
Eukaryotic cells are subject to tens of thousands of DNA lesions per day. In order to overcome the accumulation of DNA damage and maintain genomic integrity, cells are equipped with machinery that detects and repairs these lesions, termed the 'DNA damage response' (DDR). DNA double strand breaks (DSBs) are among the most cytotoxic lesions, and are detected by the MRN complex (MRE11-RAD50-NBS1), which senses DSBs and participates in activation of the central DDR kinase ATM. In turn, ATM phosphorylates numerous substrates, including the histone variant H2AX to generate γ-H2AX that is required for the recruitment of MDC1 and the E3 ubiquitin ligases RNF8 and RNF168. RNF8 together with RNF168 subsequently orchestrate the histone ubiquitylation changes that are recognized by 53BP1, through its 'Ubiquitylation-Dependent Recruitment (UDR)' motif. In parallel, 53BP1 recognizes mono and dimethylated H4K20 via its Tudor domain, and the combined action of these binding modalities controls the accumulation of 53BP1 at DSBs. As a consequence, recruitment of 53BP1 together with its binding partners Rif1 and PTIP blocks DNA-end resection to promote non-homologous end-joining (NHEJ) repair of DSBs.Citation1
While virtually all data on DSB processing come from studies on interphase cells, cells may occasionally encounter DNA breaks during mitosis. When this occurs, the processing of DSBs appears fundamentally different, and cells can apparently progress through mitosis without the repair of these lesions.Citation2 Previous studies have already shown that DDR components, including 53BP1, are post-translationally modified during mitotic entry by Cdk1/cyclin B and Polo-like kinase-1 (Plk1), as part of the inactivation of the G2/M cell cycle checkpoint.Citation3,4 Two recent studies, of which one published in Cell Cycle, now demonstrate how recruitment of 53BP1 to sites of DNA damage is mechanistically blocked during mitosis.Citation5,6 Specifically, phosphorylation of RNF8 on Thr-198 by Cdk1 prevents RNF8 from binding phosphorylated MDC1 and subsequently blocks its recruitment to DSBs. In addition, phosphorylation of 53BP1 in its UDR motif on Thr-1609 and Ser-1618 by Cdk1 and Plk1 respectively, further prevents 53BP1 localization to DNA breaks during mitosis by interfering with binding to H4K20-methylated nucleosomes. Besides Cdk1-mediated phosphorylation on Ser-1609, Benada et al also report that Cdk1 phosphorylates Ser-1678 within the C-terminus of 53BP1.Citation6 The additional Cdk1 phosphorylation site Ser-1678 found in this study might provide an additional regulatory step in controlling 53BP1 binding to sites of DSBs.
Why would cells inhibit the recruitment of DNA repair factors during mitosis? Orthwein and co-workers demonstrate that the inhibition of DSB repair in mitosis is required to prevent erroneous fusion of sister telomeres, which would otherwise contribute to aneuploid daughter cells.Citation5 Apparently, DNA repair (at least through NHEJ) seems to be deleterious during mitosis, and therefore mechanisms that suppress DSB repair are required. Indeed, by assessing DNA repair kinetics directly, Benada and coworkers now show that Plk1 indeed plays an active role in suppressing DNA repair, since inhibition of Plk1 increased the DNA repair capacity in mitotic cells.Citation6
The research described above reveals the complexity of DNA repair regulation and the important role that cell cycle kinases play in orchestrating DNA repair. Also, mitotic cells, in which DNA repair is suppressed, may be very susceptible to DNA breaks-inducing agents and this could serve as a starting point to design improved anti-cancer therapies.Citation3 Although detailed insight is now provided on the regulation of end-joining repair during mitosis, other important questions remain. For instance, it is unclear whether other DNA repair pathways, including homologous recombination, are similarly blocked during mitosis. Previous studies showed that damage induced by DNA replication stress needs to be processed during mitosis, arguing that not all types of DNA repair are suppressed during mitosis.Citation7 Clearly, additional investigation is required to further uncover the complex regulation of DNA repair during mitosis and to determine which pathways need to get blocked or are required to remain active.
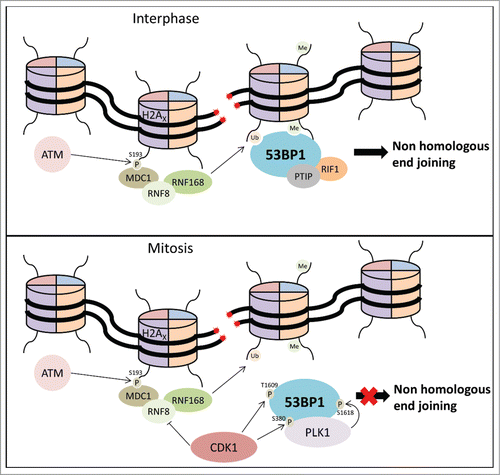
Figure 1. During Interphase DSBs trigger ATM-mediated phosphorylation of H2AX, and subsequent MDC1, RNF8 and RNF168 recruitment. Subsequently, 53BP1 is recruited to chromatin in an ubiquitilation- and methylation-dependent fashion to promote repair through NHEJ. During mitosis, Cdk1 phosphorylates RNF8, and 53BP1 is phosphorylated by Cdk1 and Plk1 to prevent 53BP1 recruitment to sites of DNA damage and block NHEJ repair of DSBs.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Panier S, Boulton SJ. Nature Reviews Molecular Cell Biology 2014; 15:7-18; PMID:24326623; http://dx.doi.org/10.1038/nrm3719
- Heijink AM et al. Mutation Research 2013; 750:45-55; PMID:23880065; http://dx.doi.org/10.1016/j.mrfmmm.2013.07.003
- Giunta S et al. The Journal of Cell Biology 2010; 190:197-207; PMID:20660628; http://dx.doi.org/10.1083/jcb.200911156
- van Vugt MA et al. PLoS Biology 2010; 8:e1000287; PMID:20126263; http://dx.doi.org/10.1371/journal.pbio.1000287
- Orthwein A et al. Science 2014; 344:189-93; PMID:24652939; http://dx.doi.org/10.1126/science.1248024
- Benada J et al. Cell Cycle 2015; 14:219-31; PMID:25607646; http://dx.doi.org/10.4161/15384101.2014.977067
- Lukas C et al. Nature Cell Biology 2011; 13:243-53; PMID:21317883; http://dx.doi.org/10.1038/ncb2201