
Abstract
Endogenous retroviruses (ERVs) and LTR retrotransposons (LRs) occupy ∼8% of human genome. Deep sequencing technologies provide clues to understanding of functional relevance of individual ERVs/LRs by enabling direct identification of transcription factor binding sites (TFBS) and other landmarks of functional genomic elements. Here, we performed the genome-wide identification of human ERVs/LRs containing TFBS according to the ENCODE project. We created the first interactive ERV/LRs database that groups the individual inserts according to their familial nomenclature, number of mapped TFBS and divergence from their consensus sequence. Information on any particular element can be easily extracted by the user. We also created a genome browser tool, which enables quick mapping of any ERV/LR insert according to genomic coordinates, known human genes and TFBS. These tools can be used to easily explore functionally relevant individual ERV/LRs, and for studying their impact on the regulation of human genes. Overall, we identified ∼110,000 ERV/LR genomic elements having TFBS. We propose a hypothesis of “domestication” of ERV/LR TFBS by the genome milieu including subsequent stages of initial epigenetic repression, partial functional release, and further mutation-driven reshaping of TFBS in tight coevolution with the enclosing genomic loci.
Introduction
Human endogenous retroviruses (ERVs) and LTR retrotransposons (LRs) are remnants of retroviral infections from millions of years ago.Citation1,2 These elements fixed in the genome and became inheritable because their insertions occurred in the germ cell lineage.Citation3-6 Genomic ERVs and LRs are of particular interest because they harbor DNA regions functioning as promoters,Citation7-9 enhancers,Citation10,11 polyadenylation signalsCitation12,13 insulatorsCitation14,15 and binding sites for various nuclear proteins.Citation8,16-19 Many families of ERV/LRs exhibit high transcriptional activity in human tissues.Citation7,20-23
Over the last decade there has been an exponential accumulation of experimental data in molecular biology.Citation24-28 Investigation of the DNA variations itself cannot explain most of the aspects of functional genetics and phenotypes, and additional large-scale studies are needed to investigate the activities of the genomic regulatory regions.Citation29-30 DNaseI hypersensitivity sites (DHS) and transcription factor binding sites (TFBS) are probably the most important genomic landmarks for regions of open (functionally active) chromatin and those regions of DNA with nuclear protein binding properties, respectively.Citation31,32
In order to accomplish analysis on a genomic scale, we devised a bioinformatic algorithm that can map relevant TFBS identified by annotating all the inserts of ERV/LRs in the human DNA (504 families and ∼720,000 copies). We used the ENCODE project repository http://genome.ucsc.edu/ENCODE/ as the source database for human TFBS. The ENCODE data were the sets of deep-sequenced chromatin immunoprecipitated DNA. The results of our studies are freely available online through our Web-based service http://herv.pparser.net. Individual human genomic ERV/LRs and their whole families may be ranged by the user according to their structural nomenclature, representation in the genome, and according to the numbers and densities of the TFBS. We identified ∼110.000 individual ERV/LR inserts having TFBS, some of them were highly saturated by the TFBS having 50 TFBS or more. Our study provides new clues for identification and functional validation of tens of thousands of previously unknown regulatory sequences of the human genome.
Results
In this study, we performed a genome-wide mapping of all the available sequenced TFBS regions for all the human ERV/LR elements. In addition we created a Web-based interface for user-friendly interpretation of this information.
Our database allows individual human genomic ERV/LRs and their whole families to be ranged by the user according to their structural nomenclature, representation in the genome, and according to the numbers and densities of the TFBS. The database is supplemented by our novel human genome browser that may be used to visualize ERV/LR inserts and their genomic location specific to functional genes and TFBS. The database and genome browser enable downloading datasets in the form of raw FASTA DNA sequences or as a table including genomic coordinates of the individual insert, its divergence from the respective consensus sequence, the number of mapped TFBS, and the links to our ERV/LR genome browser http://herv.pparser.net/GenomeBrowser.php or UCSC genome browser https://genome.ucsc.edu/.
Genome-wide identification of the human ERV/LR elements
To extract genomic sequences of the human ERV/LR elements, we used the RepeatMasker algorithmCitation33 and the database of genomic repeat consensus sequences RepBase Update.Citation34 We identified in the human genome a total of 717,612 inserts of the different ERV/LR elements, which cover approximately 8% of the human DNA, in agreement with the previous reports for ERV/LRs.Citation35,36 These inserts represented 504 different ERV/LR families.
Genome-wide mapping of TFBS and DHS
We used the ENCODE project database http://genome.ucsc.edu/ENCODE/ to extract the full list of TFBS and DNase I hypersensitivity sites (DHS) of the human genome. Using our original PostParser algorithm, these entries were further mapped to human ERV/LR sequences. We found that for the entire set of ERV/LR elements, ∼140,000 inserts (∼19%) include at least one mapped DHS and ∼110,000 inserts (∼15%) have at least one mapped TFBS. The total numbers of all DHS and TFBS in all ERV/LR elements were ∼155,000 and ∼320,000, respectively. We next characterized specific ERV/LR families with regard to their TFBS content. We introduced the following characteristic values: (i) RT+, Ratio of the TFBS-containing elements among all the family members, which is calculated according to the formula NTFBS+ / Ntotal, where NTFBS+ is the number of the ERV/LR family members with TFBS, and Ntotal is the total number of the particular ERV/LR family members; (ii) NDT, Normalized density of TFBS, evaluated according to the formula ∑TFBS/ Ntotal, where ∑TFBS is the sum of TFBS in all the particular ERV/LR family members; (iii) TD+, TFBS density among the TFBS-positive members of the ERV/LR family, expressed by the formula ∑TFBS/ NTFBS+.
