further analysis revealed that the mean fold change of LC3 in Ecd+Ras group was significantly higher than that of vector or Ras or Ecd group (all 3 cases P < 0.0001; Table S2).
Figure 7. Co-overexpression of Ecd and Ras induces upregulation of autophagy marker LC3. (A) Cells were growth factor deprived by culture in PBS for the indicated time points. Lysates were blotted with the indicated antibodies. (B) Cells were growth factor deprived by culture in PBS for 2 hours and treated with or without Bafilomycin A1. Lysates were blotted with the indicated antibodies. (C) The indicated cell lines were analyzed for LC3B mRNA expression by q-RT-PCR. Fold change relative to vector expressing cell lines are shown. Mean +/− SD and p-values are shown for triplicate samples from 3 independent experiments (N = 3).
Further analyses using immunofluorescence assays confirmed presence of more LC3 puncta in Ecd+Ras-overexpresssing cells (Fig. S4A). Also, electron microscopy confirmed the higher number of autophagic vacuoles in Ecd+Ras-overexpressing cells (Fig. S4B). Taken together, these results demonstrate a cooperative increase in basal autophagic program in Ecd+Ras-overexpressing hMECs, suggesting a potential contribution of autophagy in Ecd+Ras overexpression-dependent survival that promotes oncogenesis.
Discussion
Precise regulation of cell cycle is a requisite for normal tissue development and homeostasis. Aberrations that promote uncontrolled transit through cell cycle lead to tumorigenesis. We have previously identified mammalian Ecdysoneless (Ecd) as a novel cell cycle regulator that acts through functional antagonism of Rb-family proteins and is essential for embryonic development in mice.Citation4 Cre-mediated deletion of Ecd in MEFs led to G1/S arrest that was rescued by hEcd, strongly supporting an essential role of mammalian Ecd to promote cell cycle progression.Citation4 Based on the original identification of Ecd from a hMEC cDNA library,Citation3 and further findings that Ecd is overexpressed in human breast cancers,Citation5 we undertook studies that reveal Ecd as an essential regulator of cell cycle in hMEC, and as a cooperating oncogene in the oncogenesis when overexpressed.
Consistent with our previous work in which Ecd was conditionally deleted in Ecdflox/flox MEFs,Citation4 siRNA-mediated Ecd KD in hMECs exhibited loss of hyperphosphorylated Rb and Rb accumulation in its hypophosphorylated form () together with a reduction in the level of DHFR (), a cell cycle-related transcriptional target of Rb-repressed E2F transcriptional activators. Notably, microarray-based gene expression profiling demonstrated that several of the the top 40 genes whose expression was downregulated upon Ecd KD were E2F target genes. These findings strongly support our conclusion that Ecd is required for cell cycle progression in hMECs and that this role relies on the ability of Ecd to counter the Rb-mediated negative regulation of E2Fs, as we have delineated in a MEF system.Citation4
The above conclusion, together with our recent findings that Ecd is overexpressed in human breast cancers,Citation5 raised the possibility that Ecd could function as an oncogene in hMECs. Overexpression of Ecd alone in hMECs led to only a modest increase in cell cycle progression or proliferation under reduced growth factor conditions (; Table S3). Thus, it appears unlikely that Ecd can function as an oncogene by itself, consistent with its overexpression in the context of other oncogenic alterations that are known to drive human breast cancers.Citation5 To test if Ecd can alternatively serve as a cooperating oncogene, we utilized experimental oncogenesis of hMECs with activated H-Ras as an oncogene. While oncogenic mutations of Ras are less frequent in breast cancer,Citation27 overexpression of mutant Ras in hMEC and other epithelial transformation models is well-established as a strategy to reveal cooperating oncogenic roles of other genes.Citation28-30 Notably, mutations in components of the Ras pathways, such as MAPK, AKT and ERK, are frequently altered in breast cancer,Citation31-34 and Ras functions downstream of receptor tyrosine kinases such as ErbB2 that are major drivers of breast cancers.Citation35 In this context, Ecd overexpression together with ErbB2 signifies an especially poor prognosis.Citation5
We demonstrate that co-overexpression of Ecd and Ras in hMECs leads to a significant increase in cell cycle transit through S to G2/M under conditions of growth factor withdrawal (), which is not seen in cells overexpressing either gene alone. Importantly, under conditions where normal growth factor-containing medium is provided to cells that have been synchronized in G1 following growth factor withdrawal, Ecd+Ras overexpressing hMECs exhibit a significantly higher proportion of cells that transit through the G1-phase and at substantially earlier times (). Furthermore, when cell proliferation was monitored under conditions where growth factors had been removed from the medium, hMECs with Ecd+Ras overexpression continued to proliferate at substantially higher levels compared to vector controls, while only minor enhancement was seen in Ras or Ecd overexpressing cells (). While enhanced, pERK signals in hMECs expressing Ras was expected, it is notable that Ecd+Ras overexpressing hMECs showed a significant upregulation of phospho-ERK compared to Ras alone expressing cells (). As activated MAPK/ERK signaling pathway is known to relax the restriction point of cell cycle and promote progression through cell cycle,Citation36 it is likely that ERK pathway activation cooperates with the aberrant Rb function as a result of Ecd overexpression to deregulate cell cycle in Ecd+Ras overexpressing hMECs.
