
Abstract
PPARγ and Wnt signaling are central positive and negative regulators of adipogenesis, respectively. Here we identified that, eicosapentaenoic acid (EPA) could effectively induce the transdifferentiation of myoblasts into adipocytes through modulation of both PPARγ expression and Wnt signaling. During the early stage of transdifferentiation, EPA activates PPARδ and PPARγ1, which in turn targets β-catenin to degradation and down-regulates Wnt/β-catenin signaling, such that the myogenic fate of myoblasts could be switched to adipogenesis. In addition, EPA up-regulates the expression of PPARγ1 by activating RXRα, then PPARγ1 binds to the functional peroxisome proliferator responsive element (PPRE) in the promoter of adipocyte-specific PPARγ2 to continuously activate the expression of PPARγ2 throughout the transdifferentiation process. Our data indicated that EPA acts as a dual-function stimulator of adipogenesis that both inhibits Wnt signaling and induces PPARγ2 expression to facilitate the transdifferentiation program, and the transcriptional activation of PPARγ2 by PPARγ1 is not only the key factor for the transdifferentiation of myoblasts to adipocytes, but also the crucial evidence for successful transdifferentiation. The present findings provided insight for the first time as to how EPA induces the transdifferentiation of myoblasts to adipocytes, but also provide new clues for strategies to prevent and treat some metabolic diseases.
Abbreviations
| BSA | = | bovine serum albumin |
| C/EBP | = | CCAAT/enhancer-binding protein |
| DHA | = | docosahexaenoic acid |
| DMEM | = | Dulbecco's modified Eagle's medium |
| EPA | = | eicosapentaenoic acid |
| IMF | = | intramuscular fat |
| PPAR | = | peroxisome proliferator-activated receptor |
| PPRE | = | peroxisome proliferator responsive element |
| PUFA | = | polyunsaturated fatty acids |
| RXR | = | retinoid X receptor. |
Introduction
Intramuscular fat (IMF), which presents in connective tissue surrounding muscle fibers and muscle fiber bundles, is composed of adipocytes interspersed among fiber fascicules (intramuscular adipocytes),Citation1,2 and plays very important roles in the physiologic function of muscle tissues, such as maintaining lipid homeostasis and insulin sensitivity.Citation1,3 Previous studies indicate that IMF tissue is not a simple ectopic extension of other fat locations; instead, it displays specific biological features in developmental and metabolic regulations. In particular, the developmental origin of IMF differs from other depositions. Besides being differentiated from mesodermal derived multipotent stell cells like other fat depositions, it is suggested that intramuscular adipocytes might also be transdifferentiated from the myoblasts in muscle under certain stimulations.Citation4 However, very little is known about this event, especially the regulation and molecular mechanisms of the transdifferentiation (from myoblasts into adipocytes, the same below).
Adipogenesis is the process by which mesodermal precursor cells convert into adipocytes, where lipid deposits and serves as central regulators of metabolism.Citation5,6 The adipogenesis process is controlled by both positive and negative regulators.Citation5,6
Nuclear receptor peroxisome proliferator-activated receptor (PPAR) γ is the chief positive and central regulator of adipogenesis.Citation7 It has been demonstrated that PPARγ induced during adipocyte differentiation is both necessary and sufficient for the process.Citation8 Furthermore, ectopical expression of PPARγ in nonadipogenic cells (fibroblasts or myoblasts) induces adipogenic transdifferentiation.Citation9,10 Notably, the PPARγ gene is transcribed from alternative promoters, yielding 2 major protein isoforms, PPARγ1 and PPARγ2.Citation11 PPARγ1 is expressed in many tissues and cell types, including adipose tissue, skeletal muscle, liver, pancreatic β-cells, macrophages, colon, bone, and placenta, whereas PPARγ2 expression is restricted almost exclusively to adipocytes under physiological conditions.Citation12 Moreover, PPARγ2 has the more adipogenic potential than PPARγ1, and is essential for effective adipogenesis in vitro.Citation13,14
As the main negative regulator of adipogenesis, Wnt/β-Catenin signaling serves as an adipogenic switch and thus is important for the maintenance and proliferation of preadipocytes. Adipogenesis will be repressed when it is on, while myoblasts will be spontaneously transdifferentiated to adipocytes when it is off.Citation15,16 Adipogenic differentiation is accompanied by the suppression of Wnt signaling and the concurrent activation of PPARγ.Citation15 However, the mechanisms underlying this switch are poorly understood. In particular, it is unclear how the Wnt pathway is shut off.Citation17
It is worth mentioning that another transcription factor PPARδ is also involved in adipogenesis.Citation18 Despite that it is not directly involved in the regulation of adipose terminal differentiation, PPARδ is implicated in the initial steps of the adipogenic program by inducing PPARγ expression in response to various adipogenic stimulators.Citation19,20 However, the detailed mechanism still remains to be addressed.
The n-3 polyunsaturated fatty acids (n-3 PUFAs) which belong to one of the major classes of long chain fatty acids are potential activators of PPARs.Citation21 Our previous study has shown that n-3 PUFA enrichment in muscle increases IMF content in pigs by influencing the expression of adipogenesis related genes.Citation21 There was also evidence that n-3 PUFAs inhibit Wnt/β-catenin signaling pathway in cell cultures.Citation22 However, the molecular mechanisms of n-3 PUFA induce both PPARs expression and Wnt signaling is still largely unknown during adipogenesis, especially in the transdifferentiation of myoblasts into adipocytes.
In the present study, for the first time, we discovered that a representative n-3 PUFA, eicosapentaenoic acid (EPA; 20:5n-3) could effectively induce the transdifferentiation of myoblasts into adipocytes. During the transdifferentiation process, EPA serves as a dual-function stimulator. It both inhibits Wnt signaling at the early stage by targeting PPARδ and PPARγ1, and subsequently activates PPARγ2 expression by promoter activation though PPARγ1. These findings provide us evidence that EPA could induce myoblasts to transdifferentiate into intramuscular adipocytes, further to the increase the IMF tissue and maintain the insulin sensitivity in skeletal muscle.
Results
EPA induces transdifferentiation of myoblasts to adipocytes
Our previous study discovered n-3 PUFAs increased intramuscular fat deposition in muscle of pigs,Citation21 and we speculated the intramuscular adipocytes might be partly transdifferented from the myoblasts under the stimulation of n-3 PUFAs. In order to test this hypothesis, a representative n-3 PUFA, EPA, was selected for the induction assays.
