
Abstract
The mammalian E3 ubiquitin ligases RNF8 and RNF168 facilitate recruitment of the DNA damage response protein 53BP1 to sites of DNA double-strand breaks (DSBs). The mechanism involves recruitment of RNF8, followed by recruitment of RNF168, which ubiquitinates histones H2A/H2AX on K15. 53BP1 then binds to nucleosomes at sites of DNA DSBs by recognizing, in addition to methyl marks, histone H2A/H2AX ubiquitinated on K15. We report here that expressing H2AX fusion proteins with N-terminal bulky moieties can rescue 53BP1 recruitment to sites of DNA DSBs in cells lacking RNF8 or RNF168 or in cells treated with proteasome inhibitors, in which histone ubiquitination at sites of DNA DSBs is compromised. The rescue required S139 at the C-terminus of the H2AX fusion protein and was occasionally accompanied by partial rescue of ubiquitination at sites of DNA DSBs. We conclude that recruitment of 53BP1 to sites of DNA DSBs is possible in the absence of RNF8 or RNF168, but still dependent on chromatin ubiquitination.
Introduction
The mechanisms by which cells recognize the presence of DNA damage to activate repair and checkpoint pathways are of considerable importance, because these pathways are critical for maintaining genomic integrity and for preventing cancer development.Citation1-5 One of the proteins involved in recognizing a particular type of DNA damage, DNA double-strand breaks (DSBs), is p53 Binding Protein 1 (53BP1), a protein that participates both in checkpoint activation by inducing cell cycle arrest and in DNA DSB repair by stimulating non-homologous end joining.Citation6-15
53BP1 contains a tandem tudor domain, between amino acids 1485–1602, which is critical for recruitment to sites of DNA DSBs.Citation16-17 The tudor domain binds to methylated lysines in the histone core; it can recognize histone H3 dimethylated on lysine 79 (H3K79me2) or histone H4 dimethylated on lysine 20 (H4K20me2) and inhibition of methylation of either of these residues partially compromises 53BP1 recruitment to sites of DNA DSBs.Citation18-21 It appears that H3K79me2 facilitates recruitment mainly in the G1 and G2 phases of the cell cycle, while H4K20me2 is more important in S phase.Citation22 Because both H3K79 and H4K20 map to the histone core and are thought to be inaccessible in higher order chromatin structure, it has been proposed that DNA DSBs facilitate 53BP1 recruitment by opening up chromatin structure.Citation18,23
A second domain required for recruitment of 53BP1 to sites of DNA DSBs maps to residues 1231–1277 and mediates 53BP1 homo-oligomerization.Citation24 Yet, a third fragment corresponding to residues 1614–1629 (region C-terminal to the tudor domain; RCTD) is also essential for recruitment of 53BP1 to sites of DNA DSBs.Citation24 As mentioned below, the RCTD can recognize ubiquitinated nucleosomes.
53BP1 recruitment to sites of DNA DSBs is dependent on histone H2AX, the DNA damage response protein MDC1 and the ubiquitin ligases RNF8 and RNF168.Citation25-32 Key aspects regarding how these proteins function to recruit 53BP1 have been elucidated. Activation of the ATM kinase at sites of DNA DSBs leads to phosphorylation of histone H2AX, which acts as a platform for recruitment of MDC1. Once recruited, MDC1 becomes phosphorylated by ATM, creating binding sites for RNF8, which then ubiquitinates histones H2A and H2AX and possibly other targets. RNF168 is subsequently recruited and this is followed by recruitment of 53BP1. The recruitment of RNF168 and 53BP1 depends on UBC13 E2 ubiquitin-conjugating enzyme and ubiquitin-activation enzyme UBA1.Citation27,28,32,33 Importantly, RNF168 recruitment depends on its ability to bind ubiquitin conjugates,Citation30-32 and overexpression of RAD18 ubiquitin-binding domain blocks RNF168 and 53BP1 accumulation at DNA damage sites.Citation34 However, 53BP1 lacks any obvious ubiquitin binding motif.
The exact mechanism by which RNF8 and RNF168 facilitate 53BP1 recruitment to sites of DNA DSBs has been a matter of debate. According to one model, histone ubiquitination changes chromatin structure and provides 53BP1 with access to methylated histones.Citation27,29 A second model proposes that ubiquitination results in the removal from chromatin of proteins that mask the 53BP1-binding epitopes. Specifically, L3MBTL1, a Polycomb protein that binds H4K20me2, is ubiquitinated and released from chromatin upon induction of DNA damage in an RNF8, RNF168 and VCP-dependent manner.Citation35 JMJD2A, another H4K20me2-binding protein, is also degraded in an RNF8 and RNF168-dependent manner following DNA damage.Citation36
Yet, a third model argues that the RCTD of 53BP1 interacts with the epitope formed when histone H2A is ubiquitinated on K15 and a new name, ubiquitination-dependent recruitment (UDR) motif, was proposed for the RCTD.Citation37 According to this model, 53BP1 is a bivalent histone modification reader, recognizing both methylation and ubiquitination, via its tudor domain and RCTD/UDR motif, respectively.
In an effort to better understand how 53BP1 is recruited to sites of DNA DSBs, we investigated ways, in which we could rescue 53BP1 recruitment in RNF8- or RNF168-deficient cells. Our results suggest that it is possible to recruit 53BP1 to sites of DNA DSBs in the absence of RNF8 or RNF168. However, our findings are still consistent with a model in which the RCTD/UDR motif recognizes ubiquitinated histones.
Results
Rescue of 53BP1 ionizing radiation-induced foci in RNF8-/- MEFs
In response to ionizing radiation (IR), RNF168 ubiquitinates histones H2A and H2AX on lysines 13 and 15.Citation38,39 The importance of this modification is strengthened by the observation that overexpression of USP3, a de-ubiquitinating enzyme that targets histones H2A and H2AX, abolishes 53BP1 recruitment.Citation40 Accordingly, we wondered whether expressing a ubiquitin-histone H2AX fusion protein in cells deficient for RNF8 or RNF168 would rescue 53BP1 recruitment to IR-induced foci (IRIF). We first examined RNF8-/- mouse embryo fibroblasts (MEFs). In these cells, expression of GFP-tagged RNF8 restored 53BP1 IRIF, confirming the previously published observations that loss of RNF8 is responsible for the defect in 53BP1 recruitment to sites of DNA DSBs (Fig. S1A). To attempt to bypass the RNF8 requirement for 53BP1 IRIF formation, we generated a fusion protein containing a FLAG tag at its N-terminus, then a ubiquitin molecule and finally a histone H2AX molecule (ubiq-H2AX). Strikingly, expression of this fusion protein rescued 53BP1 focus formation in RNF8-/- MEFs (). Importantly, the observed 53BP1 foci were IR-dependent (Figure S1B) and co-localized with γH2AX (Figure S1C). Expression of 2 control proteins, FLAG-tagged histone H2AX without a ubiquitin moiety (H2AX) or FLAG-tagged H2AX with a ubiquitin molecule fused to the C-terminus of histone H2AX (H2AX-ubiq) did not rescue 53BP1 IRIF ().