As uncontrolled cell cycle entry is well-established as a hallmark of oncogenesis, we characterized the Ecd+Ras overexpressing hMECs for evidence of oncogenic transformation in vitro. Significantly, Ecd+Ras overexpressing cells exhibited several in vitro traits of oncogenesis, including increased anchorage independence when grown in soft agar, increased branching when cultured in 3D matrigel cultures, and increased invasion and migration. Notably, overexpression of Ecd or Ras alone does not promote these traits significantly, consistent with cooperative oncogenesis by Ecd when co-expressed with an established oncogene Ras. Ecd+Ras overexpression is sufficient to promote full oncogenic transformation is demonstrated by their ability to form tumors when orthotopically implanted in the mammary fat pads of immunocompromised mice. Consistent with our in vitro findings, Ecd or Ras alone overexpressing hMECs did not form tumors under identical conditions, nor were any tumors observed with vector control cells. Thus, overexpressed Ecd functions as a cooperating oncogene in fully transforming immortal, non-tumorigenic, hMECs. These results are consistent with other reports where Ras together with other oncogenes has been shown to induce the transformation of TERT-immortalized hMECs.Citation37,38
Notably, the mouse tumors formed by implanted Ecd+Ras overexpressing hMECs showed both epithelial and metaplastic components, consistent with stem/progenitor properties of TERT-immortalized hMECs.Citation7 Consistent with relaxation of cell cycle transit observed in Ecd+Ras overexpressing hMECs in vitro, the tumors arising from these cells had high proliferation index as measured by the proportion of Ki-67 positive cells, a known and commonly used marker of cell proliferation in tumors.Citation15 Taken together, these results demonstrate the cooperative role of Ecd and Ras to promote full oncogenesis of hMECs by promoting deregulated cell cycle progression.
Given our observation that Ecd+Ras overexpressing hMECs continue to proliferate at a higher rate when growth factors are withdrawn, we investigated if Ecd plus Ras overexpression promotes survival under nutrient deprivation by elevating autophagy. Upregulation of basal autophagy is thought to help cancer cells to cope with oncogenesis-associated stress.Citation19,39,40 Furthermore; Ras is known to promote an autophagic response.Citation19,22 We observed increased basal levels of microtubule-associated protein1 lightchain3 (LC3) in Ecd+Ras overexpressing hMECs under nutrition deprivation conditions. Indeed, immunofluorescence analyses showed increased LC3 puncta Citation24 in Ecd+Ras overexpressing hMECs (Fig. S4A). Furthermore, increased LC3 conversion into LC3-II was seen upon autophagy inhibition with H+/ATPase inhibitor baflomycin A1.Citation23,24 Finally, electron microscopy confirmed the increase in autophagic vacuoles in Ecd+Ras overexpressing hMECs (Fig. S4B). LC3 may accumulate in cells through stabilization of protein due to reduced lysosomal turnover or by upregulation of transcription.Citation20,Citation22-26 Recent reports suggest that LC3 is transcriptionally regulated by the activation of pathways downstream of Ras.Citation41,42 We observed significantly higher levels of LC3B mRNA expression, suggesting a possible role of Ecd plus Ras to upregulate basal autophagy.