The C2C12 myoblasts have been extensively used to investigate the cellular and molecular mechanisms of muscle differentiation.Citation23,24 In our experiments, C2C12 cells were treated with 5% FBS supplemented with different levels of EPA for 10 d (). Without EPA, most cells were differentiated into myotubes as shown by microscopic analysis. With the elevation of the EPA concentration, the formation of myotubes was acutely disrupted, while the percentage of oil red O positive cells strongly increased (), indicating transdifferentiation process occured. In the 400 μM and 600 μM EPA treated cells, majority of cells were converted into lipid-laden adipocytes, but no myotube formation was observed. Since cell death was noticeablein 600 μM EPA group, 400 μM EPA was thus chosen for subsequent experiments.
Figure 1 (See previous page). EPA induces transdifferentiation of myoblasts to adipocytes. (A) Analysis of transdifferentiation by oil red O (ORO)-staining of C2C12 cells. The C2C12 myoblasts were treated with EPA of indicated concentrations for 10 d before ORO-staining. Top: plate view of ORO-stained cultures; bottom: microscopic view. Bars represent 50 μm. (B) Real-time PCR analysis of the expression patterns of myogenic marker genes during transdifferentiation of C2C12 myoblasts. The cells were shifted form medium supplemented with 10% FBS to control medium supplemented BSA (Control) or treatment medium supplemented 400 μM EPA (EPA) at 60% confluence. On Day 0, 3, 5, 7 and 9, the cultures were harvested for analysis. mRNA expressions in this and all subsequent figures were normalized to that of β-actin. All values are represented as mean ± SD from 3 independent experiments. The variance analysis was performed between the same time point of “Control” and “EPA.” The significance is presented as **P < 0.01. (C) Real-time PCR analysis of the expression patterns of adipogenic marker genes during transdifferentiation from cells treated as in (B).
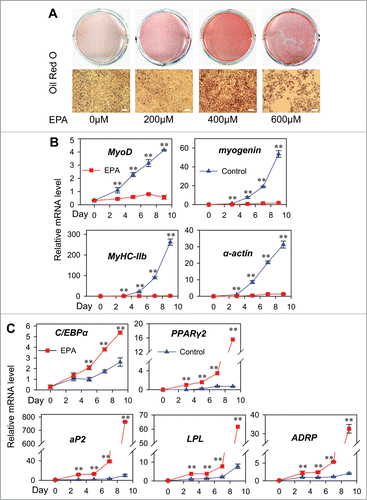
Transdifferentiation assay was performed using 400 μM EPA, and the expression pattern of myogenic and adipogenic marker genes during transdifferentiation were assessed by qRT-PCR. In the control group without addition of EPA, the expression of the early and later myogenic transcriptional factors (MyoD and myogenin) and structural protein of muscle fiber (MyHC-IIb and α-actin) continued to rise in a time-dependent manner, whereas the expression of these genes were greatly suppressed in EPA group (). In contrast, CCAAT/enhancer-binding protein (C/EBP) α and PPARγ2, 2 key adipogenic transcriptional factors and their target genes, aP2, LPL and ADRP, were highly expressed after EPA treatment, and elevated as the incubation time with EPA prolongs during transdifferentiation (). The gene expression data were all in agreement with the morphological changes.
Transdifferentiation is defined as an irreversible switch of one type of already differentiated cell to another type of normal differentiated cell.Citation25 A true transdifferentiation event of myoblasts to adipocytes must meet 2 important characteristics, i.e, the discrete change in cellular morphology and change in the expression of master regulatory (master switch) genes.Citation26 In the current study, after 10 d of induction with EPA, the conversion of myotubes to adipocytes was accompanied by suppressed expression of myogenic master genes (MyoD and myogenin) and induced expression of adipogenic master genes (C/EBPα and PPARγ2), indicating that EPA can successfully induce transdifferentiation of myoblasts to adipocytes.
PPARγ and Wnt/β-catenin signaling are regulated during transdifferentiation
n-3 PUFAs are known to affect target gene expression by directly acting at the level of the nucleus, in conjunction with some nuclear receptors. This is considered as the major mechanism of n-3 PUFAs in regulating gene expression.Citation27 So far, the nuclear receptors involved in adipogenesis, PPARs and retinoid X receptor (RXR) α, were shown to bind to EPA,Citation28,29 suggesting that EPA may induce transdifferentiation of myoblasts through these nuclear receptors.
We first examined the basal expression of these nuclear receptors in 80% confluence C2C12 cells (). Among these nuclear receptors, PPARδ, PPARγ1 and RXRα were expressed at similar level, while PPARα was expressed at low level, and PPARγ2 was hardly detected.
Figure 2. Regulation of nuclear receptors expression during transdifferentiation. (A) Absolute quantitative realtime PCR analysis of the expression of PPARs and RXRα in C2C12 cells. The C2C12 cells were cultured in 10% FBS DMEM medium, and harvested at 80% confluence for analysis. The copy number of genes was all normalized to that of β-actin. All values are represented as mean ± SD from 3 independent experiments. (B) Real-time PCR analysis of the expression patterns of nuclear receptors during transdifferentiation from cells treated as in (). All values are represented as mean ± SD from 3 independent experiments. The variance analysis was performed between the same time point of “Control” and “EPA.” The significance is presented as *P < 0.05, **P < 0.01. (C) Real-time PCR analysis of the expression patterns of cyclin D1 and nuclear receptors during transdifferentiation from cells treated as in ().