RT+ indicates the measure of abundances of functionally relevant (TFBS-positive) family members, NDT indicates the mean density of TFBS among the family members, and TD+ denotes the density of TFBS among the functionally relevant family members. These factors were used to rate all the 504 ERV/LR families, results available at http://herv.pparser.net/TotalStatistic.php an option is enabled to sort the families list according to each of the above characteristic indexes. Of note is that the individual ERV/LR families differ dramatically in their copy number, ranging from just few copies as for the HERV-F family, to more than 22,000 members as for the THE1B family. The total number of TFBS was also strikingly different for the different families – from 0 (families LTR5, LTR7A) to ∼13,000 (MLT1K family). The maximum absolute number of the TFBS-positive members was also seen for the MLT1K family (∼4,000).
RT+ values varied from 0 (families LTR5, LTR7A) to 0.72 (family LTR12), which indicates that the impact of the individual ERV/LR families in the genome regulatory interplay is markedly different. This finding was confirmed by the assessment of the NDT index, which also varied greatly from 0 (LTR5, LTR7A) till ∼8 (LTR13). Finally, TD+ indicator varied from 0 (LTR5, LTR7A) to ∼12 (LTR13), which means that for some families like LTR13, the TFBS+ members are highly involved in functional activities.
The families with the greatest densities of TFBS may be regarded as the most functionally active ones among the ERV/LR. However, it is also important to take into account the absolute numbers of TFBS contributed by each family. For example, the family LTR12 has the highest proportion of TFBS-positive members, which is reflected by an RT + value of 0.72, and contributed a total of ∼1,300 TFBS to the human genome, whereas the family MLT1K contributed the greatest number of TFBS (∼13,000), but has a rather small density of TFBS-positive members (RT+ = 0.22).
For every ERV/LR family, we also plotted the distribution of family members having different numbers of TFBS. Using these graphs allows zooming into the zones of interest and obtaining detailed information about the individual family members by clicking on the data points. Complete data is available at http://herv.pparser.net/FamilyInfo.php. This option enables quick and easy navigation of all ERV/LR family members having the desired number of TFBS. Each record is cross-linked with both our original and the UCSC genome browsers, which facilitates direct mapping of each ERV/LR on the human genome.
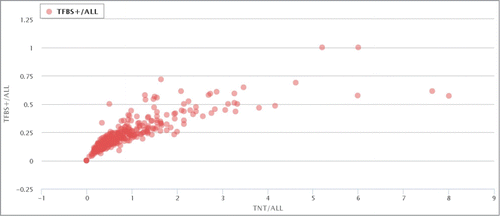
Further analysis shows a clear positive correlation between the ratio of TFBS-containing elements (RT+) and the normalized densities of TFBS (NDT) in the ERV/LR families (; http://herv.pparser.net/TNTandTFBS.php) This shows that although different ERV/LR families have very different proportions of the TFBS-positive members, there is a correlation between prevalence and density of TFBS that is common for both relatively TFBS-enriched and TFBS-poor families.
Figure 1. Correlation between NDT and RT+ for all ERV/LR families. Each data point represents a separate ERV/LR family. NDT is the normalized density of TFBS, and RT+ is the proportion of TFBS-containing elements in a family. “TFBS+/all” means RT+, and “TNT/all” means NDT.
Correlation between the TFBS and DHS data
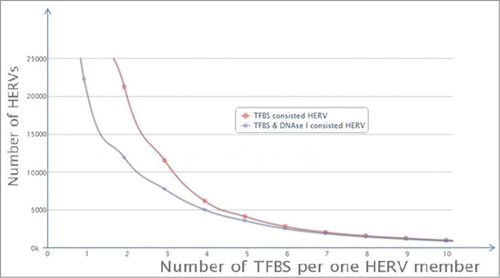
We next compared the TFBS mapping results with information on the DNase hypersensivity sites (DHS) within the ERV/LR elements. DHS are landmarks for the active fraction of eukaryotic genomes.Citation37-39 We identified a definite trend. The probability that an individual ERV/LR element has DHS increases proportionate to the increase in mapped TFBS (). More than 80% of the elements containing 4 TFBS also contained DHS. This proportion was increased as the number of TFBS per element increased, with >90% of the elements having 5 TFBS also containing DHS, etc. (). The correlation is validated by previous findings that DHS are most often located in promoter regions, which ensure the gene is accessible for transcription factor binding. Our findings reveal a practical application for quantification of the number of TFBS as a measure of functional activity of ERV/LR elements.
Figure 2. Distribution of the individual TFBS and DHS-containing ERV/LR elements is dependent on the number of TFBS per single element. “TFBS consisted HERV” means individual ERV/LR elements containing TFBS, shown in red; “TFBS & DNase I consisted HERV” means individual ERV/LR elements containing both TFBS and DNase I hypersensitivity site(s), shown in blue.
Experimental analysis of the enhancer activity for the elements having different density of TFBS
We next attempted to investigate if the enrichment in TFBS number correlates with increased functional activity of ERV/LR elements. Since ERV/LR elements may have diverse functional activities like promoters, enhancers, silencers and insul-atorsCitation14,40-43 a number of various functional essays may be used to study their activity. In this study, we used a specific family of ERV/LR, the LTR5Hs family, which represents the LTRs of the HERV-K(HML-2) family of human endogenous retroviruses. The LTR5Hs elements are the most recently inserted ERV/LR elements of the human genome,Citation44 ∼150 individual LTR5Hs family members inserted into the human DNA after the human-chimpanzee ancestor radiation and represent human specific elements,Citation2,45 many of which are polymorphic in the human population.Citation46 The ERV family is largely uniform in length (∼1 kb per element) and has relatively low heterogeneity in nucleotide sequence.Citation2,47 The LTR5Hs family is also reported to be one of the most functionally active groups of human ERV/LR elements.Citation48-50 Most of the literature deals with the enhancer activity of the LTR5Hs members.Citation10,11,18,51 In this study, we investigated the enhancer activities of the individual family members with different densities of TFBS, using the luciferase reporter assay.