Figure 1. Rescue of 53BP1 IRIF in RNF8-/- MEFs. (A)RNF8-/- MEFs transiently expressing the indicated FLAG-tagged H2AX proteins were exposed to IR (9 Gy) and 4 h later processed for immunofluorescence. More than one hundred cells with high level of FLAG signal were scored for 53BP1 IRIF. The percentages of cells with more than 10 53BP1 foci (means ± 1 SD) from 3 to 4 independent experiments are indicated. Scale bar = 10 μm. K1315R, K13R/K15R double substitution. (B)RNF8-/- MEFs transiently expressing the indicated FLAG-tagged H2AX proteins were exposed to IR (9 Gy) and 4 h later processed for immunofluorescence using antibodies reacting with conjugated ubiquitin (FK2) or K63-linked polyubiquitin chains (K63). (C)RNF8-/- MEFs transiently expressing GFP-tagged RNF8 were exposed to IR (9 Gy) and 4 h later processed for immunofluorescence using antibodies reacting with GFP, conjugated ubiquitin (FK2) or K63-linked polyubiquitin chains (K63).
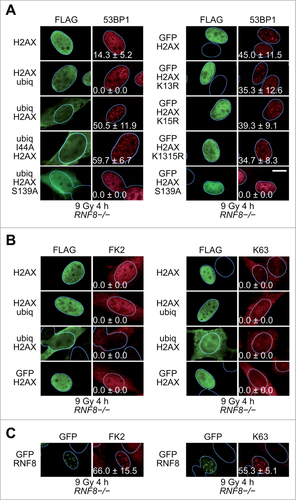
All the ectopically expressed H2AX proteins described above were incorporated into chromatin, as revealed by immunoblotting of chromatin pellets solubilized by acid (Fig. S2). Interestingly, a fraction of H2AX-ubiq was polyubiquitinated, when present in chromatin, and high amounts of polyubiquitinated H2AX-ubiq were also found in whole cell extracts. In contrast, most of the ubiq-H2AX protein present in chromatin was not polyubiquitinated, whereas in whole cell extracts ubiq-H2AX was polyubiquitinated (Fig. S2).
Although these results confirm that H2AX N-terminal ubiquitination is critical for 53BP1 recruitment to IRIF, they do not inform us on whether the ubiquitin-histone fusion protein itself provides a binding site for the 53BP1 RCTD/UDR motif or has a more indirect effect, such as, for example, opening up chromatin to provide access of the methyl marks to 53BP1. The interaction of many proteins with ubiquitin involves a hydrophobic patch on ubiquitin itself. Substitution of I44 at the center of this patch with alanine abolishes many of the known ubiquitin-protein interactions, including the interaction of 53BP1 with nucleosome core particles (NCPs) ubiquitinated on K15 of histone H2A.Citation37,41 Accordingly, we reasoned that expression of an I44A ubiquitin-H2AX fusion protein in RNF8-/- cells would not rescue 53BP1 IRIF. However, the I44A mutant was as efficient as wild-type ubiquitin in rescuing 53BP1 foci (). Guided by these results, we next asked if any bulky moiety fused to the N-terminus of H2AX could rescue 53BP1 IRIF. Strikingly, a GFP-H2AX fusion protein expressed in RNF8-/- MEFs restored 53BP1 recruitment to sites of DNA DSBs ( and Fig. S3). Similar results were obtained with every other H2AX N-terminal fusion studied; AcGFP, SUMO1 and SUMO2 fused to H2AX all rescued 53BP1 IRIF in RNF8-/- MEFs (Fig. S4A).
Immunoblotting of the chromatin fraction, indicated that the majority of the ectopically expressed GFP-H2AX fusion protein incorporated into chromatin migrated at the expected molecular size (Fig. S2A). However, a minor species, most likely corresponding to monoubiquitinated GFP-H2AX (see below) was also observed. This could be GFP-H2AX ubiquitinated on K119 (most of the monoubiquitinated endogenous H2A in cells is ubiquitinated on this residue) or GFP-H2AX ubiquitinated on K13 or K15 (since the N-terminal tail of H2AX is intact in the GFP-H2AX protein). If the latter were true, then this could explain the rescue of 53BP1 recruitment. To examine this possibility we expressed GFP-H2AX fusion proteins bearing K13R or K15R single substitutions or a K13R/K15R double substitution or a K13R/K15R/K119R triple substitution. All these mutant proteins were able to rescue 53BP1 IRIF ( and Fig. S4B). These results suggest that H2AX fusion proteins containing bulky modifications at the N-terminus can rescue 53BP1 IRIF in RNF8-/- cells without the need for the ectopic proteins themselves being ubiquitinated on residues K13, K15 or K119 of H2AX.
The H2AX fusion proteins with the N-terminal bulky moieties that rescue 53BP1 recruitment have an intact H2AX C-terminus that is, most likely, capable of being phosphorylated on S139 in response to DNA damage. Thus, the question arises whether phosphorylation of this residue is required for rescue of 53BP1 recruitment. A S139A substitution in the context of either ubiq-H2AX or GFP-H2AX, abolished the 53BP1 recruitment rescue in RNF8-/- cells (). Thus, both a bulky moiety at the N-terminus of H2AX and a C-terminal S139 are required for 53BP1 recruitment in RNF8-/- cells.
One mechanism by which the ectopically expressed H2AX fusion proteins could rescue 53BP1 IRIF could be by facilitating ubiquitination of endogenous H2A/H2AX molecules at sites of DNA DSBs. By immunofluorescence, ubiquitination can be observed at sites of DNA DSBs using antibodies specific for conjugated ubiquitin (FK2 antibody) or for K63-linked polyubiquitin chains. Thus, we examined whether expression of the H2AX fusion proteins rescued ubiquitination at sites of DNA DSBs. The RNF8-/- MEFs expressing the various H2AX fusion proteins were stained by immunofluorescence for conjugated ubiquitin (FK2 antibody), as well as for the presence of K63-linked polyubiquitin chains. None of the H2AX fusion constructs tested in RNF8-/- MEFs rescued FK2 or K63 IRIF ( and Fig. S4C), despite rescuing 53BP1 foci. In contrast, as a positive control, expression of GFP-RNF8 rescued both FK2 and K63 IRIF ( and Fig. S4D). However, we note that the background staining with the FK2 antibody was quite high () and, therefore, the possibility that some ubiquitination was present at sites of DNA DSBs in these cells cannot be excluded.