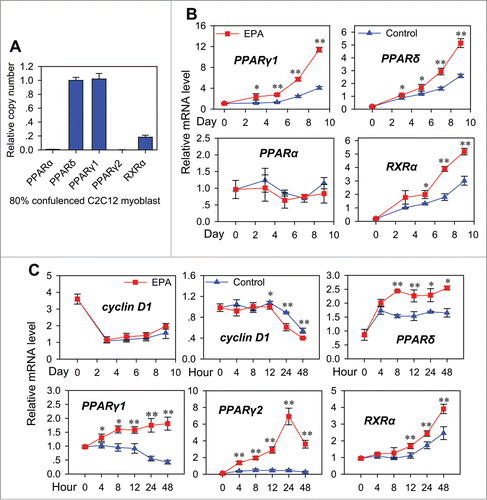
Next we determined the expression pattern of PPARα, PPARδ, PPARγ1 and RXRα during transdifferentiation (). In comparison with the control group, PPARα expression in the EPA group did not change significantly, while the expression of PPARδ, PPARγ1 and RXRα all elevated over the course of transdifferentiation in EPA treated groups. In particular, the expression level of PPARγ2 after EPA induction was 23-fold higher than that in the control group (). As mentioned before, PPARγ2 was hardly expressed at basal level in C2C12 cells and kept at a low level in the control group. This is in agreement with the fact that PPARγ2 is an adipocyte specific gene, suggesting that transcriptional activation of PPARγ2 was crucial in transdifferentiation of myoblasts to adipocytes. Additionally, PPARδ, RXRα and PPARγ1 all had similar expression trends to PPARγ2, indicating that PPARδ, RXRα and PPARγ1 might be involved in the up-regulation of PPARγ2 or the consequence of PPARγ2 upregulation.
In order to identify the regulation pattern of Wnt/β-catenin signaling during transdifferentiation, one of the well-known Wnt target genes, cyclin D1, was assessed (). During 9 d of transdifferentiation after EPA induction, the mRNA level of cyclin D1 remained unchanged as compared to the control group. It is known that it is indispensible to turn off Wnt/β-catenin signaling and down-regulate its target genes such cyclin D1 for adipogenesis,Citation15 this led us to speculate that the effect of EPA on Wnt/β-catenin signaling emerged at the early stage of transdifferentiation. To test this hypothesis, the mRNA level of cyclin D1 within 48 hours after induction were measured (). From 24 h to 48 h after EPA treatment, the mRNA level of cyclin D1 dropped significantly. This is in line with our hypothesis. The expression of PPARδ, RXRα, PPARγ1 and PPARγ2 within 48 hours after induction were also measured (). In contrast to cyclin D1, the expression of PPARδ, RXRα, PPARγ1 increased, implying that PPARδ, RXRα and PPARγ1 might be involved in the shutdown process of Wnt/β-catenin signaling.
EPA inhibits Wnt/β-catenin signaling through PPARδ and PPARγ1
To confirm the inhibition ability of EPA on Wnt/β-catenin signaling, a TCF-reporter was used to monitor the Wnt/β-catenin signaling. After treatment with EPA, the TCF-reporter activity reduced significantly in C2C12 cells (), and this is in accordance with the decline of cyclin D1 expression (). To investigate the function of β-catenin which is the central regulator of Wnt/β-catenin signaling after EPA treatment, the wild type or β-catenin with GSK3β phosphorylation sites mutation was co-transfected with the TCF-reporter. The luciferase assay showed that EPA inhibited Wnt/β-catenin signaling, while the inhibition activity was disrupted by mutatant β-catenin, which resists proteasomal degradation (). These results suggest that EPA may suppress Wnt/β-catenin signaling through β-catenin. To explore this possibility, the protein level of β-catenin was detected by western blot. After EPA treatment, the protein level of β-catenin significantly decreased (), indicating that EPA inhibits Wnt/β-catenin signaling by inducing the proteasomal degradation of β-catenin. Similar results were observed in cancer cells treated with EPA and other n-3 PUFA,Citation22,30,31 suggesting that n-3 PUFAs have the general effect to inhibit Wnt/β-catenin signaling in both normal and tumor cells.
Figure 3. PPARδ and PPARγ1 inhibits Wnt/β-catenin signaling. (A) Effect of EPA on Wnt signaling reporter in wild, β-catenin transfected or β-catenin mutant transfected C2C12 cells. Twelve hours after transfection, the cells were treated with control medium supplemented with BSA or treatment medium supplemented with 400 μM EPA for another 24 hours before harvest for luciferase reporter activity determination. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as (NS, not significant; **P < 0.01). (B) Western blot analysis of total cell lysates of C2C12 cells treated with control medium supplemented with BSA or treatment medium supplemented with 400 μM EPA for 24 hours. (C) Effect of PPARδ, PPARγ1 and RXRα on Wnt signaling reporter. Wnt reporter was co-transfected into C2C12 cells with pCMV-PPARδ, pCMV-PPARγ1, or pCMV-RXRα. The luciferase reporter activity was measured 24h after transfection. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as (NS, not significant; **P < 0.01). (D) Effect of PPARδ and PPARγ1 on Wnt signaling reporter under LiCl treatment. Wnt reporter was co-transfected into C2C12 cells with pCMV-PPARδ or pCMV-PPARγ1. The cells were treated with 25 mM LiCl after transfection. The luciferase reporter activity was measured 24 h after transfection. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as NS, not significant. (E) Western blot analysis of total cell lysates of C2C12 cells transfected with vector, pCMV-PPARδ or pCMV-PPARγ1 24 hours after transfection. (F) Real-time PCR analysis of the expression of cyclin D1 after knocking down of PPARδ or PPARγ1 in C2C12 cell by shRNA. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as *P < 0.05, **P < 0.01.