Basing on the distribution of TFBS, we selected 12 elements, each having 5–23 TFBS per element (). For the assay, we took 5 human cell lines of different tissue origins: Tera-1 (embryonal non-differentiated testicular carcinoma), NT2/D1 (testicular carcinoma partly differentiated into neural cells), A549 (lung adenocarcinoma), NGP127 (neuroblastoma) and HepG2 (hepatocarcinoma). Notably, in the previous reports the Tera-1 cell line was shown to exhibit the strongest enhancer activity for most of the LTR5Hs elements tested. The individual genomic sequence for each LTR5Hs element was cloned into a luciferase-reporter construct for each of the 12 elements under investigation (, ) and used in a luciferase assay (). For better accuracy, we employed a Dual-Luciferase Reporter Assay (Promega). The LTR5Hs inserts were cloned upstream of the standard SV40 promoter for the luciferase gene, and their enhancer activities were compared with the control vector lacking any inserts upstream of the SV40 promoter (). Following transfections, normalized luciferase activity was measured and the LTR5Hs-containing/control ratios were calculated for each type of insert. With a fold2- cut-off factor, we detected statistically-significant enhancer activities for 10 individual elements () (except Elements 9 and 12). The extent of this ratio varied from ∼2 till 27.2 fold for the different elements and the different cell lines. For the Tera-1, NT2/D1, A549, NGP127 and HepG2 cell lines there were 7, 2, 7, 1 and 3 enhancer-active elements, respectively. These results are in agreement with previous findings that the enhancer activity of LTR5Hs is largely tissue-specific.Citation10,52,53 The 2 elements for which the enhancer activity was not detected in this assay may be active in other cell types.
Table 1. List of the LTR5Hs elements selected for the experimental luciferase assay
Figure 3. Profiling of LTR enhancer activity in luciferase reporter experiments. (A) Schematic representation of the luciferase reporter constructs. Filled arrow – individual LTR elements tested in this assay; empty arrow – SV40 promoter; black bar – luciferase gene; (B) relative enhancer activities for LTR elements 1–12, established in a dual-luciferase assay. Data show means ± standard deviations of 4 independent experiments. Data is shown for the cell lines Tera-1, NT2/D1, A549, NGP127 and HepG2.
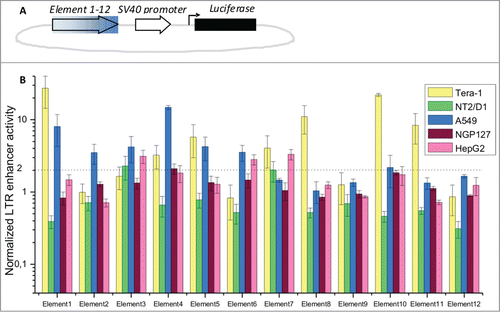
Interestingly, the LTR5Hs elements that did not show any enhancer activity (elements 9 and 12) have relatively low abundance of TFBS compared to the other elements tested (). Taken together, our data suggest that the density of TFBS may be an overall measure of the functional activity of ERV/LR elements. Our web-based database, therefore, may be used to efficiently capture the new functional regulatory elements of the human genome created by the ERV/LR elements.
Correlation of TFBS density relatively to the divergence of the ERV/LR families
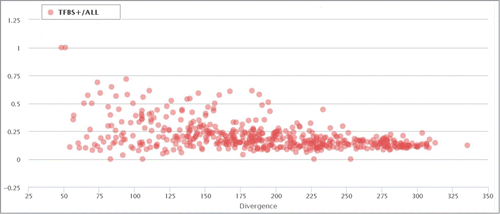
We next analyzed the distribution of the TFBS-containing elements in all ERV/LR families relative to the average divergence of the particular family members from their consensus sequence (divergence from a consensus sequence positively reflects evolutionary age of the inserted ERV/LR element).Citation54,55
We found a significant difference between the proportions of TFBS-positive family members in the evolutionally “young” families, e.g. those with an average divergence from their consensus sequence less than 10% (; each point represents an individual ERV/LR family). However, the dispersion of the TFBS+ family members drops as the divergence increases (>15%) and further tends to lay within an interval of 0,12–0,18. This clearly demonstrates that the low-divergence (evolutionally recent) ERV/LR families are characterized by a significantly lower likelihood of functional activity in human DNA compared to the evolutionally “older” groups. This may suggest that genomic “domestication” of the newly integrated ERV/LR sequences involves reshaping of their active TFBS profiles and their final “standardization” upon accumulation of the significant proportion of mutations.
Figure 4. Proportion of TFBS-containing elements in correlation with the divergence of each ERV/LR family from their consensus sequences. Each data point represents a separate ERV/LR family. “TFBS+/all” means RT+. The divergence is shown as a millidiv score, with each unit equal to one substitution per 1000 nucleotides.
Another aspect of this concept was uncovered when we compared the distributions of TFBS for 2 developmentally important transcription factors, NF-YA and Rad21. We found very different TFBS accumulation profiles for the different transcription factors (). For example, for the protein NF-YA there were highly abundant TFBS in the evolutionarily young, low-diverged ERV/LR elements (divergence 5–8% from the consensus sequence). Whereas for the evolutionarily older elements, the TFBS ratio was significantly lower. In contrast, for the protein Rad21 there was a relatively low ratio of TFBS for the evolutionarily “young” elements followed by a subsequent increase for the evolutionarily older elements, reaching a maximum value at ∼22% divergence. This example shows, that for the NF-YA protein, the recently inserted ERV/LR elements are enriched in TFBS, whereas further genomic domestication and mutation pressure significantly decreased the TFBS proportion for the evolutionarily older elements. In contrast, for the Rad21 protein, the evolutionarily older families showed increased ratio of TFBS-positive elements.