Ecd was recently found to interact with PIH1D1 in a phosphorylation dependent manner,Citation43 resulting in its incorporation into the R2TP multi protein chaperone complex Citation44,45 which regulates multiple biological processes, including the phosphatidylinositol-3 kinase-related protein kinase (PIKK) signaling and assembly of the mTORC1 complex.Citation45 R2TP complex involvement in multiple cellular functions, and upregulation of some of the complex members in cancers suggest a significant role of this complex in transformation. Given the fact that Ecd is upregulated in cancers it is possible that Ecd helps to functionally regulate activity of this complex and mediates Ecd+Ras induced oncogenesis. Future studies will be needed to assess this potential link of Ecd-R2TP complex and oncogenesis.
A recent study in Drosophila model system showed Ecd directly interacts with core components of the U5 snRNP spliceosomal complex, and Prp8 protein.Citation46 Deletion of Ecd prevented splicing of a large intron from CYP307A2/spookier (spok) pre-mRNA which inhibited the synthesis of Ecdysone hormone. Notably, human Ecd rescued this deletion phenotype, suggesting this function of Ecd is conserved in humans. Given the important role of pre-RNA splicing in cancer formation, it remains possible that this function of Ecd may contribute in Ecd+Ras mediated oncogenesis, an area of future investigation in our laboratory.
In conclusion, the present study establishes a requirement of Ecd for cell cycle progression in hMECs and demonstrates that overexpression of Ecd together with Ras deregulates cell cycle and provides survival advantage to promote hMEC oncogenesis. Future biochemical and cell biological studies, together with transgenic models, should help further delineate the mechanisms by which Ecd functions as a cooperating oncogene in mammary tumorigenesis.
Materials and Methods
Cell culture and generation of cell lines
Previously established hTERT- immortalized hMEC line 76N.TERT Citation7 was cultured at 37°C and 5% CO2 in the DFCI-1 medium.Citation8The human Ecd cDNA sequence was cloned in the pMSCV-puro retroviral vector (Clonetech) to yield pMSCV-puro-Ecd. pBABE-hygro-H-RasQ61L (mutant Ras) was used to overexpress Ras. The retroviral expression constructs and their parent vectors were transfected into amphotropic packaging cell line phoenix to generate replication-deficient retroviral particle. Stable retroviral transdunctants of 76N.TERT cell line were generated and maintained in medium containing 0.5 μg/ml of puromycin and 5 μg/ml of hygromycin.
Chemicals and reagents
The following chemicals were used as per manufacturer's protocols unless specifically mentioned. Bafilomycin A1 was obtained from SIGMA (# B-1793), puromycin (SIGMA# P8833), hygromycin (Invitrogen # 10687-010), TRIzol reagent (Invitrogen #15596-018), SYBR green PCR Master Mix (Applied Bio systems # 4309155).
Western blotting and antibodies
Western blotting was performed as previously described.Citation4 Ecd monoclonal antibody used for western blotting was developed in our laboratory.Citation4 The following antibodies were obtained from commercial sources: Anti-Ras (BD Transduction labs #61001), Cyclin B1 (BD PharMingen #554179), Retinoblastoma (BD PharMingen #554136), CD49f (BD PharMingen #555734), α-tubulin (SIGMA #T5168), LC3B (Cell signaling #2775S), anti-p63 (Neomarkers #MS-1081-P), anti-Ki67 (DAKO #7240). Santa Cruz Biotechnology: p-ERK(sc-7383), ERK1(sc-94), p-AKT (sc-7985-R), AKT1 (sc-5298), AKT2 (sc-5270), Cyclin D1 (sc-20044), Cyclin E (sc-481), Cdk2 (sc-6248), Cdk4 (sc-23896), Hsc70 (sc-7298), anti-E-cadherin (sc-8426), anti-Vimentin (sc-6260) and DHFR (sc-14778).
Microarray analyses
Total RNA was isolated from triplicate cultures of 76N cells expressing scrambled siRNA control or Ecd siRNA,Citation3 and were analyzed using Affymetrix HG-U133 Plus 2 array to determine global transcriptional changes after Ecd knockdown. MAS 5.0 normalization was performed on the raw data obtained. An empirical Bayes shrinkage moderated t-statistics was adopted to estimate the significance of differential gene expression. Normalized and raw expression data were deposited in the Gene Expression Omnibus at the National Center for Biotechnology Information (GSE61277).
