
Abstract
The ribosome is a structurally and functionally conserved macromolecular machine universally responsible for catalyzing protein synthesis. Within eukaryotic cells, mitochondria contain their own ribosomes (mitoribosomes), which synthesize a handful of proteins, all essential for the biogenesis of the oxidative phosphorylation system. High-resolution cryo-EM structures of the yeast, porcine and human mitoribosomal subunits and of the entire human mitoribosome have uncovered a wealth of new information to illustrate their evolutionary divergence from their bacterial ancestors and their adaptation to synthesis of highly hydrophobic membrane proteins. With such structural data becoming available, one of the most important remaining questions is that of the mitoribosome assembly pathway and factors involved. The regulation of mitoribosome biogenesis is paramount to mitochondrial respiration, and thus to cell viability, growth and differentiation. Moreover, mutations affecting the rRNA and protein components produce severe human mitochondrial disorders. Despite its biological and biomedical significance, knowledge on mitoribosome biogenesis and its deviations from the much-studied bacterial ribosome assembly processes is scarce, especially the order of rRNA processing and assembly events and the regulatory factors required to achieve fully functional particles. This article focuses on summarizing the current available information on mitoribosome assembly pathway, factors that form the mitoribosome assembly machinery, and the effect of defective mitoribosome assembly on human health.
Introduction
Synthesis of all proteins is catalyzed by the ribosome. Functional ribosomes are macromolecular ribonucleoprotein nanomachines that are assembled from rRNA (rRNA) and proteins. Ribosomes are present in the cytoplasm of bacterial and eukaryotic cells. They are also present within semiautonomous eukaryotic organelles of bacterial ancestry, namely mitochondria and chloroplasts. In this review we will focus on discussing the current knowledge on the biogenesis of mitochondrial ribosomes (mitoribosomes), from yeast to mammals.
Mammalian and yeast mitoribosomes were first isolated in the late 1960s/early 1970s.Citation1-3 Mitoribosomes reside in the matrix of the organelle and associate with the inner membrane to facilitate co-translational insertion of highly hydrophobic nascent polypeptides. Mitoribosomes are specialized in the synthesis of only a handful of polypeptides (8 in yeast, 13 in human cells), which are essential for oxidative phosphorylation (OXPHOS) that facilitates ATP production aerobically. As remnants of their eubacterial origin, mitoribosomes are sensitive to similar antibiotics as bacteria and have many conserved proteins and RNA moieties. However, owing to evolution, mitoribosomes differ significantly in structure and composition, not only in comparison to their bacterial ancestors but also among different speciesCitation4-7
In all organisms, mature ribosomes are composed of 2 subunits, the large subunit (LSU) and the small subunit (SSU). The LSU is involved in catalyzing the peptidyl-transferase reaction, while the SSU provides the platform for mRNA binding and decoding. The LSU includes 3 rRNAs (rRNAs) in eukaryotes (28S, 5.8S, 5S in Saccharomyces cerevisiae) and 2 rRNAs in prokaryotes (23S and 5S in Escherichia coli). The SSU has instead only one rRNA in all kingdoms (18S in S. cerevisiae and 16S in E. coli). S. cerevisiae mitochondria contain 74S ribosomes composed of a 37S small subunit (mtSSU) formed by a 15S rRNA and 38 mitoribosomal proteins (MRPs), and a 54S large subunit (mtLSU) formed by a 21S rRNA and 46 proteins.Citation6,8 Mammalian mitoribosomes sediment as 55S particles, constituted of a 28S mtSSU, formed by a 12S rRNA and 29 MRPs, and 39S mtLSU, formed by a 16S rRNA and 50 MRPs.Citation9,10 These two mitochondrial rRNAs are universally transcribed from mtDNA genes. The rRNA content of the mtLSU, however, varies among species. For instance, yeast mtLSU lacks a 5S rRNA and the region where the cytoplasmic and bacterial 5S rRNA is occupied by mito-specific RNA and protein extensions. In that position, the mammalian mtLSU contains a structural mtDNA-encoded tRNA,Citation11 which is tRNAVal in the case of the human mitoribosome.Citation12
