
Abstract
Eukaryotic cells respond to DNA breaks, especially double-stranded breaks (DSBs), by activating the DNA damage response (DDR), which encompasses DNA repair and cell cycle checkpoint signaling. The DNA damage signal is transmitted to the checkpoint machinery by a network of specialized DNA damage-recognizing and signal-transducing molecules. However, recent evidence suggests that DNA repair proteins themselves may also directly contribute to the checkpoint control. Here, we investigated the role of homologous recombination (HR) proteins in normal cell cycle regulation in the absence of exogenous DNA damage. For this purpose, we used Chinese Hamster Ovary (CHO) cells expressing the Fluorescent ubiquitination-based cell cycle indicators (Fucci). Systematic siRNA-mediated knockdown of HR genes in these cells demonstrated that the lack of several of these factors alters cell cycle distribution, albeit differentially. The knock-down of MDC1, Rad51 and Brca1 caused the cells to arrest in the G2 phase, suggesting that they may be required for the G2/M transition. In contrast, inhibition of the other HR factors, including several Rad51 paralogs and Rad50, led to the arrest in the G1/G0 phase. Moreover, reduced expression of Rad51B, Rad51C, CtIP and Rad50 induced entry into a quiescent G0-like phase. In conclusion, the lack of many HR factors may lead to cell cycle checkpoint activation, even in the absence of exogenous DNA damage, indicating that these proteins may play an essential role both in DNA repair and checkpoint signaling.
Abbreviations
| ATM | = | ataxia telangiectasia mutated |
| Brca1 | = | breast cancer susceptibility protein 1 |
| Chk2 | = | cell cycle checkpoint kinase 2 |
| CHO | = | Chinese Hamster Ovary |
| CtIP | = | CtBP-interacting protein |
| DSBs | = | double-stranded breaks |
| DDR | = | DNA damage response |
| Fucci | = | Fluorescent ubiquitination-based cell cycle indicator |
| HR | = | homologous recombination |
| hGem | = | human Geminin |
| MDC1 | = | mediator of DNA-damage checkpoint 1 |
| MMEJ | = | microhomology mediated end joining |
| mAG | = | monomeric version of Azami green |
| mKO2 | = | monomeric version of Kusabira Orange 2 |
| MRN | = | Mre11/Rad50/Nbs1 complex |
| NHEJ | = | non-homologous end-joining |
| Rad | = | radiation-repair gene |
| siRNA | = | short interfering RNA |
| Xrcc | = | X-ray repair cross-complementing |
Introduction
DNA double strand breaks (DSBs), one of the most deleterious types of DNA lesions, can result from ionizing radiation or chemical agents, or from natural cellular processes such as DNA replication or maturation of the immune system genes. If left unrepaired, they constitute a major threat to genetic integrity and stability, possibly leading to cell death or carcinogenesis.Citation1 In response to DSBs, cells activate a network of DNA repair and signaling pathways, collectively termed the DNA damage response (DDR).Citation2-Citation4 To allow time for DNA repair, the DDR machinery activates cell cycle checkpoints that arrest cell cycle progression until genome integrity is restored. The DDR-activated checkpoints include the G1/S, the intra-S and the G2/M transitions. The G1/S checkpoint, the one most sensitive to DNA damage, is defective in most human cancer cells.Citation5,6
The Mre11/Rad50/Nbs1 (MRN) complex is among the first sensors of DSBs, subsequently activating Ataxia telangiectasia mutated (ATM).Citation7 ATM, a key protein kinase in the DDR network, is responsible for phosphorylation of many downstream DNA repair and cell cycle factors, including tumor suppressor p53, mediator of DNA-damage checkpoint 1 (MDC1), cell cycle checkpoint kinase 2 (Chk2), and breast cancer susceptibility protein 1 (Brca1).Citation8,9 The activation of these factors results in signaling cascades ultimately leading to cell cycle arrest. ATM-dependent phosphorylation of histone H2AX also induces global changes in the chromatin structure, leading to the recruitment of DNA repair proteins to the sites of damage.
Several specialized pathways act to repair DNA breaks in higher eukaryotic cells. One of the main pathways responsible for DSB repair is non-homologous end-joining (NHEJ). NHEJ is a fast process, based on a simple ligation of the 2 broken DNA ends, active throughout the entire cell cycle.Citation10 In the absence of functional NHEJ, cells were shown to use a highly error-prone, backup mechanism termed microhomology mediated end joining (MMEJ).Citation11,12 The third pathway, considered to be the most precise of all DSB repair mechanisms, is based on homologous recombination (HR).Citation13 HR requires extensive homology for repair, and thus is primarily used in late S and G2 phases of the cell cycle, when the genetic material has been replicated and sister chromatids are available as repair template.