Figure 5. Distribution of TFBS for the particular transcription factor proteins among all the ERV/LR elements, in correlation with the divergence of the respective ERV/LR elements from their consensus sequence. The distribution is shown for NF-YA (red) and Rad21 (blue) transcription factor proteins. The divergence is shown as a millidiv score, with each unit equal to one substitution per 1000 nucleotides. The Y-axis is arbitrary and is customized for each transcription factor.
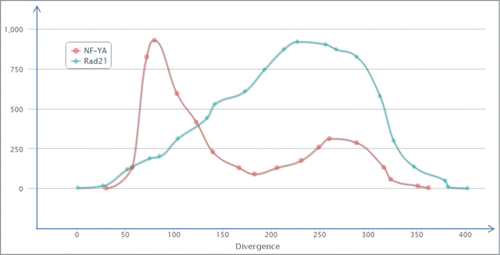
Interestingly, many transcriptional factors for which the TFBS distribution was profiled in ERV/LRs, had one common feature in their TFBS evolutionary dynamics: a decrease in the proportion of TFBS in the divergence interval around 8–15%. This indicates that functional adaptation and modification of an ERV/LR insert includes strong initial silencing of the TFBS which originally came from this element and further accumulation of the new functional TFBS in tight co-evolution with the host genome. The full set of TFBS distribution profiles is accessible online at http://herv.pparser.net.
Support for our hypothesis comes from studies showing that the newly integrated inserts are initially under strong DNA methylation repression.Citation56,57 This preserves the human genome from the deleterious influence of these elements on the gene regulatory networks. Upon de- methylation, a number of the ERV/LR copies release their regulatory potential and provide functional TFBS to the human genome. However, de-methylation is followed by mutation of the ERV/LR sequence, which is reflected by a further decrease in the TFBS density. We propose that this stage corresponds to a sharp reduction in TFBS coming directly from the ERV/LR inserts which were active in the initial intact elements. However, mutations do not only cause removal of these sites, but they also create new TFBS in the ERV/LR inserts that fall under selection pressure according to their implication in the overall genomic context. Finally, the highly diverged ERV/LR elements become equilibrated with the genomic location and show little difference compared to the average genomic sequence. This theoretical model is supported by our findings from this study, but needs further experimental and bioinformatic validation.
The Web-based interface and genome browser for the presentation of ERV/LR data
For easy navigation through the results of ERV/LR mapping on the human genome and quantification of the related functional features, we created a Web-based interface termed “PostParser HERV Browser," available through the link http://herv.pparser.net. It has the options for performing detailed analysis of the individual ERV/LR families, mapping its individual members on the human genome, or ranging of the individual family members depending on the TFBS density and divergence from the consensus sequence. This information can be obtained and downloaded for whole ERV/LR families and for the individual members as well. It is also possible to analyze TFBS distributions for transcription factors of interest in several ways, including TFBS density with relation to ERV/LR divergence from consensus sequence. Using the PostParser HERV Browser (available at http://herv.pparser.net/GenomeBrowser.php), it is possible to select a family of interest, to identify the individual elements having TFBS and to map them onto the human genome, relative to known human genes and other functional elements. It is also possible to directly extract the DNA sequence(s) for the element(s) of interest, or switch to the navigation mode through the UCSC Human Genome Browser (). Our HERV browser supports multiple regimes of scaling, from full-chromosome to 250-bp screen view. Enabling the “refGene” option shows mapped human genes. Furthermore, clicking on every individual ERV/LR element produces an output in table format, providing detailed information on this individual element including chromosome coordinates, divergence from the consensus sequence, number of TFBS, and links to the element views in our HERV browser and in the UCSC Genome Browser.
Figure 6. Screen shot of the representative HERV/LR browser output page. The user settings were “LTR5Hs” as the repeat family, “chr1” as the chromosome number. The Browser displays all the LTR5Hs inserts on the 1st chromosome, featuring TFBS – positive elements. An option is shown “show list of browser HERVs” that enables listing all the elements of a selected category on the browser screen, as a table supplemented by hyperlinks to the structure of particular each element. The zooming tool is enabled to facilitate navigation.

Discussion
In this study, we used a number of bioinformatic approaches including mapping of ∼720,000 individual ERV/LR elements representing 504 families on the human genome. Using published ENCODE project data, we identified the TFBS that can be unambiguously mapped on the above ERV/LR elements. For the family LTR5Hs, we showed that the densities of TFBS roughly correlate with the enhancer activities of the respective elements. Among the 12 tested elements, each with 5–23 mapped TFBS, 10 showed considerable enhancer activity. None of these individual elements has been previously reported as an active enhancer. These results show that our database may be used as a reliable source to identify new potential regulatory elements of the human genome. In parallel with our data, analogous databases created for the SINE, LINE and other repetitive elements of the human DNA will make it possible to create a comprehensive map of the human functional regulatory elements created by the genomic repeats, which will cover >50% of human genome http://herv.pparser.net/GenomeBrowser.php.