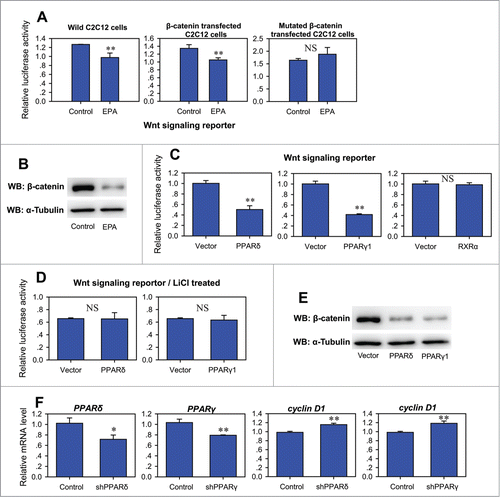
We next examined the effect of PPARδ, PPARγ1 and RXRα on Wnt/β-catenin signaling by co-transfection of PPARδ, PPARγ1 or RXRα expression plasmid with TCF-reporter. Both PPARδ and PPARγ1 suppressed the activity of TCF-reporter in C2C12 cells, whereas RXRα had no effect (). Interestingly, neither PPARδ nor PPARγ1 possessed the ability to suppress Wnt/β-catenin signaling in C2C12 cells after treatment with LiCl, a potent inhibitor of GSK3β and activator of β-catenin dependent transcription (). This led us to speculate that the inhibition ability of PPARδ and PPARγ1 was dependent on β-catenin. To test this hypothesis, the protein level of β-catenin was assessed by western blot after overexpression of PPARδ or PPARγ1, and the results showed that both PPARδ and PPARγ1 increased the degradation of β-catenin protein (), which mimicked the effect of EPA. These results demonstrated that EPA inhibits Wnt/β-catenin signaling through PPARδ and PPARγ1. Previous study done on Swiss mouse fibroblasts also indicated that PPARγ could inhibit Wnt signaling by targeting β-catenin for degradation.Citation32,33 However, to date, the effect of PPARδ on Wnt signaling is still not clear, owing to different results obtained in different cells or under experimental designs.Citation34
To provide additional evidence for the inhibition of PPARδ and PPARγ1 on Wnt/β-catenin signaling, the plasmids encoding inhibitory shRNAs were transfected to knock down PPARδ and PPARγ1 (). Along with the depressed expression of PPARδ or PPARγ1, the mRNA level of cyclin D1 was elevated, indicating that endogenous PPARδ and PPARγ1 have already contributed to the inhibition of Wnt/β-catenin signaling, and that these 2 nuclear receptors might be essential for the inhibition of Wnt/β-catenin signaling by EPA. However, the manners of inhibition by PPARδ and PPARγ1 may be different from each other (see Supplementary data and Supplementary and ).
PPARγ1 and PPARγ2 promoter activity are regulated by different nuclear receptors
To further validate the regulation of PPARγ1 and PPARγ2 by EPA, reporters with 2700 bp long PPARγ1 promoter and 2500 bp long PPARγ2 promoter were used. After EPA treatment, the activities of both PPARγ1 and PPARγ2 promoter reporters significantly enhanced in C2C12 cells (), which is in agreement with the mRNA change of PPARγ1 and PPARγ2 during the course of transdifferentiation ().
Figure 4. Regulation of PPARγ promoter activity by nuclear receptors. (A) EPA enhanced the promoter activity of PPARγ1 and PPARγ2 in C2C12 cells. C2C12 cells were transfected with pGL2-basic, mG1p2700, pGL3-basic or mG2p2500 to detect basal activity of PPARγ1 and PPARγ2 promoters (up). The cells transfected with mG1p2700 or mG2p2500 plasmid were cultured in control medium supplemented with BSA or treatment medium supplemented with 400 μM EPA (down). The luciferase reporter activity was measured 48 h after transfection. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as **P < 0.01. (B) Effect of PPARδ, PPARγ1 and RXRα on the promoter activity of PPARγ1. PPARγ1 promoter reporter mG1p2700 was co-transfected into indicated C2C12 cells with pCMV-PPARδ, pCMV-PPARγ1 and/or pCMV-RXRα. The luciferase reporter activity was measured 48 h after transfection. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as **P < 0.01. (C) Effect of PPARδ, PPARγ1 and RXRα on the promoter activity of PPARγ2. PPARγ2 promoter reporter mG2p2500 was co-transfected into indicated C2C12 cells with pCMV-PPARδ, pCMV-PPARγ1 and/or pCMV-RXRα. The luciferase reporter activity was measured 48 h after transfection. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as **P < 0.01. (D) Real-time PCR analysis of the expression change of PPARγ1 or PPARγ2 in the C2C12 cells transfected with pCMV-RXRα (left), pCMV-PPARδ or pCMV-PPARδ DN (middle), and pCMV-PPARγ1 or pCMV-PPARγ1 DN (right). Measurements were performed 48 h after transfection. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as **P < 0.01.
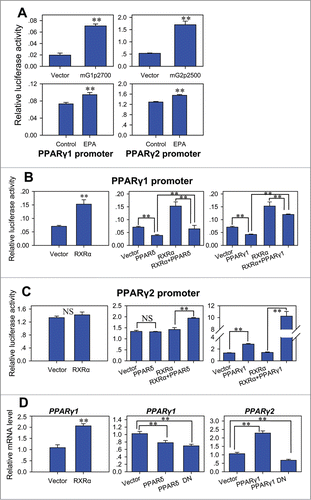
The effect of PPARδ, PPARγ1 and RXRα on the promoter activity of PPARγ1 and PPARγ2 were further assessed. The promoter activity of PPARγ1 was enhanced by RXRα whereas decreased by PPARδ and PPARγ1 (). In contrast, the promoter activity of PPARγ2 was only enhanced by PPARγ1, while PPARδ and RXRα had no significant effect on the promoter activity of PPARγ2 (). Interestingly, the promoter activity of PPARγ2 was further enhanced by co-expression of PPARγ1 and RXRα, suggesting that PPARγ1 may directly bind to the promoter of PPARγ2 to regulate its expression. The effect of PPARδ, PPARγ1 and RXRα were also measured by qRT-PCR (). Importantly, the dominant-negative PPARγ1 blocked the expression of PPARγ2, while its expression was enhanced by the wild-type PPARγ1. This is because the PPARγ1 dominant-negative mutant retains both ligand and DNA binding, and exhibits markedly reduced transactivation and further silences basal gene transcription,Citation35 the different actions of wild and dominant-negative PPARγ1 again suggest that PPARγ1 may function by directly binding to PPARγ2 promoter.
In our study, transient expression of PPARδ did not enhance the promoter activity of neither PPARγ1 nor PPARγ2, while some other studies suggested that PPARγ beinduced by stably expressed PPARδ.Citation19,20 These might seem confusing. However, we have already showed that transient expression of PPARδ suppresses Wnt/β-catenin signaling. These data suggest that the primary effect of PPARδ is on Wnt/β-catenin signaling, and the elevated level of PPARγ observed in cells stably expressing PPARδ is secondary to increase adipocyte differentiation.