A key role in eukaryotic HR is played by the Rad51 recombinase, which coats ssDNA ends resulting from the initial processing of the DSB.Citation14,15 The DNA-bound Rad51 then searches for sequence homology along a cDNA strand and mediates pairing between the 2 strands. The Rad51 protein is essential, as the targeted knock-out of its gene leads to embryonic lethality in mice.Citation16 Other proteins involved in HR include CtIP, Brca2, Rad52, Rad54 and the 5 Rad51 paralogs: Rad51B, Rad51C, Rad51D, Xrcc2, and Xrcc3.Citation17–Citation21 Rad51B, Rad51C, Rad51D and Xrcc2 together form the BCDX2 complex, which was proposed to facilitate the formation and stabilization of the Rad51 nucleofilament.Citation22 Rad51C also participates in the formation of a second complex with Xrcc3 termed CX3, which was reported to play an essential role in the final resolution of recombination intermediates.Citation23 The MRN complex, MDC1 and Brca1, which are components of the DDR response, also play a role in the initial steps of HR.Citation24-Citation26
It was recently proposed that HR proteins may also directly contribute to cell cycle control, in addition to their role in DNA repair.Citation27-Citation29 The knock-down of Rad51 was shown to induce G2/M arrest, suggesting that this protein is required for the progression from the G2 phase to mitosis,Citation30-Citation32 and Brca1 was reported to play a role in the regulation of the G2/M and intra-S checkpoints.Citation29,33 Rad51C was also proposed to contribute to cell cycle regulation, although there are conflicting reports as to its exact role. Rodrigue and others observed that the knock-down of Rad51C in human cells leads to arrest at the G2/M checkpoint, similarly to Rad51.Citation27 In another study, knock-down of Rad51C caused cells to escape the intra-S and G2/M checkpoints, thus allowing entry into mitosis.Citation28 Several other DSB repair proteins have also been proposed to participate in cell cycle progression.Citation34,35
Here, we systematically assessed the role of HR factors in cell cycle regulation in the absence of exogenous DNA damage by silencing HR genes of Chinese Hamster Ovary (CHO) cells expressing the fluorescent ubiquitination-based cell cycle indicator (Fucci) probes.Citation36 We show that the knock-down of many HR factors, including Rad51, MDC1, Brca1, several Rad51 paralogs, CtIP, and Rad50 significantly affected cell cycle progression, albeit differentially. The knock-down of MDC1, Rad51 and Brca1 caused the cells to arrest at the G2/M checkpoint, suggesting that these factors may be required for the transition through the G2 phase and entry into mitosis. In contrast, the absence of the remaining HR proteins increased the proportion of G1/G0 phase cells, indicating that their deficiency may cause the cells to escape the G2/M checkpoint, divide and subsequently become arrested in the G1 phase. We also observed that knock-down of Rad51B, Rad51C, CtIP and Rad50 increased the proportion of G0 cells, suggesting that the absence of these factors may cause cells to enter a quiescent state. We conclude that many HR proteins may regulate cell cycle progression in addition to their known role in DSB repair.
Results
Characterization of CHO Fucci cells
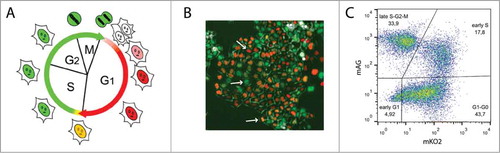
The fluorescent ubiquitination-based cell cycle indicator (Fucci) system enables the simultaneous observation of multiple cell cycle phases in living cells.Citation36 It is based on the expression, ubiquitination and degradation of the cell cycle-dependent human Cdt1 and Geminin proteins fused to fluorescent markers, the monomeric Kusabira Orange 2 (mKO2) and monomeric Azami Green (mAG), respectively. Depending on the fluorophore levels, 4 main cell subpopulations can be visualized, namely early G1 phase cells, G1/G0 cells, early S cells, and late S, G2 and M phase cells (). The freshly divided early G1 phase cells are non-fluorescent, but they start to express and accumulate the mKO2-hCdt1(30/120) chimeric protein as they progress through the G1 phase. In the early S phase, as the mKO2-hCdt1(30/120) becomes ubiquitinated and degraded, expression of the mAG-hGem(1/110) fusion protein yields yellow cells. Expression of mAG-hGem(1/110) and green fluorescence is then maintained from late S until the end of M phase.
Figure 1. Characterization of CHO Fucci cells. (A) Scheme of the Fucci cell cycle indicator assay.Citation36 (B) Typical fluorescence image of non-synchronized CHO Fucci cells. Arrows point to cells in G1/G0 (top), late S, G2, and M (middle) and early S phase cells (bottom). (C) Flow cytometry analysis of CHO Fucci cells with 4 subpopulations of cells: mKO2-positive G1/G0 cells (mKO2), mAG-positive late S, G2 and M cells (mAG), double positive (mKO2+mAG+) early S cells, and a double negative (mKO2-, mAG-) early G1 cells.