FACS-based cell cycle analyses
Various 76N.TERT transductants were plated at 5 × 105 cells per 100-mm2 dishes for 1 day prior to cell cycle synchronization using growth factor deprivation. The cells were cultured for 72 hours in growth factor-deficient DFCI-3 mediumCitation47 to arrest cells in G1 phase, and initiated into cell cycle by culture in growth factor-containing DFCI-1 medium. Half of the cells were fixed in chilled 70% ethanol followed by propidium iodide staining for FACS-based cell cycle analysis.Citation4 The remaining cells were harvested for protein gel blot analysis.
Survival proliferation assay
76N.TERT transductants were seeded in triplicates in 6-well plates at a density of 1 × 105 cells/ well. The cells were growth factor deprived in DFCI-3 medium beginning the day after plating and this was considered day 0. The cells were trypsinized and counted on day 1, 3, and 5. Three samples were collected for each cell type. The outcome cell count was measured for each sample at starving day 0, 1, 3, and 5 respectively. The average cell count measurement of 3 technical replicates per sample at each starving time was recorded. The cell count was log-transformed to meet the normality assumptions of the mixed linear model. Mixed linear models were used to assess the difference in log cell count among cell type accounting for the correlated measurement within a sample. If the interaction between cell type and starving time was significant, the cell type effect at each starving time was determined; if the cell type effect at one starving time was significant, pair wise comparisons for each pair cell types at that starve time was made with Tukey-Kramer's correction.
Anchorage-independent growth
2 × 104 cells per well were plated in triplicates in 6-well plates in 2 ml of 0.3% agarose in DFCI-1 on top of a bottom layer of 2 ml of 0.6% agarose in DFCI-1 medium . Cultures were fed every 2 days with 2 ml DFCI-1 medium. Twenty-one days after cell seeding, the plates were fixed and stained with 0.05% crystal violet in 25% methanol and colonies in 5 random fields per well were counted.
3-D Matrigel cultures
Laminin-rich, reduced growth factor basement membrane matrix (Matrigel®) for 3D cultures was obtained from BD Biosciences (San Jose, CA). Cells were suspended in DFCI-1 medium and seeded at 2 × 104 per well in 6-well plates over a layer of polymerized 100% Matrigel (400 μl/well). The cells were cultured for 10–12 days and images were taken under 4× magnification on a Nikon Eclipse TS100 microscope. Immunoflorescence was performed for 3D colonies using antibody against polarity marker CD49f. DAPI was used as nuclear stain.
Transwell migration and invasion assays
The migration assay was done using BD BioCoatTranswell chambers (#354578). Overnight growth factor deprived cells were trypsinized and plated at 2 × 104 per well in growth factor deprived DFCI-3 medium Citation47in the top chambers. After 2 h, growth factors containing medium (DFCI-1) was added to the lower chamber and incubation carried out for 24 h. Cells were then fixed with ice-cold methanol and stained with propidium iodide. Non-migrated cells on the upper surfaces of filters were removed by scraping with cheese cloth. The migrated cells on the bottom surface of filters were observed at 10× magnification under rhodamine filter. Three random fields were counted from each well of triplicates. Invasion assay was performed using BD Matrigel invasion chambers (#354480). Cells were plated, processed and counted similar to migration assay.
Tumorigenicity assays
Six-week-old female NOD/SCID mice (Jackson Laboratories) were anesthetized and 2 × 106 cells suspended in 50 μl of 1:1 matrigel and saline were implanted in the mammary fat pad of ninth mammary gland, and animals were monitored for tumor growth. Animals were euthanized, and necropsies were performed when tumors reached 1 to 1.5 cm diameter, or after about 4 months if no tumors were formed. Each cell line was tested in 5 animals. All animal-related procedures were approved by the Institutional Animal Care and Use Committee.
Immunohistochemistry
Tumor tissues were fixed in 10% Neutral buffered Formalin (NBF) and processed into paraffin blocks. 4 μm sections were cut and stained with H&E or indicated antibodies. The standard staining procedure was performed using DAKO kit as per the manufacturer's protocol (# K4007) and as described in (5). The tissue sections were incubated with polyclonal or monoclonal primary antibodies for overnight unless otherwise mentioned. Antibodies used were: Monoclonal anti-Ecd Citation4,5; anti-Ras, anti-p63, anti-E-cadherin, anti-Vimentin and anti-Ki67.