Expression of canonical histones with terminal bulky moieties in RNF8-/- cells does not rescue 53BP1 IRIF
An unresolved question, from the experiments presented so far, is whether histone H2AX, when fused to bulky moieties, is unique in its ability to rescue defects in 53BP1 recruitment or whether similar effects can be achieved by fusing the same bulky moieties to other histones. To address this question we fused ubiquitin to the N-termini or C-termini of the 4 canonical histones H2A, H2B, H3 and H4 or GFP to the N-termini of these same histones and examined the ability of the fusion proteins to rescue 53BP1 recruitment in RNF8-/- cells. Interestingly, none of the generated fusion proteins rescued 53BP1 recruitment (Fig. S5), in agreement with the observation that a S139A substitution within H2AX renders ubiq-H2AX and GFP-H2AX fusion proteins incapable of rescuing 53BP1 IRIF in RNF8-/- cells ().
Rescue of 53BP1 ionizing radiation-induced foci in RIDDLE cells
The experiments presented above indicate that placing a bulky moiety at the N-terminus of histone H2AX can rescue 53BP1 IRIF in RNF8-/- MEFs. RNF8 facilitates recruitment of a second ubiquitin ligase, RNF168, to chromatin, which then ubiquitinates lysines 13 and 15 of histones H2A and H2AX.Citation30,32,38,39 The ubiquitin modifications induced by RNF168 are the ones that are critical for 53BP1 recruitment. Thus, to further probe the mechanism by which RNF8 and RNF168 recruit 53BP1 to sites of DNA DSBs, we examined whether H2AX fusion proteins can rescue 53BP1 IRIF in RIDDLE cells, which do not retain a wild-type RNF168 gene.Citation42
Expression of ubiq-H2AX rescued 53BP1 IRIF formation in RIDDLE cells, while expression of H2AX-ubiq or FLAG-tagged H2AX did not () paralleling the results obtained with RNF8-/- MEFs. As expected, the 53BP1 foci in the RIDDLE cells expressing ubiq-H2AX were dependent on IR (Fig. S6A) and all the H2AX fusion proteins were incorporated into chromatin (Fig. S6B).
Figure 2. Rescue of 53BP1 IRIF in RIDDLE cells. (A)RIDDLE cells were transduced with lentiviral particles directing the expression of FLAG-tagged H2AX fusion proteins. Four hours after exposure to IR (9 Gy) the cells were processed for immunofluorescence. More than one hundred cells with high level of FLAG signal were scored for 53BP1 IRIF. The percentages of cells with more than 10 53BP1 foci (means ± 1 SD) from 3 to 4 independent experiments are indicated. Scale bar = 10 μm. (B)RIDDLE cells were transduced with lentiviral particles directing the expression of GFP-tagged RNF168. The cells were processed for immunofluorescence 4 h after exposure to IR (9 Gy) using antibodies reacting with GFP, conjugated ubiquitin (FK2) or K63-linked polyubiquitin chains (K63). (C)RIDDLE cells were transduced with lentiviral particles directing the expression of FLAG-tagged H2AX fusion proteins. The cells were processed for immunofluorescence 4 h after exposure to IR (9 Gy) using antibodies reacting with GFP, conjugated ubiquitin (FK2) or K63-linked polyubiquitin chains (K63).
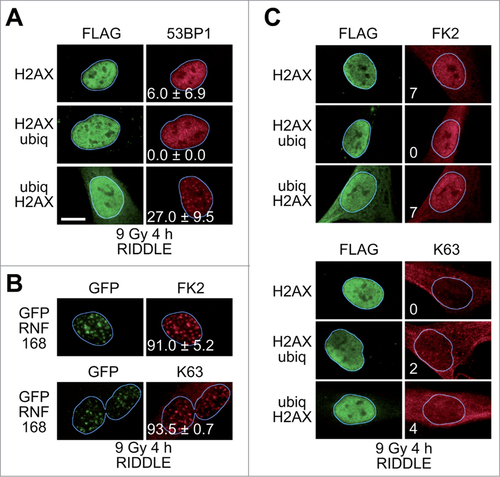
We also expressed GFP-H2AX in RIDDLE cells, but the level of expression was very low compared to its expression in RNF8-/- cells (Fig. S6B). As a result, we could not score significant number of cells expressing high levels of GFP-H2AX and were unable to ascertain whether GFP-H2AX rescues 53BP1 IRIF in RIDDLE cells. As a positive control for all the experiments mentioned above, expression of GFP-RNF168 in RIDDLE cells rescued 53BP1 IRIF (Fig. S6C).
We next examined if rescue of 53BP1 IRIF in RIDDLE cells expressing H2AX fusion proteins was accompanied by rescue of polyubiqutination at sites of DNA DSBs. As a positive control, expression of GFP-RNF168 rescued the formation of IR-induced foci reactive with the FK2 and K63-linked polyubiquitin chain antibodies ( and Fig. S6D). Expression of the H2AX fusion proteins did not appear to rescue polyubiquitination at sites of DNA DSBs ( and Fig. S6E). However, as with the RNF8-/- MEFs, the level of nuclear FK2 staining was quite high (), so again we cannot exclude the possibility of low levels of ubiquitination at sites of DNA DSBs in these cells.
Expression of H2AX fusion proteins rescues 53BP1 IRIF in cells treated with a proteasome inhibitor
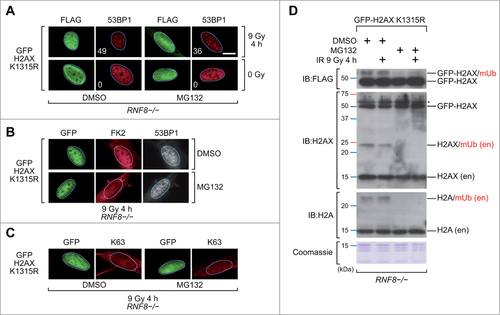
The experiments presented above raise the possibility that polyubiquitination at sites of DNA DSBs may not be required for 53BP1 recruitment. To further explore this premise, we treated RNF8-/- cells with MG132, a proteasome inhibitor.Citation43 Treatment of cells with MG132 exhausts the nuclear pool of free ubiquitin, resulting in failure of 53BP1 to be recruited to sites of DNA DSBs.Citation29 Interestingly, the GFP-H2AX K13R/K15R fusion protein could still rescue 53BP1 recruitment in RNF8-/- cells treated with MG132 (). Under these conditions ubiquitination at sites of DNA DSBs was not observed, although again the background levels of FK2 and K63 staining were not negligible (). Immunoblot analysis of chromatin fractions prepared from the MG132-treated RNF8-/- cells verified that MG132 diminished the nuclear pool of ubiquitin, since the monoubiquitinated species of endogenous H2AX and ectopically-expressed GFP-H2AX were depleted ().