To confirm that the CHO Fucci cells express the fluorescent probes in a cell cycle dependent manner, we synchronized the culture by serum deprivation, to induce a cell cycle arrest in the G1/G0 phase, or by contact inhibition, which can cause G1 arrest.Citation37,38 Over 90% of serum-deprived cells displayed red fluorescence indicative of the G1/G0 phase (Figs. S1A, S2A). In cultures grown to confluence, a lower proportion (50%) of the red fluorescent G0/G1 cells was obtained, indicating that this method did not efficiently synchronize cells into the same phase of the cell cycle (Figs. S1B, S2B). We subsequently released the cells from cycle arrest and monitored the cell cycle phase distribution 16–24 h and 40–48 h following release. Within 20 h, the serum deprived cells had progressed through the S phase into the G2 and M phases, as evidenced by the accumulation of green fluorescent cells. Most serum deprived cells remained synchronized until 48 h post release, whereas the cells grown to confluence became desynchronized. Overall, the CHO Fucci cells appeared to express the fluorescent probes in a cell cycle dependent manner and therefore may serve as a model to study cell cycle regulation.
Knock-down of HR factors differentially influences cell cycle distribution
To assess the role of recombination factors in cell cycle regulation, we subsequently treated the CHO Fucci cells with a panel of short interfering RNAs (siRNAs) targeting HR protein mRNAs. The efficiency of siRNA knock-down was assessed by qPCR (Fig. S3A). Rad51 and Rad51D knock-down was also confirmed by western blot (Fig. S3B). A non-targeting siRNA, as well as an siRNA targeting Cyclin D1, which is required for the progression through the G1/S checkpoint,Citation39 were used as a negative and positive control, respectively.
The siRNA treatments had little effect on the proportion of freshly divided early G1 phase cells, except for a decrease of this sub-population, albeit not statistically significant, with Rad51C siRNA (). However, the number of G1/G0 cells was significantly increased upon treatment with the Cyclin D1 siRNA, as expected from a G1/S checkpoint arrest (). Interestingly, the knock-down of 3 Rad51 paralogs – Rad51B, -C, and -D, as well as Rad50, one of the MRN components, and to a lower extent CtIP, had a similar effect. The accumulation of G1/G0 cells upon the knock-down of these factors suggests that the progression through the G1 phase may be perturbed in their absence, possibly involving the G1/S checkpoint. This is surprising, as these proteins are thought to operate primarily in the late S and G2 phases of the cell cycle, when HR is most active. However, it is also possible that these factors may be necessary for the G2/M checkpoint activation in response to DNA damage. In this scenario, the absence of these factors would cause the cells to circumvent arrest and enter mitosis despite the presence of unrepaired breaks, which later on would activate the G1/S checkpoint.
Figure 2. Knock-down of HR factors affects cell cycle distribution of CHO Fucci cells. Graphs show relative numbers of cells in (A) early G1 phase, (B) G1/G0 phase, (C) early S phase, (D) late S, G2 and M phases. Results are shown as fold change over the data obtained from mock-treated cells (mock). Mean of ≥ 3 experiments, error bars show s.e.m. Asterisks indicate significant differences between siRNA-treated samples and mock control. Statistical significance relative to mock was determined by unpaired Student's t-test with Benjamini-Hochberg correction; significance level P < 0.05 (*), P < 0.01 (**).
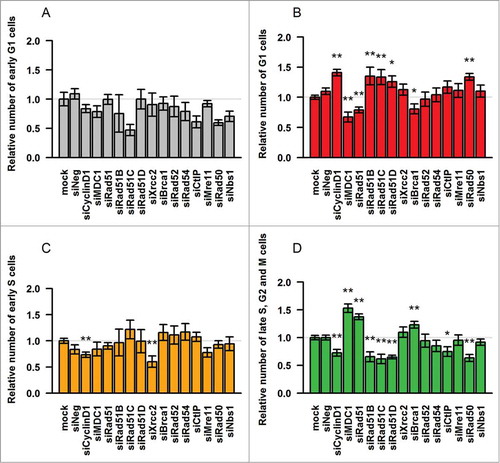
As expected, the proportion of early S phase cells was significantly decreased by the knock-down of Cyclin D1, due to the defective G1/S transition (). We also noted a decrease in this subpopulation in the presence of Xrcc2 siRNA, which, however, did not correlate with an increase in the number of G1/G0 phase cells. Distinctly, the knock-down of MDC1, Rad51 and Brca1 resulted in a significant accumulation of green-fluorescent late S, G2 or M phase cells (). This further supported the view that these proteins are required for the progression through the G2/M checkpoint.Citation29–31,40,41 The percentage of green fluorescent cells was also slightly increased in the presence of Xrcc2 siRNA. In contrast, the silencing of Cyclin D1 as well as Rad51B, -C, -D, Rad50 and CtIP, resulted in a significant decrease in this subpopulation, which may result from the aforementioned lack of G2/M checkpoint activation upon DNA damage and in the following accumulation of G1/G0 phase cells.
In conclusion, several HR proteins appear to be involved in cell cycle regulation, albeit differentially. MDC1, Rad51 and Brca1 seem to be essential for the progression from S and G2 phases into mitosis, while Rad51 paralogs, Rad51B, -C and -D, as well as the DNA end resection enzymes, Rad50 and CtIP may be required for activating the G2/M checkpoint in response to damage and/or progression through the G1/S checkpoint.