PPARγ2 is a direct target gene of PPARγ1
In order to verify whether PPARγ1 binds to the promoter of PPARγ2, multiple softwares (PPRESearch, Genomatix MatInspector, TRRD, TESS and TFSEARCH) were used to predict the PPARγ1 binding sites (peroxisome proliferator responsive element(PPRE) in 2500 bp PPARγ2 promoter. In order to confirm the binding site, 6 putative PPRE and 7 5′-deletions of PPARγ2 promoter (P1-P7) were constructed accordingly (), and further co-transfected to C2C12 cells with PPARγ1 expression plasmid. Though PPARγ1 significantly up-regulated the activity of P1, P2, P3 and P4 (), it did not level up that of P5, implying that the putative functional PPRE is located between P4 and P5 (−966˜−837). Consistent with this finding, a putative PPRE (−890˜−878) was identified within this region, which is highly conserved among multiple species ().
Figure 5 (See previous page). PPARγ2 is a direct target gene of PPARγ1. (A) Effect of PPARγ1 on the activity of truncated PPARγ2 promoters. The truncated PPARγ2 promoter reporters (P1˜P7) were co-transfected into C2C12 cells with pCMV-PPARγ1 or empty vector. The luciferase reporter activity was measured 48 h after transfection. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as (NS, not significant; **P < 0.01). (B) Conserved sequences of the PPRE in PPARγ2 promoter of different species. The PPARγ2 promoter sequences of human (Homo sapiens), troglodyte (Pan troglodytes), monkey (Macaca mulatta), mouse (Mus musculus), rat (Rattus norvegicus), cow (Bos taurus), dog (Canis lupus familiaris) and pig (Sus scrofa) were aligned for conserved domain analysis. All the PPARγ2 promoter sequences are from Genbank database. (C) The effect of PPRE mutations and deletions on PPARγ2 promoter activity. Wild type (P1, P4), mutation type (P1 mut, P4 mut) or deletion type (P1 del, P4 del) of PPARγ2 promoter reporter was co-transfected into C2C12 cells with pCMV-PPARγ1 or empty vector. The luciferase reporter activity was measured 48 h after transfection. All values are represented as mean ± SD from 3 independent experiments. The significance is presented as (NS, not significant; **P < 0.01). (D) CHIP analysis of the PPARγ1-DNA binding activity with PPARγ2 promoter. C2C12 cells were maintained in control medium supplemented with BSA or treatment medium supplemented with 400 μM EPA for 48 hours prior to CHIP assays. After immunoprecipitation, PPRE region was amplified by PCR. Total chromatins were indicated as ‘input’. Pre-immune IgG was used as a negative control (left). The normal and pCMV-PPARγ1 transfected C2C12 cells were also treated with BSA or 400 μM EPA for 48 hours, followed by CHIP assays. PPRE region was amplified by realtime PCR (right). All values are represented as mean ± SD from 3 independent experiments. The significance is presented as **P < 0.01.
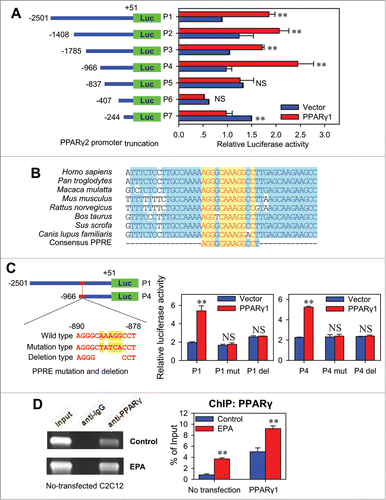
To further confirm the putative PPRE is functional, modifications of the conserved sites of this PPRE was introduced into P1 and P4 (). As expected, mutation or deletion of this PPRE both eliminated the enhancement of PPARγ1 on promoter activity of P1 and P4 (), strongly supporting the notion that this PPRE (−890˜−878) is functional.
To determine whether PPARγ1 directly interacts with the PPARγ2 promoter, ChIP assays were performed in C2C12 cells. Chromatin was immunoprecipitated by PPARγ-specific antibody, and the DNA fragments of expected size were amplified. Normal rabbit IgG did not result in immunoprecipitation of DNA fragments detectable by PCR amplification (). This result indicated that PPARγ1 specifically binds to the functional PPRE located on PPARγ2 promoter. Further ChIP-qPCR assay showed that EPA treatment highly strengthened the binding of PPARγ1 with this PPRE in C2C12 cells, as indicated by the enhanced binding ability after the transfection of PPARγ1 expression plasmid (). These results demonstrated that PPARγ1 activates the expression of PPARγ2 by binding to the functional PPRE located on the −890∼−878 bp of PPARγ2 promoter, and PPARγ2 is thus a direct target gene of PPARγ1.
Discussion
Skeletal muscle accounts for about 40% of body mass, and it is the major tissue contributing nearly 80% of whole body insulin-stimulated glucose disposal in humans.Citation36 It has been documented that accumulation of excess lipids in myocytes play an important role in the development of lipotoxicity and insulin resistance in humans because of its limited capacity in lipid storage.Citation37,38 Lipids could be stored either intramyocellularly or in intramuscular adipocytes. Adipocytes have unique capacity to store large amounts of lipids in the form of triglyceride, so as to prevent the accumulation of deleterious lipid species such as ceramides and diacylglycerol,Citation36,37 thus lipids storage in intramuscular adipocytes in lieu of intramyocellularly may prevent lipotoxicity and ensure insulin resistance.Citation1,3,36 Therefore, it is easy to understand that the increase of intramuscular adipose tissue could contribute to the maintainance of insulin sensitivity in skeletal muscle.Citation1,39
n-3 PUFA, especially those from marine oil, i.e. EPA and docosahexaenoic acid (DHA, 22:6 n-3), are reported to increase insulin sensitivity of muscle due to their beneficial effects on inflammation and obesity.Citation30,40 Here, we identified that EPA could effectively induce transdifferentiation of myoblasts to adipocytes, which may further lead to the increase of intramuscular adipose tissue. This finding might provide another point of view for understanding the benefit effect of EPA to insulin sensitivity, and also provides us a reasonable explanation for our previous results.Citation21
In our study, EPA was found having dual-effect during the transdifferentiation from myoblasts to adipocytes: while inhibiting Wnt signaling at early stage, it subsequently induces PPARγ2 expression constantly. On one hand, EPA could target β-catenin to degradation and down-regulate Wnt/β-catenin signaling through PPARδ and PPARγ1. On the other hand, EPA could induce PPARγ2 expression through the binding of PPARγ1 to PPARγ2 promoter.