Figure 3. Rescue of 53BP1 IRIF in MG132-treated RNF8-/- MEFs. (A)RNF8-/- MEFs transiently expressing the FLAG-tagged GFP-H2AX K13R/K15R protein were pretreated with DMSO or MG132 for 1 h, exposed to IR (9 Gy) or not-irradiated and 4 h later processed for immunofluorescence. More than one hundred cells with high level of FLAG signal were scored for 53BP1 IRIF. The percentages of cells with more than 10 53BP1 foci per cell are indicated. Scale bar = 10 μm. K1315R, K13R/K15R double substitution. (B)RNF8-/- MEFs transiently expressing the FLAG-tagged GFP-H2AX K13R/K15R (K1315R) protein were pretreated with DMSO or MG132 for 1 h, exposed to IR (9 Gy) and 4 h later processed for immunofluorescence using antibodies reacting with conjugated ubiquitin (FK2) and 53BP1. (C)RNF8-/- MEFs transiently expressing the FLAG-tagged GFP-H2AX K13R/K15R (K1315R) protein were pretreated with DMSO or MG132 for 1 h, exposed to IR (9 Gy) and 4 h later processed for immunofluorescence using antibodies reacting with K63-linked polyubiquitin chains (K63). (D)Immunoblots (IB) and Coomassie blue stained gel images of acidic histone extracts prepared from RNF8-/- MEFs transiently expressing the FLAG-tagged GFP-H2AX K13R/K15R (K1315R) protein. Cells were pretreated with DMSO or MG132 for 1 h and exposed to IR (9 Gy) or not-irradiated 4 h before preparing the extracts. H2AX (en), endogenous H2AX protein; H2A (en), endogenous H2A protein; mUb, monoubiquitinated. # indicates non-specific band.
To extend these observations, U2OS cells transfected with plasmids expressing H2AX fusion proteins were also treated with the proteasome inhibitor MG132 and then monitored for 53BP1 focus formation after exposure to IR. Expression of ubiq-H2AX or GFP-H2AX fusion proteins rescued 53BP1 IRIF in MG132-treated U2OS cells () and the rescued foci co-localized with γH2AX foci (Fig. S7) confirming that they correspond to sites of DNA breaks. In contrast, expression of H2AX-ubiq or FLAG-tagged H2AX did not rescue focus formation (). Similar results were obtained with HeLa cells (Fig. S8).
Figure 4. Rescue of 53BP1 IRIF in MG132-treated U2OS cells. (A)U2OS cells transiently expressing the indicated FLAG-tagged H2AX fusion proteins were pretreated with DMSO or MG132 for 1 h, exposed to IR (9 Gy) and 4 h later processed for immunofluorescence. More than one hundred cells with high levels of FLAG signal were scored for 53BP1 IRIF. More than one hundred cells with no FLAG signal (control cells) from the same slides were also scored. The percentages of cells with more than 10 53BP1 foci per cell (mean ± 1 SD) from 3 independent experiments are indicated. ctl, control cells; Scale bar = 20 μm. (B)U2OS cells transiently expressing FLAG-tagged GFP-H2AX were pretreated with MG132 for 1 h, exposed to IR (9 Gy) and 4 h later processed for immunofluorescence for GFP, endogenous 53BP1 and conjugated ubiquitin (FK2). (C)U2OS cells transiently co-expressing a FLAG-tagged ubiq-H2AX fusion protein and a GFP-tagged 53BP1 polypeptide with a wild-type (WT) or mutant (L1622E) RCTD/UDR motif were pretreated with DMSO or MG132 for 1 h, exposed to IR (5 Gy) and 2 h later processed for immunofluorescence with antibodies specific for the FLAG tag and GFP. Scale bar = 10 μm. (D)U2OS cells transiently expressing FLAG-tagged GFP-H2AX were pretreated with DMSO or MG132 for 1 h, exposed to IR (9 Gy) and 4 h later processed for immunofluorescence. More than one hundred cells with high levels of FLAG signal were scored for Rif1 or RAP80 IRIF. More than one hundred cells with no FLAG signal (control cells) from the same slides were also scored. The percentages of cells with more than 10 Rif1 foci per cell or more than 5 RAP80 foci per cell (mean ± 1 SD) from 3 ndependent experiments are indicated. ctl, control cells; Scale bar = 10 μm.
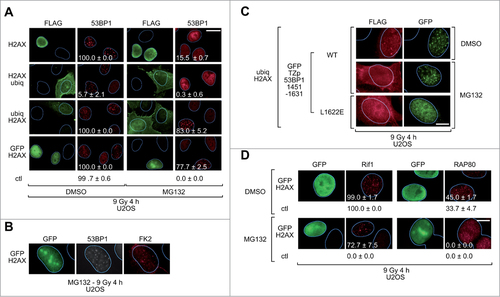
An interesting observation in the controls of this experiment was that ectopic expression of H2AX with ubiquitin fused at its C-terminus suppressed the recruitment of endogenous 53BP1 to DNA damage sites. This effect was quite strong in the U2OS cells (), but almost undetectable in the HeLa cells (Fig. S8). The underlying mechanism for this effect is beyond the scope of this study, but we note that H2A-ubiq fusion proteins have been previously shown to induce chromatin compaction and to suppress transcription.Citation44,45
Immunoblot analysis of chromatin fractions prepared from MG132-treated U2OS cells verified that MG132 depleted the monoubiquitinated species of endogenous H2AX and ectopically expressed GFP-H2AX (Fig. S9). However, the polyubiquitinated species of ectopically expressed ubiq-H2AX and H2AX-ubiq fusion proteins persisted (Fig. S9). This is consistent with accumulation of polyubiquitinated species in cells, in which the proteasome has been inhibited. The presence of polyubiquitinated species of H2AX-ubiq and ubiq-H2AX proteins, detected by immunoblotting, prompted us to monitor ubiquitination at sites of DNA DSBs by immunofluorescence. Surprisingly, in MG132-treated cells expressing ectopic ubiq-H2AX or GFP-H2AX proteins we could detect low levels of polyubiquitination at sites of DNA DSBs, whereas no such staining was evident in the adjacent cells not expressing H2AX fusion proteins ( and Fig. S10).
The observation that GFP-H2AX expression in MG132-treated U2OS partially rescued ubiquitin IRIF ( and Fig. S10) raises the possibility that the ability of the H2AX fusion proteins to rescue 53BP1 recruitment may be secondary to their ability to rescue chromatin ubiquitination. To explore this possibility we examined whether the RCTD/UDR motif is needed for 53BP1 recruitment to IRIF in MG132-treated U2OS cells. We expressed ubiq-H2AX together with a GFP-tagged fragment of 53BP1 that encompasses the native tudor domain and the RCTD/UDR motif, but which has a heterologous oligomerization domain (TZp) in place of the native oligomerization domain and, optionally, a L1622E substitution within the RCTD/UDR motif. The L1622E substitution abolishes recruitment of otherwise native 53BP1 to sites of DNA DSBs.Citation24 Interestingly, the L1622E mutant failed to form IRIF in MG132-treated cells expressing ubiq-H2AX, arguing that an intact RCTD/UDR motif is still required for 53BP1 focus formation under these conditions (). The simplest interpretation of these findings is that the rescue of 53BP1 recruitment in MG132-treated cells is secondary to the rescue of chromatin ubiquitination.