Knock-down of specific HR proteins induces entry into G0 phase and cell cycle arrest
In addition to increasing the number of mKO2-positive cells, we also observed that the knock-down of Cyclin D1 and several HR factors increased the level of mKO2 fluorescence (Fig. S4). A detailed analysis of the cells knocked-down for Rad51B, Rad51C, CtIP, and Rad50 revealed a sub-population of cells with distinctly higher mKO2 fluorescence patterns (). A recently published report identified low- and high mKO2-expressing cells as cycling G1 and quiescent G0 cells, respectively.Citation42 Consistently, we observed an increase in the number of these bright red fluorescent cells upon serum starvation (Fig. S5), confirming the view that this sub-population represents non-cycling G0 cells.
Figure 3. Knock-down of Cyclin D1, Rad51B, Rad51C, CtIP and Rad50 induces G0-like quiescence. (A) FACS plots of siRNA-treated cells. (B) Percentages of cells in G1 and G0 phases. The mean of values from ≥ 3 experiments is displayed, and error bars indicate the standard errors of the mean. Statistical significance relative to mock was determined by unpaired Student's t-test with Benjamini-Hochberg correction; significance level P < 0.05 (*), P < 0.01 (**).
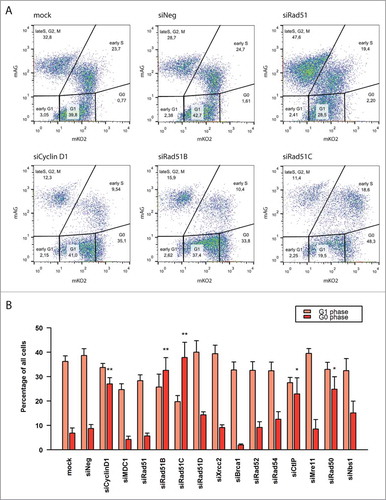
We therefore set out to quantify the G1 and G0 sub-populations in cells transfected with HR siRNAs. In controls, as well as in most siRNA-treated samples, the G0 phase cells constituted only approximately 5–10% of the population (). However, treatment with Cyclin D1 siRNA increased the number of quiescent cells to 30%. This is consistent with previous studies showing that Cyclin D1 deficiency causes entry in the G0 phase.Citation43 Interestingly, we also observed a very significant increase in the number of G0 cells upon the knock-down of Rad51B, Rad51C, CtIP, and Rad50. This was especially striking in the presence of Rad51B and Rad51C siRNAs, where G0 cells comprised up to 40% of the entire population. This implied that the absence of these HR factors may constitute a signal to enter the quiescent state.
We next sought to investigate whether the altered cell cycle distribution observed upon siRNA knock-down of HR factors results from a cell cycle arrest or from a delayed cycle progression. We focused our attention on Rad51 and Rad51C, the 2 HR proteins with pronounced, but distinct effects on the cell cycle. We synchronized the siRNA-treated cells in early S phase by sorting double positive mKO2+mAG+ cells, and subsequently analyzed their cell cycle distribution () and the presence of cell doublets and quadruplets (Fig. S6A) 1 or 2 d after sorting. Since the doubling time of CHO DG44 cells is approximately 12–14 h,Citation37,44 the cells should have completed a first cell cycle and be nearly completing a second one during the 18 h between sorting and the first measurements. Accordingly, the majority of cells should be green fluorescent at the beginning of the analysis, 40 h post transfection, and green fluorescence should decrease thereafter upon mitosis completion. This was indeed the case for untreated cells, and for cells transfected with the non-targeting siRNA ( ; Fig. S6A, B). Both populations also showed a similar cell cycle distribution over time, with another cell division around 64 h to 72 h post transfection. The time between these mitosis was also consistent with the normal CHO cell cycle duration.
Figure 4. Effect of Rad51 and Rad51C knock-down on cell cycle progression of CHO Fucci cells synchronized in early (S)phase. (A) General outline of the procedure. (B F) Percentages of cells in a given cell cycle phase at day 3 (D3) and day 4 (D4) post siRNA transfection. Arrows indicate the estimated average time of cell division, dotted arrow indicates a cell division of a subpopulation of cells.
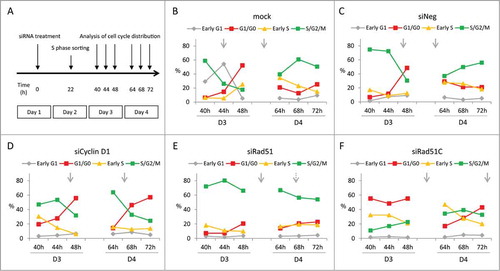
Cells treated with the Cyclin D1 siRNA initially displayed a cell cycle pattern similar to that of the cells treated with the non-targeted control siRNAs, with one division 44–48 h post transfection (; Fig. S6C). However, the next division to an 8-cell stage was delayed to 64–68 h, after which the accumulation of G1 and G0 phase red-fluorescent cells occurred (; Figs. S6B and S6C). In Rad51-depleted cells, the first division was notably delayed compared to the controls (48–64 h), despite the cell enlargement, after which most cells appeared not to divide anymore, except for a small portion of cells (approx. 20%) which underwent a second division at around 64–68 h after transfection. (; Fig. S6D). The percentage of green fluorescent cells stayed high throughout the time course, implying that most cells were arrested in late S, G2 or M phase. Thus, a near complete Rad51 deficiency may have caused the cells to arrest in the late phases of the cycle, likely at the G2/M checkpoint, whereas a milder Rad51 depletion, as due to lower knock-down efficiency, may have resulted in delayed cell cycle progression.