Wnt/β-catenin signaling is well established as the adipogenic switch. When it is on, adipogenesis will not occur.Citation7 In addition, Wnt/β-catenin signaling is also the essential signaling for myogenesis.Citation41 Therefore, the turn-off of Wnt/β-catenin signaling may be the premise of transdifferentiation. Consistant with this speculation, our study confirmed that the Wnt target gene, cyclin D1, was down-regulated only in the first 48 h after EPA treatment, which was accompanied by a substantial increase in the expression of adipogenic marker gene PPARγ2, suggesting that the cell differentiation fate changes at the early stage of transdifferentiation after EPA treatment, and the shutdown of Wnt/β-catenin signaling is essential for effective transdifferentiation.
EPA appears to be a ligand of nuclear receptors, rather than work directly.Citation27 In our study, EPA induced the transdifferentiation of myoblasts to adipocytes through activating the nuclear receptors PPARδ and PPARγ1. Thus, we next elucidated the inhibition mechanism of EPA on Wnt/β-catenin signaling through PPARδ and PPARγ1. Previous studies have demonstrated that β-Catenin as the central factor of Wnt signaling must be recruited to Wnt target genes by TCF/LEFs to start Wnt signaling, and that phosphorylation-dependent degradation of β-catenin is often a key step in turning off Wnt signals in many situations.Citation42,43 We thus turned our attention to the status of β-catenin. Our results showed that both PPARδ and PPARγ1 could inhibit Wnt/β-catenin signaling by targeting β-catenin for degradation, mimicking the action of EPA. This demonstrated that the action of EPA in suppressing Wnt/β-catenin signaling may be through PPARδ and PPARγ1.
The expression of 2 PPARγ isoforms, i.e., PPARγ1 and PPARγ2, were both elevated during transdifferentiation, whereas the adipocyte-specific isoform PPARγ2 was hardly expressed in untreated C2C12 cells. Interestingly, the extent of the change in PPARγ2 expression is more dramatic than that of PPARγ1 during transdifferentiation, indicating that the activation of PPARγ2 expression is not only the key factor for the transdifferentiation of myoblasts to adipocytes, but also the crucial evidence for successful transdifferentiation. Medina-Gomez et al. also find that PPARγ2 is crucial to increase the lipid-buffering capacity of nonadipose tissues,Citation39 which is in agreement with our result.
Promoter truncation, mutation and deletion assays all proved that the PPRE located in −890˜−878 of the promoter of PPARγ2 is essential for PPARγ1 to enhance the promoter activity of PPARγ2. In addition, CHIP assays showed that the binding ability of PPARγ1 to the promoter of PPARγ2 significantly improved after EPA treatment. The above results demonstrated that EPA induces PPARγ2 expression through the binding of PPARγ1, to the functional PPRE located in the promoter of PPARγ2, thus leading to the conversion of myoblasts to adipocytes. The transcriptional activation of PPARγ2 by PPARγ1 is crucial for successful transdifferentiation, and this event might be also involved in the transdifferentiation processes induced by other PPARγ ligands such as linolenic acid, arachidonic acid and rosiglitazone.Citation23
In summary, EPA could effectively induce the transdifferentiation of myoblasts into adipocytes by inhibiting Wnt signaling at the early stage though PPARδ and PPARγ1 and subsequently activating PPARγ2 expression though PPARγ1. These findings provided evidence that EPA could induce myoblasts to transdifferentiate into intramuscular adipocytes, which may increase the lipid-buffering capacity of skeletal muscle and have significant positive effect on insulin sensitivity.
Materials and Methods
Cell culture and transdifferentiation assay
C2C12 mouse myoblasts were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal calf serum (FBS). For transdifferentiation assay, the cells were shifted at 60% confluence to medium supplemented with 5% FBS and BSA (control medium) or 5% FBS and EPA at indicated concentrations (treatment medium). EPA was first adsorbed to fatty acid-free bovine serum albumin (BSA) in a 4:1 molar ratio (EPA/BSA). Control medium and treatment medium were changed every 2 d After 10 d of induction, the cells were stained with oil red O to determine the transdifferentiation phenotype.
RNA extraction, reverse Transcription, and qPCR
RNA preparation, cDNA synthesis, and qPCR were performed as described in Luo et al.Citation21 Expression level was normalized to that of β-actin. Relative copy numbers of nuclear receptors were determined in qPCR as described in Whelan et al.Citation44 Primers are listed in Supplementary Table 1.
Western blot
Western blot was performed using 30 mg of total cell lysates. The antibodies used in this study include mouse anti-Tubulin IgM (1:1000; sc-8035, Santa Cruz), rabbit anti-β-Catenin (1:1000; #9587, Cell Signaling). Secondary antibodies, goat anti-IgM-HRP (sc-2064, Santa Cruz) and goat anti-IgG-HRP (Santa Cruz) were used at 1:10000 dilutions.
Transient transfection assays
For transient transfection assays, C2C12 cells were seeded to 24-well plate at 0.4−0.6 × 105 cells/well 18–24 h before transfection. The cells were transiently transfected with plasmids at 70% confluence using Lipofectamine™ 2000 reagent (Invitrogen) according to the manufacturer's instructions. The DNA/reagent ratio was 1 μg/2 μL. Cells were harvested 24 h or 48 h after transfection for subsequent analysis.
Luciferase reporter assay
The TOPflash plasmid (Millipore) was used to monitor the Wnt/β-catenin signaling. This plasmid contains 6 copies of the TCF binding site upstream of a TK minimal promoter and firefly luciferase open reading frame. Renilla luciferase encoded by the pRL-TK plasmid (Promega) was used as an internal control for firefly luciferase normalization. Luciferase activity was determined with the Dual-Luciferase® Reporter Assay System (Promega) and a GLOMAX luminometer (Promega) according to the manufacturer's instructions.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation assays (CHIP) were performed with Pierce® Agarose ChIP Kit (2162216, Pierce) according to the manufacturer's instructions. After Micrococcal Nuclease digestion, the digested chromatin was immunoprecipitated with the antibody against PPARγ (H-100, Santa Cruz) and normal rabbit IgG (Pierce) overnight at 4°C in the presence of Protein A beads (Pierce). DNA enrichment was quantified by real-time PCR. Primers used for CHIP are shown in Supplementary Table 1. Occupancy was quantified using a standard curve and normalized to input DNA.