The approach of mapping TFBS on the ERV/LRs has limitations. Due to the repetitive nature of these elements, it is impossible in many cases to directly map TFBS to any particular element, especially to unrevealed sequences. Those TFBS that were successfully mapped, were mapped mostly for the ERV/LR 5’- or 3’-terminal regions, relatively close to the border with the unique flanking genomic sequence. Our results, therefore, are likely an underestimation of the TFBS pool that ideally could be attributed to the particular ERV/LR elements. The TFBS that can be mapped using the available ChIP-seq NGS data, like those released by the ENCODE project team,Citation58 are typically located no further than ∼200 bp from the border of a ERV/LR element. This feature minimizes the impact of a particular element's length on a TFBS count. It is also the reason why we did not normalize the number of TFBS found for a particular element, to its length, especially considering almost all ERV/LR elements are longer than 400 bp. Further studies are required to fully validate our data, using economically-viable sequencing platforms that generate longer sequence reads.Citation59
In this study, we performed the first targeted genome-wide identification of all human ERV/LRs that contain TFBS revealed by the ENCODE project. We demonstrate that the active TFBS pattern in ERV/LR elements greatly depends on the divergence of these elements from their consensus sequence, i.e. on the evolutionary age of these genomic inserts. Also, we provide evidence that the modification of the newly integrated TFBS containing ERV/LR inserts includes a stage of a sharp decrease of initial TFBS activity. We provide a novel interactive ERVs/LRs database that groups the individual inserts according to their familial nomenclature, number of mapped TFBS and divergence from their consensus sequence. Information on any particular ERV/LR element can be easily extracted by the user. To facilitate navigation through the data, we also created a genome browser tool enabling quick mapping of any ERV/LR inserts according to genomic coordinates, known human genes and TFBS. This browser is cross-linked with the UCSC Genome Browser to enable mapping of other genetic features on the ERV/LR element(s) of interest. The database and our genome browser may be widely used by researchers for quickly locating functionally relevant individual ERVs/LRs, and for studying their impact on the regulation of human genes. Finally, we propose a hypothesis of modification of the ERV/LR – derived TFBS, which includes the stages of initial epigenetic repression of functional TFBS, their further functional release, and subsequent mutation-driven termination of their activity. This is followed by the appearance of tightly regulated new TFBS, which results in a “genome-equilibrated” TFBS profile for the highly-diverged ERV/LR elements. Which in turn results in a tightly controlled regulation of gene expression.
Methods
Source databases
As the source database for downloading DNA consensus sequences of ERV/LR elements, we took RepBase Update databaseCitation60 created by the US Genetic Information Research Institute and available online at http://www.girinst.org/repbase/. For TFBS and DNaseI hypersensitivity sites, we used the ENCODE (the ENCyclopediaOf DNA Elements) project database (TFBS andDNAse I Clusters),Citation58 available at http://encodeproject.org/ENCODE/downloads.html. Approximately 3.700.000 signatures were further analyzed. To extract sequences of known human genes and transcripts including their exon-intron structure signatures, we used the GenBank at NCBI Reference Sequence Genes database available at http://www.ncbi.nlm.nih.gov/RefSeq/ and the Expressed Sequence Tags database available at http://www.ncbi.nlm.nih.gov/dbEST/index.html.
Computational facilities and software use and development
We used 4 processors (total performance 0,05 teraflops) and 10 Gb random access memory. The operation system used was Linux Ubuntu 10.04 64-bit (Apache+PHP5+MySQL+phpmyadmin). For alignment of RNA or DNA sequences, we used downloadable versions of Megablast, Dmegablast and Blastn software, available through http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=Download. For mapping of the TFBS and DHS – related sequence reads on the human genome, we used the algorithm BLAT (The BLAST-Like Alignment Tool) available through the UCSC server at http://genome.ucsc.edu/FAQ/FAQblat.html#blat3. For identification of the ERV/LR elements in thew human DNA, the RepeatMasker software was used, available at http://www.repeatmasker.org/. For creating the ERV/LR genome browser, we used our original PostParser software,Citation50 http://www.postparser.net. The codes were written using C++, PHP, bash supplemented with JavaScript, AJAX on the platform Apache+MySQL.
Cell lines
The cell lines Tera-1 (testicular embryonal germ cell tumor), NT2/D1 (partly-differentiated testicular germ cells with CNS-precursor cell characteristics), NGP127 (neuroblastoma), HepG2 (hepatocarcinoma) and A549 (lung carcinoma) were used in this study. Cells were grown on DMEM/F12 medium containing 10% fetal calf serum (Invitrogen) at 37°C and 5% CO2.
DNA extraction, PCR amplification and cloning
The overall design of this study was approved by the local ethical committee and follows the EU ethical guidelines. Genomic DNA was isolated using the Wizard Genomic DNA Purification Kit (Promega, USA) using a blood sample taken from a healthy adult male donor with his written consent. PCR amplification was conducted with the Encyclo PCR Kit (Evrogen, Russia). Sequences (Element 1–12) were PCR-amplified using 40 ng of human genomic DNA. Primer sequences are listedon . PCR conditions for amplifications were as follows: 95°C – 1.5 min; 95°C – 30 s, 60°C – 30 s, 72°C – 1.5 min; 35 cycles. PCR products (lengths 935, 1230, 381, 1444, 1390, 1210, 1209, 1200, 1221, 1282, 1277 and 1235 bp for Elements 1–12, respectively) were cloned into pGL3 basic vector (Promega) upstream the reporter gene for Luciferase using KpnI and MluI restriction sites.
Table 2. Sequences of oligonucleotides used in this study
Cell transfections and luciferase assays
Transfections were carried out in 24-well plates using Unifectin-56 transfection reagent according to the manufacturer recommendations (Rusbiolink). Dual luciferase system (Promega) was used for the luciferase activity screens. For each transfection, 0.5 mcg of 10:1 mixture of the analytical plasmid (including firefly luciferase under control of the regulatory sequence of interest) and normalization plasmid was used. We used pRL-TK normalization vector (Promega) including another reporter gene - Renillareniformis luciferase, under control of a herpes simplex virus thymidine kinase promoter to provide uniform levels of Renilla luciferase expression in co-transfected cells. Prior transfections with analytical plasmids, we have tested thymidine kinase promoter - driven Renilla luciferase expression in normalization vector on 5 human cell cultures. Renilla luciferase activity was measured in the cell cultures Tera-1, NT2/D1, NGP127, HepG2 and A549 transfected with pRL-TK normalization vector. Analytical plasmids based on pGL3 vector (Promega) had the following regulatory sequences: cloned Element 1–12 or SV40 early promoter. Twenty-four-Hours after transfection, cells were lysed and the activities of Renilla and firefly luciferases were measured using Dual-Luciferase Reporter Assay System (Promega) using luminometer «GENios Pro» (Tecan). Plasmid pRL-TK was used in all experiments as the internal control to minimize errors caused by the differences in transfection efficiencies in independant replicates. The obtained values for the fire-fly luciferase were normalized to the values for the Renilla luciferase. Each transfection experiment was done at least in quadruplicate.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank "UMA Foundation" for their support in preparation of the manuscript. The authors thank Dr. S.B.Akopov (Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Moscow) for providing cell lines.