Cells treated with Rad51C siRNA also divided later than the controls (between 48 and 64 h) pointing to a delay in the cell cycle (; Fig. S6E). However, Rad51C-deficiency resulted in a steadily elevated proportion of cells in G1 and G0 phases, resembling the effect of the Cyclin D1 knock-down. Indeed, cluster analysis demonstrated that Rad51C-depleted cells grouped together with Cyclin D1 siRNA-treated cells (data not shown). However, the majority of these red-fluorescent cells appeared to be quiescent, reaching 50% of the entire population (). This was more than observed in the presence of Cyclin D1 siRNA (10–20%). After the delayed first cycle, most of the Rad51C-depleted cells underwent a second division within a normal time of 12–14 h, although some cells remained arrested in a prolonged G1, G0-like phase (; Fig. S6E). This indicated that, out of all the cells initially arrested in a quiescent G0-like phase, some were later released from the block and re-entered the cell cycle. At 64 h post transfection, most of the cells had reverted to a G1 profile, indicating that the cell Rad51C depletion-mediated cell cycle arrest is reversible.
Figure 5. Effect of HR protein depletion on the percentages of quiescent cells in CHO Fucci cells synchronized in early S phase. Numbers below the bars represent time (in hours) post siRNA transfection. The siRNA targets are indicated below the plot. Mock indicates cells treated with the transfection reagent only.
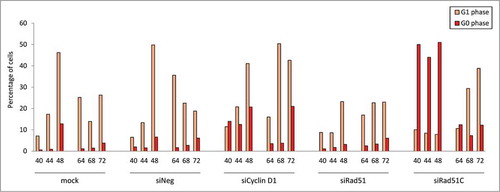
Overall, we concluded that Rad51 knock-down causes an arrest at the late phases of the cell cycle, most probably at the G2/M checkpoint, or delayed cell cycle progression in cells circumventing the arrest. In contrast, Rad51C depletion causes entry into a quiescent G0-like phase, most likely due to the activation of the G1/S checkpoint.
Discussion
The DNA damage response encompasses many functionally interconnected pathways, including cell cycle checkpoint signaling and DNA repair. Many proteins participating in cell cycle regulation are also known to control DNA repair, whereas the converse was not known to be true for the DNA repair factors. Recent evidence, however, suggested that some DSB repair proteins may also be implicated in cell cycle regulation in response to exogenous DNA damage.Citation28,29,34 To further explore this connection between DNA repair proteins and cell cycle machinery, we analyzed the effect of HR protein depletion on the CHO cell cycle in the absence of induced DNA damage. We found that the knock-down of several HR factors altered cell cycle progression, suggesting that they may play a direct role in cell cycle control. However, the lack of these proteins affected the cell cycle differentially, despite their being part of the same DNA repair pathway.
In the late S and G2 phases, the presence of unrepaired DNA breaks results in the activation of the G2/M cell cycle checkpoint by DNA damage sensing factors, e.g. ATM, ATR, p53, and Chk1.Citation45,46 We anticipated that the absence of crucial HR factors might lead to the accumulation of unrepaired DSBs, thereby stimulating the DDR to arrest the cells before division. Surprisingly, the knock-down of the majority of HR proteins, including 3 Rad51 paralogs, CtIP, and Rad50, failed to arrest the cells at the G2/M checkpoint. Instead, the presence of these factors seemed to be necessary for the progression from G1 to S phase. Only the loss of MDC1, Rad51 and Brca1 led to an accumulation of late S, G2 and M cells, likely due to the activation of the intra-S and/or G2/M checkpoints. This could indicate that the knock-down of these 3 genes, which products are involved in the early steps of the HR pathway, led to the accumulation of enough endogenous DNA damage to trigger the G2/M checkpoint. The absence of the remaining HR factors would potentially inhibit the repair process incompletely, instead rendering it more error-prone. These cells would still enter mitosis, but due to the accumulation of imprecisely or incompletely repaired DSBs, they may become arrested at the next G1/S checkpoint, explaining the accumulation of cells in the G1/G0 phase. In a recent study, Shibata and co-workers estimated that only about 15% of DSBs occurring in the G2 phase are repaired by the HR pathway, while the remaining breaks are efficiently repaired by other mechanisms.Citation47 This, together with our results obtained in the absence of induced DNA damage, may indicate that the effects of HR protein knock-down described here are not merely due to the accumulation of unrepaired DSBs. Instead, these HR proteins may play a more direct role in the cell cycle, for instance by interacting with cell cycle signaling factors, cyclins or cyclin-dependent kinases.