Recruitment of Rif1 and RAP80 proteins to sites of DNA DSBs
53BP1 mediates recruitment of Rif1 to sites of DNA DSBs via a mechanism that involves ATM-dependent phosphorylation of the N-terminus of 53BP1. Once recruited, Rif1 functions with 53BP1 to promote repair of DNA DSBs by non-homologous end joining.Citation46-50 As expected, treatment of U2OS cells with MG132 inhibited recruitment of Rif1 to sites of DNA DSBs and expression of GFP-H2AX, which rescues 53BP1 recruitment, also rescued Rif1 recruitment (). This indicates that 53BP1 recruited to sites of DNA DSBs by ectopically expressed H2AX fusion proteins is capable of being phosphorylated by ATM and performing at least one of its physiological functions, that of recruiting Rif1.
RAP80 is another protein that is recruited to sites of DNA DSBs. Recruitment of RAP80 to IRIF is 53BP1-independent, but dependent on hybrid ubiquitin-SUMO chains that are recognized by ubiquitin and SUMO-interacting motifs present within RAP80.Citation51,52 RAP80 facilitates the subsequent recruitment of BRCA1 and repair of DNA DSBs by homologous recombination.Citation53-56 Treatment of U2OS cells with MG132 inhibited the formation of RAP80 IRIF (). However, ectopic expression of GFP-H2AX did not rescue the defect in RAP80 recruitment to sites of DNA DSBs (), thus, distinguishing RAP80 recruitment from the recruitment of 53BP1 and Rif1.
Effect of expression of H2AX fusion proteins on sensitivity of chromatin to micrococcal nuclease digestion
One possibility to explain how the H2AX fusion proteins facilitate recruitment of 53BP1 to sites of DNA DSBs is to propose that they open up chromatin structure. To address this possibility we expressed ubiquitin- and GFP-H2AX fusion proteins in HEK293 cells and monitored chromatin accessibility by limited micrococcal nuclease digestion. We did not observe any consistent differences in micrococcal nuclease digestion patterns (Fig. S11), suggesting that if the H2AX fusion proteins alter chromatin structure, the changes are localized to the sites of DNA DSBs and are, therefore, not evident when bulk chromatin is analyzed.
Discussion
The mechanism by which 53BP1 localizes to sites of DNA DSBs has been the subject of intense study.Citation27,29,30,32-37,57,58 The importance of recognition of methylated histones H3 or H4 by the tudor domain of 53BP1 is well-established.Citation18,19,22,59 Further, there is universal consensus that RNF8 and RNF168 are required for formation of 53BP1 IRIF. However, how 53BP1 recruitment is facilitated by ubiquitination events at sites of DNA DSBs has been more enigmatic, because 53BP1 lacks a canonical ubiquitin-binding domain. Ubiquitination may open up chromatin structure or target for degradation proteins that compete with 53BP1 for binding to methylated histones.Citation27,29,35,36 Alternatively, as recently proposed, a small region of 53BP1, called the RCTD/UDR motif, may serve as a non-canonical ubiquitin-binding domain that recognizes ubiquitinated nucleosomes.Citation37 Indeed, the RCTD/UDR motif exhibits remarkable specificity for ubiquitinated nucleosomal core particles (NCPs) in vitro. NCPs ubiquitinated on K15 of histone H2A are recognized by 53BP1, but NCPs ubiquitinated on K13 are not.Citation37 Ubiquitination of both these lysines is mediated by RNF168.Citation38,39
In the course of performing this study we considered multiple mechanisms by which 53BP1 can be recruited to sites of DNA DSBs. Our main finding that 53BP1 recruitment in cells with impaired RNF8 and/or RNF168 activity can be rescued by expressing fusion histone H2AX proteins with bulky moieties at their N-terminus could be interpreted as indicating that the N-terminal bulky moieties open up chromatin structure. This would make the methylated lysines in the histone core (H3K79 or H4K20) more accessible for interaction with the tudor domain of 53BP1. Supporting this interpretation, fusing bulky moieties to histone proteins interferes with chromatin fiber folding.Citation60,61 Moreover, expression of H2AX with ubiquitin fused to its C-terminus, which has been associated with transcriptional repression and chromatin compaction,Citation44-45 suppressed recruitment of endogenous 53BP1 to sites of DNA DSBs (). However, when we tested the chromatin opening hypothesis, we did not observe significant changes in chromatin susceptibility to microccocal nuclease digestion in cells expressing the H2AX fusion proteins (Fig. S11). Of course, one caveat of this negative result is that changes in chromatin compaction induced by ectopic expression of H2AX fusion proteins may be limited to the chromatin surrounding DNA DSBs and not global enough to be detected by the micrococcal nuclease digestion assay.
Another mechanism by which the ectopically expressed H2AX fusion proteins might rescue 53BP1 recruitment could be by promoting ubiquitination at sites of DNA DSBs. Rescue of 53BP1 recruitment in RNF8-/- and RIDDLE cells was not associated with obvious ubiquitin IRIF; however, in some cells there was background ubiquitin signal present, which could potentially obscure any ubiquitin IRIF. In U2OS cells treated with a proteasome inhibitor, expression of GFP-H2AX rescued partially the FK2 and K63 IRIF ( and Fig. S10). Therefore, in these cells, rescue of 53BP1 recruitment is most likely secondary to rescue of FK2 and K63 IRIF. Still, only 53BP1 recruitment, but not recruitment of RAP80, a protein that contains canonical ubiquitin-interaction motifs, was rescued in these cells. Further, depletion of JMJD1C, a protein essential for the interaction of RNF8 with MDC1, abolishes formation of FK2 and RAP80 IRIF, but does not affect 53BP1 recruitment.Citation62
Interestingly, the rescue of 53BP1 recruitment in RNF8-/- cells by N-terminal H2AX fusion proteins required an intact phosphorylation site at the H2AX C-terminus, since substitution of S139 of H2AX with alanine did not permit 53BP1 recruitment (). S139 becomes phosphorylated at sites of DNA DSBs and this phosphorylation recruits MDC1.Citation63 In turn, MDC1 recruits, in addition to RNF8, the chromatin remodeler NuA4 to sites of DNA DSBs.Citation64 Hence, expression of the ectopic H2AX fusion proteins could enhance NuA4 accumulation, favoring chromatin opening and 53BP1 recruitment.
A second mechanism by which an intact S139 might favor 53BP1 recruitment might involve direct recognition of phosphorylated S139 by the BRCT domains of 53BP1. In both budding and fission yeast, recruitment of the orthologs of 53BP1 to sites of DNA DSBs involves binding of their BRCT domains to phosphorylated histone H2A.Citation65,66 In mammals, an equivalent interaction has not been demonstrated and the BRCT domains of 53BP1 are dispensable for recruitment to sites of DNA DSBs.Citation8 However, the BRCT domains of 53BP1 are highly conserved in evolutionCitation67 and we cannot exclude the possibility that they may facilitate 53BP1 recruitment to sites of DNA DSBs when histone H2AX fusion proteins are overexpressed.