Statistical analysis
Statistical analysis was performed using SAS software package (version 8.2; SAS Institute, Cary, NC, USA). The data are presented as mean ± SD. Differences between the means of the individual groups were assessed by one-way ANOVA; means were considered statistically different at P < 0.05. All experiments were performed at least 3 times.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Author Contributions
HFL, XMH and XWP performed the experiments. HFL and YFZ designed experiments, developed analysis tools, analyzed data and wrote the paper. YFZ and HKW interpreted results. JP and SWJ contributed to the design of the experiments, conceived of the study and wrote the paper.
1033594_supplemental_files.zip
Download Zip (524.8 KB)Acknowledgments
The authors are grateful to Dr. Tony Kouzarides (University of Cambridge, UK) for β-catenin S33, 37, 45A T41A plasmid, Janardan Reddy (Northwestern University, USA) and Dietmar Spengler (Max Planck Institute of Psychiatry, Germany) for pGL2-mG1p2700 plasmid. The authors are also grateful to Dr. Cuiping Liu for her suggestion to this paper.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31272457), Natural Science Foundation of Hubei Province of China (Grant No. 2013CFA010); Fundamental Research Funds for the Central Universities (Grant No. 2013PY047); Hubei Provincial Creative Team Project of Agricultural Science and Technology (Grant No. 2007–620).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
Related Research Data
References
- Poulos SP, Hausman GJ. Intramuscular adipocytes-potential to prevent lipotoxicity in skeletal muscle. Adipocytes 2005; 1:79–94
- Verbeke W, Van Oeckel MJ, Warnants N, Viaene J, Boucque CV. Consumer perception, facts and possibilities to improve acceptability of health and sensory characteristics of pork. Meat Sci 1999; 53:77–99; PMID:22063085; http://dx.doi.org/10.1016/S0309-1740(99)00036-4
- Lelliott C, Vidal-Puig AJ. Lipotoxicity, an imbalance between lipogenesis de novo and fatty acid oxidation. Int J Obesity 2004; 28: S22-S28; http://dx.doi.org/10.1038/sj.ijo.0802854
- Harper GS, Pethick DW. How might marbling begin? Aust J Exp Agr 2004; 44:653-62; http://dx.doi.org/10.1071/EA02114
- Farmer SR. Transcriptional control of adipocyte formation. Cell Metab 2006; 4:263-73; PMID:17011499; http://dx.doi.org/10.1016/j.cmet.2006.07.001
- Zhou YF, Peng J, Jiang SW. Role of histone acetyltransferases and histone deacetylases in adipocyte differentiation and adipogenesis. Eur J Cell Biol 2014; 93:170-7; PMID:24810880; http://dx.doi.org/10.1016/j.ejcb.2014.03.001
- Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 2006; 7:885-96; PMID:17139329; http://dx.doi.org/10.1038/nrm2066
- Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 2008; 77:289-312; PMID:18518822; http://dx.doi.org/10.1146/annurev.biochem.77.061307.091829
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994; 79:1147-56; PMID:8001151; http://dx.doi.org/10.1016/0092-8674(94)90006-X
- Hu E, Tontonoz P, Spiegelman BM. Transdifferentiation of myoblasts by the adipogenic transcription factors PPAR gamma and C/EBP alpha. Proc Natl Acad Sci U S A 1995; 92:9856-60; PMID:7568232; http://dx.doi.org/10.1073/pnas.92.21.9856
- Zhu Y, Qi C, Korenberg JR, Chen XN, Noya D, Rao MS, Reddy JK. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc Natl Acad Sci U S A 1995; 92:7921-5; PMID:7644514; http://dx.doi.org/10.1073/pnas.92.17.7921
- Vidal-Puig A, Jimenez-Linan M, Lowell BB, Hamann A, Hu E, Spiegelman B, Flier JS, Moller DE. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest 1996; 97:2553-61; PMID:8647948; http://dx.doi.org/10.1172/JCI118703
- Ren D, Collingwood TN, Rebar EJ, Wolffe AP, Camp HS. PPARgamma knockdown by engineered transcription factors: exogenous PPARgamma2 but not PPARgamma1 reactivates adipogenesis. Genes Dev 2002; 16:27-32; PMID:11782442; http://dx.doi.org/10.1101/gad.953802
- Mueller E, Drori S, Aiyer A, Yie J, Sarraf P, Chen H, Hauser S, Rosen ED, Ge K, Roeder RG, et al. Genetic analysis of adipogenesis through peroxisome proliferator-activated receptor gamma isoforms. J Biol Chem 2002; 277:41925-30; PMID:12200443; http://dx.doi.org/10.1074/jbc.M206950200
- Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA. Inhibition of Adipogenesis by Wnt Signaling. Science 2000; 289:950-3; PMID:10937998; http://dx.doi.org/10.1126/science.289.5481.950
- Prestwich TC, Macdougald OA. Wnt/beta-catenin signaling in adipogenesis and metabolism. Curr Opin Cell Biol 2007; 19:612-7; PMID:17997088; http://dx.doi.org/10.1016/j.ceb.2007.09.014
- Villanueva CJ, Waki H, Godio C, Nielsen R, Chou WL, Vargas L, Wroblewski K, Schmedt C, Chao LC, Boyadjian R, et al. TLE3 is a dual-function transcriptional coregulator of adipogenesis. Cell Metab 2011; 13:413-27; PMID:21459326; http://dx.doi.org/10.1016/j.cmet.2011.02.014
- Grimaldi PA. The roles of PPARs in adipocyte differentiation. Prog Lipid Res 2001; 40:269-81; PMID:11412892; http://dx.doi.org/10.1016/S0163-7827(01)00005-4
- Matsusue K, Peters JM, Gonzalez FJ. PPARbeta/delta potentiates PPARgamma-stimulated adipocyte differentiation. FASEB J 2004; 18:1477-9; PMID:15247146
- Grimaldi PA. Peroxisome Proliferator-Activated Receptors as sensors of fatty acids and derivatives. Cell Mol Life Sci 2007; 64:2459-64; PMID:17876521; http://dx.doi.org/10.1007/s00018-007-7278-5