Our results obtained with Rad51B and Rad51C siRNAs contrast recently published observations that the knock-down of these Rad51 paralogs blocks progression through the G/M checkpoint in HeLa cells.Citation27 However, it is possible that the absence of these proteins in human cells may have a different impact on HR and/or the cell cycle than it does in CHO cells, which divide 2 times faster than HeLa cells and display different kinetics of DSB repair.Citation48,49
In the present study, we also observed that apart from a defect in the G1/S transition, the knock-down of Rad51B, Rad51C, CtIP and Rad50 also caused entry into a non-proliferative G0 phase resembling the effect of the Cyclin D1 knock-down. This suggests that the absence of these proteins may constitute a signal for the cells to withdraw from the cell cycle. This may in part explain the characteristic enhanced proliferation of cancer cells, in which HR proteins are often overexpressed. Taken together, these results indicate that many HR components are required for normal cell cycle progression, at least in CHO cells. Thus, their expression to levels that are sufficiently high to handle spontaneous DSB would act as one of the regulatory cues that control progression through cell cycle checkpoints. It will therefore be of interest to decipher how these proteins transmit the signals to the cell cycle control machinery, and what their molecular targets may be, to further understand their function in the cell cycle regulation network.
Materials and Methods
CHO cells expressing Fucci probes
Adherent CHO DG44 cellsCitation50 were cultivated in DMEM/F12+GlutaMAX™ supplemented with 1x HT and 10% fetal bovine serum (FBS) (Gibco, Invitrogen), and with antibiotic-antimycotic solution (Sigma-Aldrich, #A5955). CHO Fucci cells were constructed using lentiviral vectors carrying the red and green fluorescent ubiquitination-based cell cycle indicator (Fucci) cassettes.Citation36 The red Fucci cassette contains a monomeric version of Kusabira Orange 2 (mKO2) reporter gene fused to a truncated human Cdt1 (hCdt1, amino acids 30–120). The mKO2-hCdt1(30/120) protein is expressed in G1 phase and degraded at the onset of the S phase. The green Fucci cassette contains the monomeric version of Azami green (mAG) reporter gene fused to the 110 amino acid N-terminus of human Geminin (hGem amino acids 1–110). The mKO2-hGem(1/110) protein accumulates through S, G2 and M phases of the cell cycle and is degraded in the metaphase/anaphase transition of mitosis. The lentiviral constructs were kindly provided by M. Lutolf (EPFL, Lausanne, Switzerland). Briefly, the cells were transduced with a 1:1 ratio of mKO2 and mAG vectors at MOI 50. Three weeks after transduction double positive (mKO2+mAG+) clones were single-cell sorted by fluorescence-activated cell sorting (FACS) (FACSAria II sorter, Becton-Dickinson, Allschwil Switzerland). A single clone with similar levels of mKO2 and mAG fluorescence intensity was selected for subsequent experiments.
Cell synchronization
For starvation synchronization CHO cells were grown for 72 h in medium supplemented with 0.2% FBS (Gibco, Invitrogen). For synchronization through contact inhibition cells were grown for 3–5 d until complete confluency. Both methods synchronize the cells in G1/G0 phase. To reinitiate cell cycle progression cells were replated at lower density in complete medium. For early S phase synchronization 10,000 double positive (mAG+mKO2+) cells were sorted by FACS (FACSAria II sorter, Becton-Dickinson, Allschwil Switzerland) into each well of a 12-well plate.
siRNA transfection
Small interfering RNA duplexes were specifically designed to target the Chinese hamster homologs of HR genes. The siRNAs were designed and provided by Microsynth AG (Balgach, Switzerland). Three RNA duplexes were designed per gene to increase the probability of successful knock-down. Three negative (non-targeting) siRNAs were also designed as controls. For siRNA-mediated knock-down, CHO-Fucci cells were transfected with equimolar amounts of 3 siRNA duplexes at a final concentration of 50 nM using Lipofectamine RNAiMAX, according to the manufacturer's instructions (Invitrogen). After 72 h cells were analyzed using the Axio Observer.A1 microscope (Zeiss, Jena, Germany).
Flow cytometry
For flow cytometry cells were harvested 72 h following siRNA transfection, resuspended in 0.5 ml of PBS with 2% FBS (Gibco, Invitrogen), and analyzed using the CyAn analyzer (Beckman Coulter, Nyon, Switzerland). Acquired data was analyzed using the FlowJo software (Tree Star Inc., Ashland, OR, USA). For the time point experiment cells were harvested every 4 h at 40, 44, 48, 64, 68 and 72 h post transfection, fixed in PBS with 4% PFA (Merck) (1:2 v/v) and analyzed by flow cytometry.
Disclosure of Potential Conflicts of Interest
NM is a co-founder and owns shares of Selexis SA, a company that generates therapeutic-producing CHO cell lines. None declared by other authors.
