
ERRβ belongs to the nuclear receptors superfamily (3B subgroup, including ERRα and γ), and is closely related to estrogen receptors (ERs). Unlike ERs, which act as ligand-dependent transcription factors, ERRs display ligand-independent, constitutive transcriptional activity. Although referred to as “orphan” receptors, ERRs activity can be modulated by synthetic molecules acting as ligands, such as DY131 for ERRβ and γ. ERRs function as homo- or hetero-dimers thereby diversifying the repertoire of their target genes. ERRβ is expressed in embryonic cells, strongly silenced in many somatic cells, probably through epigenetic mechanisms, and is often re-expressed in cancer cellsCitation1. At least in some cell types, ERRs are known to activate the expression of the p21 cell cycle inhibitor, although via different mechanisms, thus regulating the G1/S transition of the cell cycleCitation1-Citation3. In this issue of Cell Cycle, Heckler and RigginsCitation4, have analyzed the specific function of two ERRβ splice variants, ERRβsf (for short form) and ERRβ2, in DY131-dependent cell cycle arrest. Both isoforms display an identical E domain (the putative ligand binding domain) and hence are expected to bind and respond equally well to DY131. Previous work has established that only ERRβsf is expressed in prostatic cells, with stronger expression in immortalized versus cancer cell lines [2]. Furthermore DY131 reduces the growth of cancer cells by directly upregulating p21 expression in an ERRβsf-dependent manner.
Heckler and Riggins show that unlike the situation in prostate cancer cells, both ERRβ isoforms are expressed in two different brain tumor (glioblastoma) cell lines. They confirm the work of Yu et al.,Citation1 demonstrating that the effect of DY131 depends upon ERRβ and not ERRγ, and show that ERRβsf cooperates with p53 to induce p21 transactivation, resulting in cell cycle delay in G1, while ERRβ2 does not transactivate the p21 promoter in transient assays. In addition, they provide evidence that ERRβsf can induce senescence independently of p53. Interestingly, they also shows that DY131 induces ERRβ2-dependent cell cycle arrest in G2 and M phases. Since this effect is not observed in non-transformed, foreskin fibroblasts (not expressing ERRβ isoforms), it suggests that this phenotype is specific to cancer cells. Moreover the authors provide evidence for a cross-talk between ERRβ splice variants by showing that a) ERRβ2 may inhibit ERRβsf activity on p21 promoter in transient assays, b) ERRβsf down-regulation increases the effect of DY131/ERRβ2 on G2/M arrest, suggesting a functional and perhaps physical interaction between these isoforms. How would this work? The authors speculate that ERRβ2 may function in the cytoplasm, where predominantly localizes, consistent with the presence of a non-functional nuclear localization signal (NLS). In contrast, ERRβsf is predominantly nuclear due to a functional NLS. Hence, one possibility is that ERRβ2 may inhibit ERRβsf function by sequestration in the cytoplasm, although a nuclear function cannot be excluded. Conversely, ERRβsf-ERRβ2 dimerization may limit the latter's ability to impact on G2/M arrest. It would be interesting to determine whether the subcellular localization of ERRβ2 changes during the cell cycle. ERRβsf appears to be mainly nuclear in interphase and excluded from mitotic chromosomes, while it is expressed mainly in G1 and S phases. Hence ERRβsf, by cooperating with p53 in p21 transactivation, could inhibit CDK2/CyclinE activity thus delaying the G1/S transition, but how ERRβ2 leads to G2/M arrest is not clear (). The authors speculate that this may occur through CDK1 or Src sequestration in the cytoplasm. Another, non-exclusive, “nuclear” possibility, would be through transactivation of the Dub3/USP17 ubiquitin hydrolase, a major regulator of the CDC25A phosphatase. Dub3 is an ERRβ target in mouse embryonic stem cellsCitation5, although it is not known whether this is the case in human cells. This hypothesis would be consistent with the phenotype of Dub3 overexpression that leads to G2 arrest in human cells and the observation that Dub3 expression is cell cycle regulated in G1/S and G2 phasesCitation6,7 (and references therein). In this scenario, ERRβ2 may lead to G2/M arrest by inducing CDC25A destabilization, leading in turn to reduced CDK1 activity. In sum, this work provides the first evidence for a specific role for ERRβ splice variants in cell cycle regulation, and provides another interesting example of regulation through differential splicing, a new emerging concept that appears to be important in cancer biology.
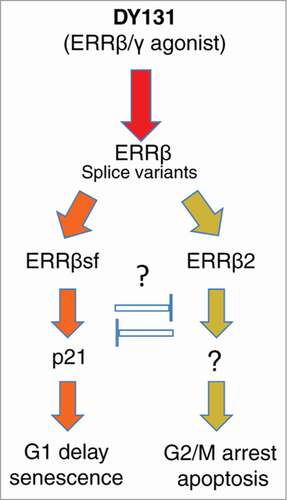
Figure 1. Schematic diagram of ERRβ splice variants function in regulation of cell cycle transitions. While ERRBsf may induce cell cycle delay in the G1 phase through transactivation of p21 gene expression, the ability of ERRβ2 to induce strong cell cycle block and apoptosis in G2 and M phases is not known, but in principle may occur through induction of the Dub3/USP17 ubiquitin hydrolase which in turn inhibits proteasomal degradation of the Cdc25A protein phosphatase leading to dephosphorylation of inhibitory tyrosine on CDK1 and subsequent G2/M arrest. Other mechanisms may also exists including cytoplasmic sequestration of CDK1 and or Src.
References
- Yu, S. et al. Oncogene 2008; 27, 3313-3328.
- Yu, S., Wang, X., Ng, C. F., Chen, S. & Chan, F. L. Cancer research 2007; 67, 4904-4914.
- Bianco, S. et al. The Journal of biological chemistry 2009; 284, 23286-23292.
- Heckler, M. M. & Riggins, R. B. Cell Cycle 2015; 14, 31-45.
- van der Laan, S., Tsanov, N., Crozet, C. & Maiorano, D. Molecular cell 2013; 52, 366-379.
- van der Laan, S., Golfetto, E., Vanacker, J. M. & Maiorano, D. PLoS One 2014; 9, e93663.
- McFarlane, C. et al. Cancer research 2010; 70, 3329-3339.