- Luo HF, Wei HK, Huang FR, Zhou Z, Jiang SW, Peng J. The effect of linseed on intramuscular fat content and adipogenesis related genes in skeletal muscle of pigs. Lipids 2009; 44:999-1010; PMID:19798528; http://dx.doi.org/10.1007/s11745-009-3346-y
- Lim K, Han C, Xu L, Isse K, Demetris AJ, Wu T. Cyclooxygenase-2-derived prostaglandin E2 activates beta-catenin in human cholangiocarcinoma cells: evidence for inhibition of these signaling pathways by omega 3 polyunsaturated fatty acids. Cancer Res 2008; 68:553-60; PMID:18199552; http://dx.doi.org/10.1158/0008-5472.CAN-07-2295
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977; 270:725-27; PMID:563524; http://dx.doi.org/10.1038/270725a0
- Teboul L, Gaillard D, Staccini L, Inadera H, Amri EZ, Grimaldi PA. Thiazolidinediones and fatty acids convert myogenic cells into adipose-like cells. J Biol Chem 1995; 270:28183-7; PMID:7499310; http://dx.doi.org/10.1074/jbc.270.47.28183
- Slack JM, Tosh D. Transdifferentiation and metaplasia–switching cell types. Curr Opin Genet Dev 2001; 11:581-6; PMID:11532402; http://dx.doi.org/10.1016/S0959-437X(00)00236-7
- Li WC, Yu WY, Quinlan JM, Burke ZD, Tosh D. The molecular basis of transdifferentiation. J Cell Mol Med 2005; 9:569-82; PMID:16202206; http://dx.doi.org/10.1111/j.1582-4934.2005.tb00489.x
- Sampath H, Ntambi JM. Polyunsaturated fatty acid regulation of genes of lipid metabolism. Annu Rev Nutr 2005; 25:317-40; PMID:16011470; http://dx.doi.org/10.1146/annurev.nutr.25.051804.101917
- Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell 1999; 3:397-403; PMID:10198642; http://dx.doi.org/10.1016/S1097-2765(00)80467-0
- Goldstein JT, Dobrzyn A, Clagett-Dame M, Pike JW, DeLuca HF. Isolation and characterization of unsaturated fatty acids as natural ligands for the retinoid-X receptor. Arch Biochem Biophys 2003; 420:185-93; PMID:14622989; http://dx.doi.org/10.1016/j.abb.2003.09.034
- Xue M, Wang Q, Zhao J, Dong L, Ge Y, Hou L, Liu Y, Zheng Z. Docosahexaenoic acid inhibited the Wnt/beta-Catenin pathway and suppressed breast cancer cells in vitro and in vivo. J Nutr Biochem 2014; 25(2):104-10; PMID:24290517; http://dx.doi.org/10.1016/j.jnutbio.2013.09.008
- Zou Z, Bellenger S, Massey KA, Nicolaou A, Geissler A, Bidu C, Bonnotte B, Pierre AS, Minville-Walz M, Rialland M, et al. Inhibition of the HER2 pathway by n-3 polyunsaturated fatty acids prevents breast cancer in fat-1 transgenic mice. J Lipid Res 2013; 54(12):3453-63; PMID:24052576; http://dx.doi.org/10.1194/jlr.M042754
- Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol 2006; 26:5827-37; PMID:16847334; http://dx.doi.org/10.1128/MCB.00441-06
- Liu JJ, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis-A glycogen synthase kinase 3 beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem 2004; 279:45020-7; PMID:15308623; http://dx.doi.org/10.1074/jbc.M407050200
- Muller R, Rieck M, Muller-Brusselbach S. Regulation of cell proliferation and differentiation by PPARbeta/delta. PPAR Res 2008; 2008:614852; PMID:18815620
- Gurnell M, Wentworth JM, Agostini M, Adams M, Collingwood TN, Provenzano C, Browne PO, Rajanayagam O, Burris TP, Schwabe JW, et al. A dominant-negative peroxisome proliferator-activated receptor gamma (PPARgamma) mutant is a constitutive repressor and inhibits PPARgamma-mediated adipogenesis. J Biol Chem 2000; 275:5754-9; PMID:10681562; http://dx.doi.org/10.1074/jbc.275.8.5754
- Watt MJ. Storing up trouble: does accumulation of intramyocellular triglyceride protect skeletal muscle from insulin resistance? Clin Exp Pharmacol Physiol 2009; 36:5-11; PMID:18986321; http://dx.doi.org/10.1111/j.1440-1681.2008.05075.x
- Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol 2003; 14:281-7; PMID:12840659; http://dx.doi.org/10.1097/00041433-200306000-00008
- Bosma M, Kersten S, Hesselink MK, Schrauwen P. Re-evaluating lipotoxic triggers in skeletal muscle: relating intramyocellular lipid metabolism to insulin sensitivity. Prog Lipid Res 2012; 51:36-49; PMID:22120643; http://dx.doi.org/10.1016/j.plipres.2011.11.003
- Medina-Gomez G, Gray SL, Yetukuri L, Shimomura K, Virtue S, Campbell M, Curtis RK, Jimenez-Linan M, Blount M, Yeo GS, et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet 2007; 3: e64; PMID:17465682; http://dx.doi.org/10.1371/journal.pgen.0030064
- Jafari T, Fallah AA, Azadbakht L. Role of dietary n-3 polyunsaturated fatty acids in type 2 diabetes: a review of epidemiological and clinical studies. Maturitas 2013; 74:303-8; PMID:23384976; http://dx.doi.org/10.1016/j.maturitas.2013.01.008
- Kim CH, Neiswender H, Baik EJ, Xiong WC, Mei L. beta-catenin interacts with MyoD and regulates its transcription activity. Mol Cell Biol 2008; 28:2941-51; PMID:18316399; http://dx.doi.org/10.1128/MCB.01682-07
- Arce L, Yokoyama NN, Waterman ML. Diversity of LEF/TCF action in development and disease. Oncogene 2006; 25:7492-504; PMID:17143293; http://dx.doi.org/10.1038/sj.onc.1210056
- Daugherty RL, Gottardi CJ. Phospho-regulation of Beta-catenin adhesion and signaling functions. Physiology (Bethesda) 2007; 22:303-9; PMID:17928543; http://dx.doi.org/10.1152/physiol.00020.2007
- Whelan JA, Russell NB, Whelan MA. A method for the absolute quantification of cDNA using real-time PCR. J Immunol Methods 2003; 278:261-9; PMID:12957413; http://dx.doi.org/10.1016/S0022-1759(03)00223-0