Author Contributions
NM conceived the first ideas leading to this project. KK and NM designed the experiments and wrote the manuscript. KK, SB, ZU performed and analyzed the experiments.
1049784_Fig_S1.pdf
Download PDF (3.5 MB)Acknowledgments
The authors thank Matthias Lutolf and Mukul Girotra for the Fucci lentivirus constructs and their help in the transduction of CHO cells. The authors also thank Ioannis Xenarios and the VitalIT group for expert advice and help with the cluster analysis.
Funding
This project was funded by the University of Lausanne, by the SUR Summer School program of the Faculty of Biology and Medicine, and by a research grant from the Swiss Government Commission for Technology and Innovation and Selexis SA.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Shiloh Y, Lehmann AR. Maintaining integrity. Nat Cell Biol 2004; 6:923-8; PMID:15459720; http://dx.doi.org/10.1038/ncb1004-923
- Hiom K. Coping with DNA double strand breaks. DNA Repair (Amst) 2010; 9:1256-63; PMID:21115283; http://dx.doi.org/10.1016/j.dnarep.2010.09.018
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012; 481:287-94; PMID:22258607; http://dx.doi.org/10.1038/nature10760
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nature08467
- Löbrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 2007; 7:861-9; PMID:17943134; http://dx.doi.org/10.1038/nrc2248
- Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 2009; 8:547-66; PMID:19568282; http://dx.doi.org/10.1038/nrd2907
- Lee J-H, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005; 308:551-4; PMID:15790808; http://dx.doi.org/10.1126/science.1108297
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 2006; 31:402-10; PMID:16774833; http://dx.doi.org/10.1016/j.tibs.2006.05.004
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197-210; http://dx.doi.org/10.1038/nrm3546
- Pastwa E, Błasiak J. Non-homologous DNA end joining. Acta Biochem Pol 2003; 50:891-908
- Iliakis G. Backup pathways of NHEJ in cells of higher eukaryotes: cell cycle dependence. Radiother Oncol 2009; 92:310-5; PMID:19604590; http://dx.doi.org/10.1016/j.radonc.2009.06.024
- Kabotyanski EB, Gomelsky L, Han J, Stamato TD, Roth DB. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res 1998; 26:27-31; PMID:9399794; http://dx.doi.org/10.1093/nar/26.23.5333
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008; 77:229-57; PMID:18275380; http://dx.doi.org/10.1146/annurev.biochem.77.061306.125255
- Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Löbrich M, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 2011; 30:1079-92; PMID:21317870; http://dx.doi.org/10.1038/emboj.2011.27
- Baumann P, West SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci 1998; 23:247-51; PMID:9697414; http://dx.doi.org/10.1016/S0968-0004(98)01232-8
- Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci U S A 1996; 93:6236-40; PMID:8692798; http://dx.doi.org/10.1073/pnas.93.13.6236
- Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol 2011; 22:898-905; PMID:21821141; http://dx.doi.org/10.1016/j.semcdb.2011.07.019
- Swagemakers SMA, Essers J, de Wit J, Hoeijmakers JHJ, Kanaar R. The Human Rad54 Recombinational DNA Repair Protein Is a Double-stranded DNA-dependent ATPase. J Biol Chem 1998; 273:28292-7; PMID:9774452; http://dx.doi.org/10.1074/jbc.273.43.28292
- Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell 2001; 7:263-72; PMID:11239455; http://dx.doi.org/10.1016/S1097-2765(01)00174-5
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, Powell SN. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci U S A 2011; 108:686-91; PMID:21148102; http://dx.doi.org/10.1073/pnas.1010959107
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature 2007; 450:509-14; PMID:17965729; http://dx.doi.org/10.1038/nature06337
- Masson JY, Tarsounas MC, Stasiak AZ, Stasiak A, Shah R, McIlwraith MJ, Benson FE, West SC. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev 2001; 15:3296-307; PMID:11751635; http://dx.doi.org/10.1101/gad.947001
- Liu Y, Tarsounas M, O'regan P, West SC. Role of RAD51C and XRCC3 in genetic recombination and DNA repair. J Biol Chem 2007; 282:1973-9; PMID:17114795; http://dx.doi.org/10.1074/jbc.M609066200
- Lamarche BJ, Orazio NI, Weitzman MD. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett 2010; 584:3682-95; PMID:20655309; http://dx.doi.org/10.1016/j.febslet.2010.07.029
- Zhang J, Ma Z, Treszezamsky A, Powell SN. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat Struct Mol Biol 2005; 12:902-9; PMID:16186822; http://dx.doi.org/10.1038/nsmb991
- Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene 2002; 21:8981-93; PMID:12483514; http://dx.doi.org/10.1038/sj.onc.1206176