In RIDDLE cells, the rescue of 53BP1 IRIF was less robust than in RNF8-/- cells (compare ; Fig. S1A and S6C). Expression of the ubiquitin-H2AX fusion protein led to partial rescue of 53BP1 foci in 27% of the cells, whereas expression of GFP-RNF168, the positive control, led to complete rescue of 53BP1 foci in 100% of the cells. Expression of GFP-H2AX did not rescue 53BP1 foci in RIDDLE cells; although one should note that GFP-H2AX was expressed at much lower levels in RIDDLE cells, as compared to RNF8-/- MEFs (Fig. S6B). One explanation for the more efficient rescue of 53BP1 IRIF in RNF8-/- versus RIDDLE cells is that the former cells retain a wild-type RNF168 gene. Thus, in RNF8-/- cells, the ectopically expressed H2AX fusion proteins might somehow promote recruitment of RNF168 to sites of DNA DSBs, resulting in ubiquitination of K15 of histone H2A/H2AX.
Taking into account all the arguments mentioned above, we conclude that our findings are consistent with the recent models proposing that ubiquitination is required for 53BP1 recruitment.Citation27,29,35,36 The requirement for the RCTD/UDR motif of 53BP1 for recruitment to IRIF in U2OS cells treated with a proteasome inhibitor is most consistent with the model proposing that 53BP1 interacts directly with histone H2A/H2AX ubiquitinated on K15.Citation37 However, our observations also leave open the possibility that, under certain conditions, for example in RIDDLE cells, recruitment of 53BP1 to sites of DNA DSBs may involve additional interactions, such as, for example, a putative interaction of its BRCT domains with C-terminally phosphorylated histone H2AX. Thus, further work will elucidate the subtleties of 53BP1 recruitment to DNA damage sites.
Materials and Methods
Cell lines and cell culture
RNF8-/- MEFs, RIDDLE cells and U2OS and HeLa cells lines were cultured in DMEM, supplemented with antibiotics and 10% FBS. To inhibit nuclear ubiquitination RNF8-/- MEFs and U2OS and HeLa cells were treated with 10, 5 and 5 μM MG132 (Sigma Aldrich, Buchs, CH, M7449), respectively, dissolved in DMSO.
Recombinant plasmids
Plasmids encoding H2AX fusion constructs were cloned into pCDZ vector with 2 N-terminal FLAG tags. Plasmids encoding RNF8, and RNF168 polypeptides fused to the C-terminus of GFP were generated from a previously-described mammalian expression plasmid.Citation18 For lentiviral transduction of RIDDLE cells the coding sequences were subcloned into the pRDI292CMV vector.
Transfections
HEK293T cells were transfected with plasmid DNA using XtremeGene HP according to the manufacturer's instructions (Roche Diagnostic, Basel, CH). RNF8-/- MEFs and U2OS cells were nucleofected with plasmid DNA using BTXpress solution according to manufacturer's instructions (Harvard Apparatus, Inc., Holliston, MA, US) with Amaxa nucleofection apparatus (Lonza Group Ltd, Basel, CH). RIDDLE cells were transduced with lentivirus produced by co-transfecting HEK293T cells with lentiviral plasmids together with the helper plasmids: pCMV-VSV-G, pRSV-Rev, pRRE. Protamine sulfate was used to enhance transduction efficiency at 8 μg/ml concentration (Sigma Aldrich, Buchs, CH; P3369). Cells were analyzed 48 h after plasmid transfection and 24 h after plasmid nucleofection or lentivirus transduction.
Immunofluorescence
Cells were exposed to IR (X-ray) in order to induce DNA DSBs (5–9 Gy) or mock treated (0 Gy). Next, cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton-X in PBS and blocked with 1% BSA in PBS. Cells were subsequently stained with primary antibodies for 1 h, washed twice with PBS, stained with secondary antibodies for 30 min, washed twice with PBS and counterstained with DAPI (Invitrogen, Carlsbad, CA, USA, D3571) at 1μg/ml concentration. All steps were performed at room temperature.
Primary antibodies used were mouse anti-53BP1 hybridoma supernatant at 1:20,Citation67 rabbit anti-53BP1 (Bethyl Laboratories, Inc., Montgomery, TX, US; A300-272A) at 1:1000, rabbit anti-Rif1 (Bethyl Laboratories, Inc., Montgomery, TX, US; A300-569A) at 1:500, rabbit anti-RAP80 (Bethyl Laboratories, Inc., Montgomery, TX, US; A300-763A) at 1:1000, mouse anti-GFP (Roche Diagnostic, Basel, CH; 11 814 460 001) at 1:500, rabbit anti-GFP (Abcam, Cambridge, UK; ab290) at 1:5000, mouse anti-FLAG M2 (Sigma Aldrich, Buchs, CH; F1804) at 1:1000, rabbit anti-FLAG (Sigma Aldrich, Buchs, CH; F7425) at 1:1000, mouse anti-γH2AX (Millipore, Billerica, MA, USA; clone JBW301, 05-636) at 1:1000, mouse anti-ubiquitin FK2 (Enzo Life Sciences, Lausen, CH; PW 8810) at 1:200, and rabbit anti-ubiquitin K63 (Millipore, Billerica, MA, USA; clone Apu3, 05-1308) at 1:2000 dilution. AlexaFluor secondary antibodies (A11034, A11029, A11032, A11037, and A21244) were used at 1:1000 dilutions (Invitrogen, Carlsbad, CA, USA).
Microscopy analysis
Images of fixed samples were acquired on a Zeiss AXIO Imager M1 fluorescent microscope with 100X Plan-A (1.4 NA) or 40X Plan-N (1.3 NA) oil immersion lenses (Carl Zeiss Microscopy, Jena, Germany), a Hamamatsu Orca ER digital camera (HAMAMATSU PHOTONICS K.K., Hamamatsu City, Japan) and Axio Vision Rel.. 4.8 software (Carl Zeiss Microscopy, Jena, Germany). Grayscale images were processed into colored images based on the pixel intensities in the grayscale image ranging from 0 (black) to 255 (white) using Imagevision software (Silicon Graphics Inc., Mountain View, CA).
Generation of DNA DSBs
DNA DSBs were induced using an X-Rad 320 irradiator (Precision X-ray, Inc., North Branford, CT, USA) operating at 320 kV and 12.5 mA.