Funding
This work was supported by the Pathway Pharmaceuticals Research Initiative (Hong-Kong), by the Russian Foundation for Basic Research grant 12-04-33094 and the Program of the Presidium of the Russian Academy of Sciences “Dynamics and Conservation of Genomes.”
References
- Sverdlov ED. Retroviruses and primate evolution. Bioessays 2000; 2:161-71; http://dx.doi.org/10.1002/(SICI)1521-1878(200002)22:2%3c161::AID-BIES7%3e3.0.CO;2-X
- Buzdin A. Human-specific endogenous retroviruses. Scientific World J 2007; 7:1848-68; PMID:18060323; http://dx.doi.org/10.1100/tsw.2007.270
- Hohn O, Hanke K, Bannert N. HERV-K(HML-2) the best preserved family of HERVs:Endogenization, expression, and implications in health and disease. Front Oncol 2013; 3:246; PMID:24066280; http://dx.doi.org/10.3389/fonc.2013.00246
- Dewannieux M, Heidmann T. Endogenous retroviruses:acquisition, amplification and taming of genome invaders. Curr Opin Virol 2013; 6:646-56; http://dx.doi.org/10.1016/j.coviro.2013.08.005
- Buzdin A, Ustyugova S, Khodosevich K, Mamedov I, Lebedev Y, Hunsmann G, Sverdlov E. Human-specific subfamilies of HERV-K (HML-2) long terminal repeats:three master genes were active simultaneously during branching of hominoid lineages. Genomics 2003; 81(2):149-56; PMID:12620392; http://dx.doi.org/10.1016/S0888-7543(02)00027-7
- Wildschutte JH, Ram D, Subramanian R, Stevens VL, Coffin JM. The distribution of insertionally polymorphic endogenous retroviruses in breast cancer patients and cancer-free controls. Retrovirology 2014; 11:6; http://dx.doi.org/10.1186/s12977-014-0062-3
- Buzdin A, Kovalskaya-Alexandrova E, Gogvadze E, Sverdlov E. At least 50% of human-specific HERV-K (HML-2) long terminal repeats serve in vivo as active promoters for host nonrepetitive DNA transcription. J Virol 2006; 80(21):10752-62; PMID:17041225; http://dx.doi.org/10.1128/JVI.00871-06
- Li F, Nellåker C, Sabunciyan S, Yolken RH, Jones-Brando L, Johansson A-S, Owe-Larsson Br, Karlsson H. Transcriptional derepression of the ERVWE1 locus following influenza A virus infection. J Virol 2014; 88(8):4328-37; PMID:24478419; http://dx.doi.org/10.1128/JVI.03628-13
- Fuchs NV, Loewer S, Daley GQ, Izsvák Z, Löwer J, Löwer R. Human endogenous retrovirus K (HML-2) RNA and protein expression is a marker for human embryonic and induced pluripotent stem cells. Retrovirology 2013; 10; PMID:24156636; http://dx.doi.org/10.1186/1742-4690-10-115
- Suntsova M, Gogvadze EV, Salozhin S, Gaifullin N, Eroshkin F, Dmitriev SE, Martynova N, Kulikov K, Malakhova G, Tukhbatova G, et al. Human-specific endogenous retroviral insert serves as an enhancer for the schizophrenia-linked gene PRODH. Proc Natl Acad Sci U S A 2013; 110(48):19472-7; PMID:24218577; http://dx.doi.org/10.1073/pnas.1318172110
- Chuong EB, Rumi MA, Soares MJ, Baker JC. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat Genet 2013; 45(3):325-9; PMID:23396136; http://dx.doi.org/10.1038/ng.2553
- Kim HS. Genomic impact, chromosomal distribution and transcriptional regulation of HERV elements. Mol Cells 2012; 33(6):539-44; PMID:22562360; http://dx.doi.org/10.1007/s10059-012-0037-y
- Yu HL, Zhao ZK, Zhu F. The role of human endogenous retroviral long terminal repeat sequences in human cancer (Review). Int J Mol Med 2013; 32(4):755-62; PMID:23900638
- Schumann GG, Gogvadze EV, Osanai-Futahashi M, Kuroki A, Münk C, Fujiwara H, Ivics Z, Buzdin AA. Unique functions of repetitive transcriptomes. Int Rev Cell Mol Biol 2010; 285:115-88; PMID:21035099; http://dx.doi.org/10.1016/B978-0-12-381047-2.00003-7
- Ling J, Pi W, Yu X, Bengra C, Long Q, Jin H, Seyfang A, Tuan D. The ERV-9 LTR enhancer is not blocked by the HS5 insulator and synthesizes through the HS5 site non-coding, long RNAs that regulate LTR enhancer function. Nucleic Acids Res 2003; 31(15):4582-96; PMID:12888519; http://dx.doi.org/10.1093/nar/gkg646
- Gogvadze E, Buzdin A. Retroelements and their impact on genome evolution and functioning. Cell Mol Life Sci 2009; 66(23):3727-42; PMID:19649766; http://dx.doi.org/10.1007/s00018-009-0107-2
- Ohnuki M, Tanabe K, Sutou K, Teramoto I, Sawamur Y, Narita M, Nakamura M, Tokunaga Y, Nakamura M, Watanabe A, Yamanaka S, et al. Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc Natl Acad Sci U S A 2014; 111(34):12426-31; PMID:25097266; http://dx.doi.org/10.1073/pnas.1413299111
- Young JM, Whiddon JL, Yao Z, Kasinathan B, Snider L, Geng LN, Balog J, Tawil R, van der Maarel SM, Tapscott SJ. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS genetics 2013; 9:e1003947 http://dx.doi.org/10.1371/journal.pgen.1003947