- Rodrigue A, Coulombe Y, Jacquet K, Gagne J-P, Roques C, Gobeil S, Poirier G, Masson J-Y. The RAD51 paralogs ensure cellular protection against mitotic defects and aneuploidy. J Cell Sci 2013; 126:348-59; PMID:23108668; http://dx.doi.org/10.1242/jcs.114595
- Badie S, Liao C, Thanasoula M, Barber P, Hill MA, Tarsounas M. RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J Cell Biol 2009; 185:587-600; PMID:19451272; http://dx.doi.org/10.1083/jcb.200811079
- Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet 2002; 30:285-9; PMID:11836499; http://dx.doi.org/10.1038/ng837
- Shtam TA, Kovalev RA, Varfolomeeva EY, Makarov EM, Kil YV, Filatov MV. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun Signal 2013; 11:88; PMID:24245560; http://dx.doi.org/10.1186/1478-811X-11-88
- Du L-Q, Wang Y, Wang H, Cao J, Liu Q, Fan F-Y. Knockdown of Rad51 expression induces radiation- and chemo-sensitivity in osteosarcoma cells. Med Oncol 2011; 28:1481-7; PMID:20625943
- Le Cigne A, Menil-Philippot V, Fleury F, Takahashi M, Thiriet C. Transient expression of RAD51 in the late G2-phase is required for cell cycle progression in synchronous Physarum cells. Genes to Cells 2014; 19:755-65; PMID:25200281; http://dx.doi.org/10.1111/gtc.12174
- Wiltshire T, Senft J, Wang Y, Konat GW, Wenger SL, Reed E, Wang W. BRCA1 contributes to cell cycle arrest and chemoresistance in response to the anticancer agent irofulven. Mol Pharmacol 2007; 71:1051-60; PMID:17229870
- Xu Z-Y, Loignon M, Han F-Y, Panasci L, Aloyz R. Xrcc3 induces cisplatin resistance by stimulation of Rad51-related recombinational repair, S-phase checkpoint activation, and reduced apoptosis. J Pharmacol Exp Ther 2005; 314:495-505; PMID:15843498
- Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos Trans R Soc Lond B Biol Sci 2011; 366:3562-71; PMID:22084383
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008; 132:487-98; PMID:18267078; http://dx.doi.org/10.1016/j.cell.2007.12.033
- Grosjean F, Batard P, Jordan M, Wurm FM. S-phase synchronized CHO cells show elevated transfection efficiency and expression using CaPi. Cytotechnology 2002; 38:57-62; PMID:19003087; http://dx.doi.org/10.1023/A:1021197830091
- Rosner M, Schipany K, Hengstschläger M. Merging high-quality biochemical fractionation with a refined flow cytometry approach to monitor nucleocytoplasmic protein expression throughout the unperturbed mammalian cell cycle. Nat Protoc 2013; 8:602-26; PMID:23449254
- Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 1993; 7:812-21; PMID:8491378
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 2006; 21:187-200; PMID:16427009
- Townsend K, Mason H, Blackford AN, Miller ES, Chapman JR, Sedgwick GG, Barone G, Turnell AS, Stewart GS. Mediator of DNA damage checkpoint 1 (MDC1) regulates mitotic progression. J Biol Chem 2009; 284:33939-48; PMID:19826003
- Tomura M, Sakaue-Sawano A, Mori Y, Takase-Utsugi M, Hata A, Ohtawa K, Kanagawa O, Miyawaki A. Contrasting quiescent G0 phase with mitotic cell cycling in the mouse immune system. PLoS One 2013; 8:1-10; http://dx.doi.org/10.1371/journal.pone.0073801
- Hsu L-C, Huang X, Seasholtz S, Potter DM, Gollin SM. Gene amplification and overexpression of protein phosphatase 1alpha in oral squamous cell carcinoma cell lines. Oncogene 2006; 25:5517-26; PMID:16619035; http://dx.doi.org/10.1038/sj.onc.1209563
- Grandjean M, Girod P-A, Calabrese D, Kostyrko K, Wicht M, Yerly F, Mazza C, Beckmann JS, Martinet D, Mermod N. High-level transgene expression by homologous recombination-mediated gene transfer. Nucleic Acids Res 2011; 39:e104; PMID:21652640
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20:1803-15; PMID:11313928; http://dx.doi.org/10.1038/sj.onc.1204252
- Lin JJ, Dutta A. ATR pathway is the primary pathway for activating G2/M checkpoint induction after re-replication. J Biol Chem 2007; 282:30357-62; PMID:17716975
- Shibata A, Barton O, Noon AT, Dahm K, Deckbar D, Goodarzi AA, Löbrich M, Jeggo PA. Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest. Mol Cell Biol 2010; 30:3371-83; PMID:20421415
- Kampinga HH, Hiemstra YS, Konings AW, Dikomey E. Correlation between slowly repairable double-strand breaks and thermal radiosensitization in the human HeLa S3 cell line. Int J Radiat Biol 1997; 72:293-301; PMID:9298109
- Posakony JW, England JM, Attardi G. Mitochondrial growth and division during the cell cycle in HeLa cells. J Cell Bol 1977; 74:468-91
- Urlaub G, Chasin LA. Isolation of Chinese hamster cell mutants deficient in dihydrofolate reductase activity. Proc Natl Acad Sci U S A 1980; 77:4216-20; PMID:6933469