Protein extracts and immunoblotting
To obtain whole cell extracts, cells were incubated at 4°C for 1 h in buffer containing 50 mM Tris pH 8.0, 120 mM NaCl, 0.5 % NP-40, 1 mM DTT, protease inhibitors and phosphatase inhibitors. After centrifugation the pellet was extracted to obtain acidic histone extract by incubation at 4°C for 1 h in buffer containing 8 mM HEPES, 1.2 mM MgCl2, 8 mM KCl, 0.5 mM DTT, 1.2 mM PMSF and 200 mM HCl. Protein concentrations were measured by Bradford protein assay (Bio-Rad, Hercules, CA, USA; 500-0205) and proteins were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore, Billerica, MA, USA, IPVH00010). Primary antibodies used were: mouse anti-FLAG M2 (Sigma Aldrich, Buchs, CH; F1804) at 1:1000, rabbit anti–H2AX (Abcam, Cambridge, UK; ab11175) at 1:5000, rabbit anti-H2A (Abcam, Cambridge, UK; ab18255) at 1:1000 and mouse anti-γH2AX (Millipore, Billerica, MA, USA; clone JBW301, 05-636) at 1:1000 dilution. For loading control, gels were stained with Coomassie brilliant blue (Bio-Rad, Hercules, CA, USA; 161-0400 or 161-0406).
Micrococcal nuclease assay
MNase sensitivity assay was carried out as described.Citation68 Briefly, after transfection, HEK293 cells were washed with cold PBS and lysed with nuclei extraction (NE) buffer (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA, 2 mM MgCl2, 2 mM CaCl2, 1 mM DTT, 0.2% (v/v) NP-40) on ice for 5 min. The resultant nuclei were washed with NE buffer twice, resuspended in NE buffer and digested at 25°C for the indicated time with 0.25 U/ml of MNase (Sigma Aldrich, Buchs, CH; N5386). The reaction was stopped by adding stop buffer (50 mM Tris-HCl pH 8.0, 25 mM EDTA, 1% (w/v) SDS). DNA was purified by incubating the nuclei with 0.6 mg/ml proteinase K for 1 h at 55°C, followed by phenol-chloroform extraction and ethanol precipitation. The DNA was resuspended in TE buffer, resolved by agarose gel electrophoresis and stained with GelRed dye (Biotium, Inc., Hayward, CA, USA). Agarose gels were scanned, and profiles representing band intensity of each line were obtained using ImageJ software (US National Institutes of Health).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
MKK performed the experiments and analyzed the data, MKK and TDH designed the experiments and wrote the manuscript, AJR performed preliminary experiments, GSS provided essential reagents and revised the manuscript, TDH supervised the research.
1010918_Supplementary_Materials.zip
Download Zip (1.4 MB)Acknowledgments
We thank Niels Mailand, Pier Giuseppe Pelicci and Manuel Stucki for sharing materials and reagents, Griet van Houwe for technical support and Nicolas Roggli for help with figures preparation.
Funding
This study was supported by a grant from the Swiss National Foundation to T.D.H.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
Related Research Data
References
- Featherstone C, Jackson SP. DNA double-strand break repair. Curr Biol 1999; 9:R759-61; PMID:10531043; http://dx.doi.org/10.1016/S0960-9822(00)80005-6
- Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003; 22:5792-812; PMID:12947387; http://dx.doi.org/10.1038/sj.onc.1206679
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3:155-68; PMID:12612651; http://dx.doi.org/10.1038/nrc1011
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432:316-23; PMID:15549093; http://dx.doi.org/10.1038/nature03097
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319:1352-5; PMID:18323444; http://dx.doi.org/10.1126/science.1140735
- DiTullio RA, Jr., Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol 2002; 4:998-1002; PMID:12447382; http://dx.doi.org/10.1038/ncb892
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, et al. 53BP1 is required for class switch recombination. J Cell Biol 2004; 165:459-64; PMID:15159415; http://dx.doi.org/10.1083/jcb.200403021
- Zgheib O, Huyen Y, DiTullio RA, Jr., Snyder A, Venere M, Stavridi ES, Halazonetis TD. ATM signaling and 53BP1. Radiother Oncol 2005; 76:119-22; PMID:16024119; http://dx.doi.org/10.1016/j.radonc.2005.06.026
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature 2008; 456:529-33; PMID:18931658; http://dx.doi.org/10.1038/nature07476
- Fitzgerald JE, Grenon M, Lowndes NF. 53BP1: function and mechanisms of focal recruitment. Biochem Soc Trans 2009; 37:897-904; PMID:19614615; http://dx.doi.org/10.1042/BST0370897
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 2010; 17:688-95; PMID:20453858; http://dx.doi.org/10.1038/nsmb.1831
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-54; PMID:20362325; http://dx.doi.org/10.1016/j.cell.2010.03.012
- Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell 2012; 46:125-35; PMID:22445484; http://dx.doi.org/10.1016/j.molcel.2012.02.015
- Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 2012; PMID:22553214
- Cescutti R, Negrini S, Kohzaki M, Halazonetis TD. TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. EMBO J 2010; 29:3723-32; PMID:20871591; http://dx.doi.org/10.1038/emboj.2010.238
- Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, Cao Y, Hamada T, Imamura K, Jeggo PA, et al. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem 2003; 278:36487-95; PMID:12824158; http://dx.doi.org/10.1074/jbc.M304066200
- Ward IM, Minn K, Jorda KG, Chen J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem 2003; 278:19579-82; PMID:12697768; http://dx.doi.org/10.1074/jbc.C300117200
- Huyen Y, Zgheib O, Ditullio RA, Jr., Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004; 432:406-11; PMID:15525939; http://dx.doi.org/10.1038/nature03114
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006; 127:1361-73; PMID:17190600
- Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev 2008; 22:2048-61; PMID:18676810; http://dx.doi.org/10.1101/gad.476008
- Hartlerode AJ, Guan Y, Rajendran A, Ura K, Schotta G, Xie A, Shah JV, Scully R. Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks. PloS One 2012; 7:e49211; PMID:23209566
- Wakeman TP, Wang Q, Feng J, Wang XF. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J 2012; 31:2169-81; PMID:22373577; http://dx.doi.org/10.1038/emboj.2012.50
- Stucki M, Jackson SP. Tudor domains track down DNA breaks. Nat Cell Biol 2004; 6:1150-2; PMID:15573092; http://dx.doi.org/10.1038/ncb1204-1150
- Zgheib O, Pataky K, Brugger J, Halazonetis TD. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol Cell Biol 2009; 29:1050-8; PMID:19064641; http://dx.doi.org/10.1128/MCB.01011-08
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 2002; 4:993-7; PMID:12447390; http://dx.doi.org/10.1038/ncb884
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003; 421:961-6; PMID:12607005; http://dx.doi.org/10.1038/nature01446
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131:901-14; PMID:18001825; http://dx.doi.org/10.1016/j.cell.2007.09.041