RNA isolation and quantitative real-time PCR
Total RNA was isolated from triplicate samples of 76N.TERT transductants using TRIzol reagent according to the manufacturer's protocol. 1 μg RNA was reverse transcribed using SuperScriptTM II reverse transcriptase (Invitrogen) and oligo-dT primers (18MER) and quantitative real-time PCR (qRT-PCR) analyses with specific primer sets for LC3B Forward: 5′-GAGCAGCATCCAACCAAAAT-3′ and Reverse: 3′- CTGTGTCCGTTCACCAACAG-5′ and PrimePCR: ACTB, Human (Biorad #100–25636) for β-actin, were carried out in an Applied Biosystems 7500 Real Time PCR system using Power SYBR Green master mix. Three technical replicates were used for each experiment. One way analysis of variance (ANOVA) was used to determine if there was a significant difference in the ΔCt among all 4 groups of cell types; if the overall p-value was significant, pair wise comparisons for each pair cell types was made using t-tests with Tukey-Kramer's correction.
Autophagy induction and LC3 analysis
76N.TERT transductants were subjected to starvation stress using 1× PBS. Bafilomycin A1 was used at 10 nM for the last 30 minutes of PBS starvation. Lysates at the indicated time points were western blotted for LC3I and LC3II. For immunofluorescence studies, cells were plated on coverslips and immunostaining was done as described in (Supplementary materials and methods) section.
Statistical analysis
For continuous outcomes, one way ANOVA was used to determine if there was a significant difference among 4 groups of cell types. Pair wise comparisons for each pair cell types were adjusted by the Tukey-Kramer's method. All data were analyzed using SAS procedure (version 9.3 provided by SAS institute Inc., Cary, NC).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1006982_Supplementary_Materials.zip
Download Zip (2.3 MB)Acknowledgments
We thank all the members of Bands laboratories for their thoughtful discussion and suggestions throughout this work. We thank Janice A. Taylor and James R. Talaska of the Confocal Laser Scanning Microscope Core Facility and Tom Bargar of the Electron microscopy Core facility at the University of Nebraska Medical Center for providing assistance with microscopy.
Funding
This research is supported by the Department of Defense predoctoral fellowship W81XWH-11–1–0020 to A.B; Susan G. Komen Postdoctoral Fellowship KG111248 to S.M; NIH grant CA96844 and CA144027 and Department of Defense grant W81XWH-11-1-0171 to V.B; the NIH grants CA87986, CA105489, CA99163, CA116552 and the NCI Core Support Grant to UNMC-Fred & Pamela Buffett Cancer Center to H.B.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432:316-23; PMID:15549093; http://dx.doi.org/10.1038/nature03097
- Zafonte BT, Hulit J, Amanatullah DF, Albanese C, Wang C, Rosen E, Reutens A, Sparano JA, Lisanti MP, Pestell RG. Cell-cycle dysregulation in breast cancer: breast cancer therapies targeting the cell cycle. Front Biosci 2000; 5:D938-61; PMID:11102317; http://dx.doi.org/10.2741/zafonte
- Zhang Y, Chen J, Gurumurthy CB, Kim J, Bhat I, Gao Q, Dimri G, Lee SW, Band H, Band V. The human orthologue of Drosophila ecdysoneless protein interacts with p53 and regulates its function. Cancer Res 2006; 66:7167-75; PMID:16849563; http://dx.doi.org/10.1158/0008-5472.CAN-06-0722
- Kim JH, Gurumurthy CB, Naramura M, Zhang Y, Dudley AT, Doglio L, Band H, Band V. Role of mammalian Ecdysoneless in cell cycle regulation. J Biol Chem 2009; 284:26402-10; PMID:19640839; http://dx.doi.org/10.1074/jbc.M109.030551
- Zhao X, Mirza S, Alshareeda A, Zhang Y, Gurumurthy CB, Bele A, Kim JH, Mohibi S, Goswami M, Lele SM, et al. Overexpression of a novel cell cycle regulator ecdysoneless in breast cancer: a marker of poor prognosis in HER2/neu-overexpressing breast cancer patients. Breast Cancer Res Treat 2012; 134:171-80; PMID:22270930; http://dx.doi.org/10.1007/s10549-011-1946-8