- Arjan-Odedra S, Swanson CM, Sherer NM Wolinsky SM, Malim MH. Endogenous MOV10 inhibits the retrotransposition of endogenous retroelements but not the replication of exogenous retroviruses. Retrovirology 2012; 9:53; PMID:22727223
- Maliniemi P, Vincendeau M, Frank O, Hahtola S, Karenko L, Carlsson E, Mallet F, Seifarth W, Leib-Mösch C, Ranki A. Expression of human endogenous retrovirus-w including syncytin-1 in cutaneous T-cell lymphoma. PLoS One 2013; 8(10):e76281; PMID:24098463; http://dx.doi.org/10.1371/journal.pone.0076281
- Zhang Y, Babaian A, Gagnier L, Mager DL. Visualized computational predictions of transcriptional effects by intronic endogenous retroviruses. PLoS One 2013; 8(8):e71971; PMID:23936536; http://dx.doi.org/10.1371/journal.pone.0071971
- Gosenca D, Gabriel U, Steidler A, Mayer J, Diem O, Erben P, Fabarius A, Leib-Mösch C, Hofmann WK, Seifarth W. HERV-E-mediated modulation of PLA2G4A transcription in urothelial carcinoma. PLoS One 2012; 7(11):e49341; PMID:23145155; http://dx.doi.org/10.1371/journal.pone.0049341
- Triviai I, Ziegler M, Bergholz U, Oler AJ, Stübig T, Prassolov V, Fehse B, Kozak CA, Kröger N, Stocking C. Endogenous retrovirus induces leukemia in a xenograft mouse model for primary myelofibrosis. Proc Natl Acad Sci U S A 2014; 111(23):8595-8600; PMID:24912157; http://dx.doi.org/10.1073/pnas.1401215111
- Chen J, Qian F, Yan W, Shen B. Translational biomedical informatics in the cloud:present and future. Biomed Res Int 2013; 2013
- Zhou X, Ren L, Meng Q, Li Y, Yu Y, Yu J. The next-generation sequencing technology and application. Protein Cell 2010; 1(6):520-36; PMID:21204006; http://dx.doi.org/10.1007/s13238-010-0065-3
- Merrifield M, Kovalchuk O. Epigenetics in radiation biology: a new research frontier. Frontiers in genetics 2013; 4:40. PMID:23577019; http://dx.doi.org/10.3389/fgene.2013.00040
- Ilnytskyy Y, Kovalchuk O. Non-targeted radiation effects-an epigenetic connection. Mutat Res 2011; 714(1-2):113-25; PMID:21784089; http://dx.doi.org/10.1016/j.mrfmmm.2011.06.014
- Shao W, Boltz VF, Spindler JE, Kearney MF, Maldarelli F, Mellors JW, Stewart C, Volfovsky N, Levitsky A, Stephens RM, Coffin JM. Analysis of 454 sequencing error rate, error sources, and artifact recombination for detection of Low-frequency drug resistance mutations in HIV-1 DNA. Retrovirology 2013; 10; PMID:23402264; http://dx.doi.org/10.1186/1742-4690-10-18
- Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D, Starmer J, Mieczkowski P, Crawford GE, Magnuson T. Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 2012; 151(5):951-63; PMID:23178118; http://dx.doi.org/10.1016/j.cell.2012.10.037
- McBride DJ, Buckle A, van Heyningen V, Kleinjan DA. DNaseI hypersensitivity and ultraconservation reveal novel, interdependent long-range enhancers at the complex Pax6 cis-regulatory region. PLoS One 2011; 6(12):e28616; PMID:22220192; http://dx.doi.org/10.1371/journal.pone.0028616
- Jacques PE, Jeyakani J, Bourque G. The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet 2013; 9(5):e1003504; PMID:23675311; http://dx.doi.org/10.1371/journal.pgen.1003504
- Ho B, Baker PM, Singh S, Shih SJ, Vaughan AT. Localized DNA cleavage secondary to genotoxic exposure adjacent to an Alu inverted repeat. Genes Chrom Cancer 2012; 51(5):501-509; PMID:22334386; http://dx.doi.org/10.1002/gcc.21938
- Wheeler TJ, Clements J, Eddy SR, Hubley R, Jones TA, Jurka J, Smit AF, Finn RD. Dfam:a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res 2013; 41(Database issue):D70-82; PMID:23203985; http://dx.doi.org/10.1093/nar/gks1265
- Jurka J. Repbase update:a database and an electronic journal of repetitive elements. Trends Genet 2000; 16(9):418-20; PMID:10973072; http://dx.doi.org/10.1016/S0168-9525(00)02093-X
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature 2001; 409(6822):860-921; PMID:11237011; http://dx.doi.org/10.1038/35057062
- Seifarth W, Frank O, Zeilfelder U, Spiess B, Greenwood AD, Hehlmann R, Leib-Mösch C. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J Virol 2005:79(1):341-52; PMID:15596828; http://dx.doi.org/10.1128/JVI.79.1.341-352.2005
- Bischof JM, Gillen AE, Song L, Gosalia N, London D, Furey TS, Crawford GE, Harris A. A genome-wide analysis of open chromatin in human epididymis epithelial cells reveals candidate regulatory elements for genes coordinating epididymal function. Biol Reprod 2013; 89(4):104; PMID:24006278; http://dx.doi.org/10.1095/biolreprod.113.110403
- Sugathan A, Waxman DJ. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol 2013; 33(18):3594-610; PMID:23836885; http://dx.doi.org/10.1128/MCB.00280-13