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007; 318:1637-40; PMID:18006705; http://dx.doi.org/10.1126/science.1150034
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887-900; PMID:18001824; http://dx.doi.org/10.1016/j.cell.2007.09.040
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435-46; PMID:19203579; http://dx.doi.org/10.1016/j.cell.2008.12.041
- Pinato S, Scandiuzzi C, Arnaudo N, Citterio E, Gaudino G, Penengo L. RNF168, a new RING finger, MIU-containing protein that modifies chromatin by ubiquitination of histones H2A and H2AX. BMC Mol Biol 2009; 10:55; PMID:19500350; http://dx.doi.org/10.1186/1471-2199-10-55
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009; 136:420-34; PMID:19203578; http://dx.doi.org/10.1016/j.cell.2008.12.042
- Moudry P, Lukas C, Macurek L, Hanzlikova H, Hodny Z, Lukas J, Bartek J. Ubiquitin-activating enzyme UBA1 is required for cellular response to DNA damage. Cell Cycle 2012 11:1573-82; PMID:22456334; http://dx.doi.org/10.4161/cc.19978
- Helchowski CM, Skow LF, Roberts KH, Chute CL, Canman CE. A small ubiquitin binding domain inhibits ubiquitin-dependent protein recruitment to DNA repair foci. Cell Cycle 2013 12:3749-58; PMID:24107634; http://dx.doi.org/10.4161/cc.26640
- Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, Dantuma NP. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat Struct Mol Biol 2011; 18:1345-50; PMID:22120668; http://dx.doi.org/10.1038/nsmb.2188
- Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J 2012; 31:1865-78; PMID:22373579; http://dx.doi.org/10.1038/emboj.2012.47
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013; 499:50-4; PMID:23760478; http://dx.doi.org/10.1038/nature12318
- Gatti M, Pinato S, Maspero E, Soffientini P, Polo S, Penengo L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle 2012; 11:2538-44; PMID:22713238; http://dx.doi.org/10.4161/cc.20919
- Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012; 150:1182-95; PMID:22980979
- Sharma N, Zhu Q, Wani G, He J, Wang QE, Wani AA. USP3 counteracts RNF168 via deubiquitinating H2A and γH2AX at lysine 13 and 15. Cell Cycle 2014; 13:106-14; PMID:24196443; http://dx.doi.org/10.4161/cc.26814
- Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol 2009; 10:659-71; PMID:19773779; http://dx.doi.org/10.1038/nrm2767
- Stewart GS, Stankovic T, Byrd PJ, Wechsler T, Miller ES, Huissoon A, Drayson MT, West SC, Elledge SJ, Taylor AM. RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc Natl Acad Sci U S A 2007; 104:16910-5; PMID:17940005; http://dx.doi.org/10.1073/pnas.0708408104
- Dantuma NP, Groothuis TA, Salomons FA, Neefjes J. A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol 2006; 173:19-26; PMID:16606690; http://dx.doi.org/10.1083/jcb.200510071
- Stock JK, Giadrossi S, Casanova M, Brookes E, Vidal M, Koseki H, Brockdorff N, Fisher AG, Pombo A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol 2007; 9:1428-35; PMID:18037880; http://dx.doi.org/10.1038/ncb1663
- Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011; 477:179-84; PMID:21901007; http://dx.doi.org/10.1038/nature10371
- Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 2013; 153:1266-80; PMID:23727112; http://dx.doi.org/10.1016/j.cell.2013.05.023
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 2013; 49:858-71; PMID:23333305; http://dx.doi.org/10.1016/j.molcel.2013.01.002
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 2013; 339:711-5; PMID:23306439; http://dx.doi.org/10.1126/science.1230624
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 2013; 49:872-83; PMID:23333306; http://dx.doi.org/10.1016/j.molcel.2013.01.001
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science 2013; 339:700-4; PMID:23306437; http://dx.doi.org/10.1126/science.1231573
- Guzzo CM, Berndsen CE, Zhu J, Gupta V, Datta A, Greenberg RA, Wolberger C, Matunis MJ. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci Signal 2012; 5:ra88; PMID:23211528
- Hu X, Paul A, Wang B. Rap80 protein recruitment to DNA double-strand breaks requires binding to both small ubiquitin-like modifier (SUMO) and ubiquitin conjugates. J Biol Chem 2012; 287:25510-9; PMID:22689573; http://dx.doi.org/10.1074/jbc.M112.374116
- Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007; 316:1202-5; PMID:17525342; http://dx.doi.org/10.1126/science.1139621
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007; 316:1198-202; PMID:17525341; http://dx.doi.org/10.1126/science.1139516
- Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci U S A 2007; 104:20759-63; PMID:18077395; http://dx.doi.org/10.1073/pnas.0710061104
- Yan J, Kim YS, Yang XP, Li LP, Liao G, Xia F, Jetten AM. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res 2007; 67:6647-56; PMID:17621610; http://dx.doi.org/10.1158/0008-5472.CAN-07-0924
- Hsiao KY, Mizzen CA. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J Mol Cell Biol 2013; 5:157-65; PMID:23329852; http://dx.doi.org/10.1093/jmcb/mjs066
- Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 2013; 20:317-25; PMID:23377543; http://dx.doi.org/10.1038/nsmb.2499
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15:7-18; PMID:24326623; http://dx.doi.org/10.1038/nrm3719
- Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol 2011; 7:113-9; PMID:21196936; http://dx.doi.org/10.1038/nchembio.501
- Dhall A, Wei S, Fierz B, Woodcock CL, Lee TH, Chatterjee C. Sumoylated human histone H4 prevents chromatin compaction by inhibiting long-range internucleosomal interactions. J Biol Chem 2014; PMID:25294883
- Watanabe S, Watanabe K, Akimov V, Bartkova J, Blagoev B, Lukas J, Bartek J. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat Struct Mol Biol 2013; 20:1425-33; PMID:24240613; http://dx.doi.org/10.1038/nsmb.2702
- Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair 2006; 5:534-43; PMID:16531125; http://dx.doi.org/10.1016/j.dnarep.2006.01.012
- Xu Y, Sun Y, Jiang X, Ayrapetov MK, Moskwa P, Yang S, Weinstock DM, Price BD. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J Cell Biol 2010; 191:31-43; PMID:20876283; http://dx.doi.org/10.1083/jcb.201001160
- Hammet A, Magill C, Heierhorst J, Jackson SP. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep 2007; 8:851-7; PMID:17721446; http://dx.doi.org/10.1038/sj.embor.7401036
- Sofueva S, Du LL, Limbo O, Williams JS, Russell P. BRCT domain interactions with phospho-histone H2A target Crb2 to chromatin at double-strand breaks and maintain the DNA damage checkpoint. Mol Cell Biol 2010; 30:4732-43; PMID:20679485; http://dx.doi.org/10.1128/MCB.00413-10
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 2000; 151:1381-90; PMID:11134068; http://dx.doi.org/10.1083/jcb.151.7.1381
- Wu J, Chen Y, Lu LY, Wu Y, Paulsen MT, Ljungman M, Ferguson DO, Yu X. Chfr and RNF8 synergistically regulate ATM activation. Nat Struct Mol Biol 2011; 18:761-8; PMID:21706008; http://dx.doi.org/10.1038/nsmb.2078