- Dey P, Rachagani S, Chakraborty S, Singh PK, Zhao X, Gurumurthy CB, Anderson JM, Lele S, Hollingsworth MA, Band V, Batra SK. Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clin Cancer Res 2012; 18:6188-98; PMID:22977192; http://dx.doi.org/10.1158/1078-0432.CCR-12-1789
- Zhao X, Malhotra GK, Lele SM, Lele MS, West WW, Eudy JD, Band H, Band V. Telomerase-immortalized human mammary stem/progenitor cells with ability to self-renew and differentiate. Proc Natl Acad Sci U S A 2010; 107:14146-51; PMID:20660721; http://dx.doi.org/10.1073/pnas.1009030107
- Band V, Sager R. Distinctive traits of normal and tumor-derived human mammary epithelial cells expressed in a medium that supports long-term growth of both cell types. Proc Natl Acad Sci U S A 1989; 86:1249-53; PMID:2919173; http://dx.doi.org/10.1073/pnas.86.4.1249
- Assoian RK, Zhu X. Cell anchorage and the cytoskeleton as partners in growth factor dependent cell cycle progression. Curr Opin Cell Biol 1997; 9:93-8; PMID:9013668; http://dx.doi.org/10.1016/S0955-0674(97)80157-3
- Datta S, Hoenerhoff MJ, Bommi P, Sainger R, Guo WJ, Dimri M, Band H, Band V, Green JE, Dimri GP. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res 2007; 67:10286-95; PMID:17974970; http://dx.doi.org/10.1158/0008-5472.CAN-07-1636
- Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A 1992; 89:9064-8; PMID:1384042; http://dx.doi.org/10.1073/pnas.89.19.9064
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30:256-68; PMID:12798140; http://dx.doi.org/10.1016/S1046-2023(03)00032-X
- Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen G, Goswami R, Fernandes N, Gao Q, Dimri GP, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res 2007; 67:4164-72; PMID:17483327; http://dx.doi.org/10.1158/0008-5472.CAN-06-2580
- Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH, Petersen OW, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol 2007; 1:84-96; PMID:18516279; http://dx.doi.org/10.1016/j.molonc.2007.02.004
- Bouzubar N, Walker KJ, Griffiths K, Ellis IO, Elston CW, Robertson JF, Blamey RW, Nicholson RI. Ki67 immunostaining in primary breast cancer: pathological and clinical associations. Br J Cancer 1989; 59:943-7; PMID:2472168; http://dx.doi.org/10.1038/bjc.1989.200
- Tse GM, Tan PH, Chaiwun B, Putti TC, Lui PC, Tsang AK, Wong FC, Lo AW. p63 is useful in the diagnosis of mammary metaplastic carcinomas. Pathology 2006; 38:16-20; PMID:16484002; http://dx.doi.org/10.1080/00313020500444625
- Koker MM, Kleer CG. p63 expression in breast cancer: a highly sensitive and specific marker of metaplastic carcinoma. Am J Surg Pathol 2004; 28:1506-12; PMID:15489655; http://dx.doi.org/10.1097/01.pas.0000138183.97366.fd
- Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 2013; 8:105-37; PMID:23072311; http://dx.doi.org/10.1146/annurev-pathol-020712-163918
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:165-78; PMID:21119005; http://dx.doi.org/10.1091/mbc.E10-06-0500
- Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov 2014; 4:466-79; PMID:24513958; http://dx.doi.org/10.1158/2159-8290.CD-13-0841
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717-29; PMID:21406549; http://dx.doi.org/10.1101/gad.2016111
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149:656-70; PMID:22541435; http://dx.doi.org/10.1016/j.cell.2012.01.058
- N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 2009; 10:173-9; PMID:19148225; http://dx.doi.org/10.1038/embor.2008.238
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/10.4161/auto.5338
- Morris CR, Stanton MJ, Manthey KC, Oh KB, Wagner KU. A knockout of the Tsg101 gene leads to decreased expression of ErbB receptor tyrosine kinases and induction of autophagy prior to cell death. PLoS One 2012; 7:e34308; PMID:22479596; http://dx.doi.org/10.1371/journal.pone.0034308
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010; 29:4424-35; PMID:20514020; http://dx.doi.org/10.1038/onc.2010.191
- Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 2001; 93:1062-74; PMID:11459867; http://dx.doi.org/10.1093/jnci/93.14.1062
- Dimri G, Band H, Band V. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res 2005; 7:171-9; PMID:15987472; http://dx.doi.org/10.1186/bcr1275
- Eckert LB, Repasky GA, Ulku AS, McFall A, Zhou H, Sartor CI, Der CJ. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res 2004; 64:4585-92; PMID:15231670; http://dx.doi.org/10.1158/0008-5472.CAN-04-0396
- Rao K, Bryant E, O'Hara Larivee S, McDougall JK. Production of spindle cell carcinoma by transduction of H-Ras 61L into immortalized human mammary epithelial cells. Cancer Lett 2003; 201:79-88; PMID:14580689; http://dx.doi.org/10.1016/S0304-3835(03)00423-3
- Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res 2007; 5:195-201; PMID:17314276; http://dx.doi.org/10.1158/1541-7786.MCR-06-0263
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012; 3:954-87; PMID:23006971
- de Vries-Smits AM, Burgering BM, Leevers SJ, Marshall CJ, Bos JL. Involvement of p21ras in activation of extracellular signal-regulated kinase 2. Nature 1992; 357:602-4; PMID:1608472; http://dx.doi.org/10.1038/357602a0
- Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science 1996; 271:810-2; PMID:8628998; http://dx.doi.org/10.1126/science.271.5250.810
- Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK, Arteaga CL. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006; 10:25-38; PMID:16843263; http://dx.doi.org/10.1016/j.ccr.2006.05.023
- Zwang Y, Sas-Chen A, Drier Y, Shay T, Avraham R, Lauriola M, Shema E, Lidor-Nili E, Jacob-Hirsch J, Amariglio N, et al. Two phases of mitogenic signaling unveil roles for p53 and EGR1 in elimination of inconsistent growth signals. Mol Cell 2011; 42:524-35; PMID:21596316; http://dx.doi.org/10.1016/j.molcel.2011.04.017
- Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, Iglehart JD, Weinberg RA. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 2007; 12:160-70; PMID:17692807; http://dx.doi.org/10.1016/j.ccr.2007.06.013
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 2001; 15:50-65; PMID:11156605; http://dx.doi.org/10.1101/gad.828901
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460-70; PMID:21317241; http://dx.doi.org/10.1101/gad.2016311
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Kim JH, Hong SK, Wu PK, Richards AL, Jackson WT, Park JI. Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp Cell Res 2014; 327:340-52; PMID:25128814; http://dx.doi.org/10.1016/j.yexcr.2014.08.001
- Kim JH, Hong SK, Wu PK, Richards AL, Jackson WT, Park JI. Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp Cell Res 2014; 327:340-52; PMID:25128814; http://dx.doi.org/10.1016/j.yexcr.2014.08.001
- Horejsi Z, Stach L, Flower TG, Joshi D, Flynn H, Skehel JM, O'Reilly NJ, Ogrodowicz RW, Smerdon SJ, Boulton SJ. Phosphorylation-dependent PIH1D1 interactions define substrate specificity of the R2TP cochaperone complex. Cell Rep 2014; 7:19-26; PMID:24656813; http://dx.doi.org/10.1016/j.celrep.2014.03.013
- Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, Krogan N, Cagney G, Mai D, Greenblatt J, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 2005; 120:715-27; PMID:15766533; http://dx.doi.org/10.1016/j.cell.2004.12.024
- Kakihara Y, Houry WA. The R2TP complex: discovery and functions. Biochim Biophys Acta 2012; 1823:101-7; http://dx.doi.org/10.1016/j.bbamcr.2011.08.016
- Claudius AK, Romani P, Lamkemeyer T, Jindra M, Uhlirova M. Unexpected role of the steroid-deficiency protein ecdysoneless in pre-mRNA splicing. PLoS Genet 2014; 10:e1004287; PMID:24722212; http://dx.doi.org/10.1371/journal.pgen.1004287
- Band V, Zajchowski D, Kulesa V, Sager R. Human papilloma virus DNAs immortalize normal human mammary epithelial cells and reduce their growth factor requirements. Proc Natl Acad Sci U S A 1990; 87:463-7; PMID:2153303; http://dx.doi.org/10.1073/pnas.87.1.463