- Schlesinger S, Meshorer E, Goff SP. Asynchronous transcriptional silencing of individual retroviral genomes in embryonic cells. Retrovirology 2014; 11:31; PMID:24742368; http://dx.doi.org/10.1186/1742-4690-11-31
- Kuzmin D, Gogvadze E, Kholodenko R, Grzela DP, Mityaev M, Vinogradova T, Kopantzev E, Malakhova G, Suntsova M, Sokov D, et al. Novel strong tissue specific promoter for gene expression in human germ cells. BMC biotechnology 2010; 10:58 PMID:20716342; http://dx.doi.org/10.1186/1472-6750-10-58
- Downey RF, Sullivan FJ, Wang-Johanning F, Ambs S, Giles FJ, Glynn SA. Human endogenous retrovirus K and cancer:Innocent bystander or tumorigenic accomplice? Int J Cancer 2014; 10:1002
- Volkman HE, Stetson DB. The enemy within: endogenous retroelements and autoimmune disease. Nature immunology 2014; 15:415-22; PMID:24747712; http://dx.doi.org/10.1038/ni.2872
- Kassiotis G. Endogenous retroviruses and the development of cancer. J Immunol 2014; 192(4):1343-9; PMID:24511094; http://dx.doi.org/10.4049/jimmunol.1302972
- Medstrand P, Mager DL. Human-specific integrations of the HERV-K endogenous retrovirus family. J Virol 1998; 72(12):9782-7; PMID:9811713
- Buzdin AA. Functional analysis of retroviral endogenous inserts in the human genome evolution. Bioorg Khim 2010; 36(1):38-46; PMID:20386577
- Mamedov I, Lebedev Y, Hunsmann G, Khusnutdinova E, Sverdlov E. A rare event of insertion polymorphism of a HERV-K LTR in the human genome. Genomics 2004; 84(3):596-9; PMID:15498467; http://dx.doi.org/10.1016/j.ygeno.2004.04.010
- Mamedov IZ, Lebedev YB, Sverdlov ED. Unusually long target site duplications flanking some of the long terminal repeats of human endogenous retrovirus K in the human genome. J Gen Virol 2004; 85(Pt 6):1485-8; PMID:15166432; http://dx.doi.org/10.1099/vir.0.19717-0
- Subramanian RP, Wildschutte JH, Russo C, Coffin JM. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 2011; 8:90; PMID:22067224; http://dx.doi.org/10.1186/1742-4690-8-90
- Chudak C, Beimforde N, George M, Zimmermann A, Lausch V, Hanke K, Bannert N. Identification of late assembly domains of the human endogenous retrovirus-K(HML-2). Retrovirology 2013; 10:140 PMID:24252269; http://dx.doi.org/10.1186/1742-4690-10-140
- Kurth R, Bannert N. Beneficial and detrimental effects of human endogenous retroviruses. Int J Cancer 2010; 126(2):306-14; PMID:19795446; http://dx.doi.org/10.1002/ijc.24902
- Chuong EB. Retroviruses facilitate the rapid evolution of the mammalian placenta. Bioessays 2013; 35(10):853-61; PMID:23873343
- Illarionova AE, Vinogradova TV. Only those genes of the KIAA1245 gene subfamily that contain HERV(K) LTRs in their introns are transcriptionally active. Virology 2007; 358(1):39-47; PMID:16997346; http://dx.doi.org/10.1016/j.virol.2006.06.027
- Ruda VM, Akopov SB, Trubetskoy DO, Manuylov NL, Vetchinova AS, Zavalova LL, Nikolaev LG, Sverdlov ED. Tissue specificity of enhancer and promoter activities of a HERV-K(HML-2) LTR. Virus Res 2004; 104(1):11-16; PMID:15177887; http://dx.doi.org/10.1016/j.virusres.2004.02.036
- Beaune J, Nony P, Chassignolle J, Loire R, Gros P, Delaye J. Aortic insufficiency caused by dystrophic aneurysm of the ascending aorta:study of development in 95 cases. Value of cutaneous biopsy in the etiologic diagnosis. Arch Mal Coeur Vaiss 1989; 82(8):1389-96; PMID:2508590
- Anisimova M, Liberles DA. The quest for natural selection in the age of comparative genomics. Heredity (Edinb) 2007; 99(6):567-79; PMID:17848974; http://dx.doi.org/10.1038/sj.hdy.6801052
- Kao TH, Liao HF, Wolf D, Tai KY, Chuang CY, Lee HS, Kuo HC, Hata K, Zhang X, Cheng X, Goff SP, et al. Ectopic DNMT3L triggers assembly of a repressive complex for retroviral silencing in somatic cells. J Virol 2014; 88(18):10680-95; PMID:24991018; http://dx.doi.org/10.1128/JVI.01176-14
- Turelli P, Castro-Diaz N, Marzetta F, Kapopoulou A, Raclot C, Duc J, Tieng V, Quenneville S, Trono D. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res 2014; 24(8):1260-70; PMID:24879559; http://dx.doi.org/10.1101/gr.172833.114
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489(7414):57-74; PMID:22955616; http://dx.doi.org/10.1038/nature11247
- Baskaev K, Garazha A, Gaifullin N, Suntsova MV, Zabolotneva AA, Buzdin AA. nMETR:technique for facile recovery of hypomethylation genomic tags. Gene 2012; 498(1):75-80; PMID:22353364; http://dx.doi.org/10.1016/j.gene.2012.01.097
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110(1-4):462-7; PMID:16093699; http://dx.doi.org/10.1159/000084979