Regarding the mitoribosomal proteins, progress in genome sequencing and proteomics technology led to the identification of the full complement of yeast and mammalian mitoribosomal proteins, which has been further confirmed by high-resolution cryo-EM structures (). It is important to note that the nomenclature of mitoribosomal proteins has been unified, following a proposal by the ribosome community, as reportedCitation13 and stated in . With the single exception of yeast Var1, which is encoded in the mtDNA, all mitoribosomal proteins are encoded in nuclear genes. In S. cerevisiae mitochondria, 26 of the 38 mtSSU proteins and 30 of the 46 mtLSU proteins are homologous to E. coli proteins, while the rest are specific to mitoribosomes.Citation6,14 In mammals, 16 of the 30 mtSSU proteins and 28 of the 50 mtLSU proteins are homologs of proteins present in E. coli while the rest are mito-specific proteins.Citation5,11,12,15,16 As a consequence, yeast 74S and particularly human 55S mitoribosomes differ from bacterial (70S) and cytoplasmic ribosomes (80S) in their lower RNA:protein ratio, where significant amounts of RNA have been replaced by mitochondrion-specific proteins. Over the course of evolution mitoribosomes in both yeasts and mammalian ribosomes have acquired multiple changes in both their RNA and protein moieties compared to bacterial ribosomes. Yeast mitoribosomes expanded into both mito-specific RNA and protein segments, compared to the bacterial ribosome, giving rise to a RNA:protein ratio of 1:1; as opposed to an RNA: protein ratio of 2:1 observed in bacteria. In comparison to the bacterial ribosome, the mammalian mitoribosome has lost a significant amount of RNA, which has been replaced by mito-specific proteins giving rise to a 1:2 RNA:protein ratio.
Table 1. Mitoribosomal proteins. The old nomenclature for mitochondrial ribosomal proteins (MRPs) has been substituted by an unifying nomenclature where proteins with a prefix “u” (for universal) are observed in all kingdoms of life, proteins with a prefix “b” are bacterial in origin and do not have a eukaryotic (or archaeal) homolog, and proteins with a prefix “m” are mitochondrion-specific. Modified fromCitation13
Recent developments in electron microscopy have allowed resolving the structure of yeast mtLSUCitation13 and both mammalian mtLSUCitation11,12,15 and mtSSUCitation16,17 at near-atomic resolution, which will be summarized in depth in this review. These structures have provided a wealth of novel information, raising the next obvious question as to how these ribonucleoprotein macromachines are assembled. The assembly of ribosomes involves coordinated processing and modification of rRNAs with the temporal association of ribosomal proteins. The process is better characterized in cytoplasmicCitation18 and bacterial ribosomes,Citation19,20 with the latter having been fully reconstituted in vitro.Citation21 In the reconstitution experiments, protein binding kinetic data and time-resolved fingerprinting have demonstrated that bacterial ribosomal assembly takes place co-transcriptionally where MRPs bind to the 23S rRNA in a 5′ to 3′ direction while the transcript is still being synthesized.Citation19,20 However, despite recent progress, knowledge of the molecular details of the mitoribosome assembly pathway, the factors involved and the compartmentalization of the process is still very limited.
In addition to their biological significance, mitoribosomes and mitochondrial translation are also relevant from a biomedical perspective, as mutations in most mtDNA-encoded tRNAs and 12S rRNA, as well as nuclear genes encoding mitoribosomal proteins, translation initiation, and elongation factors, lead to clinically and genetically heterogeneous infantile multisystemic diseases, such as Leigh's syndrome, sensorineural hearing loss, encephalomyopathy and hypertrophic cardiomyopathy.Citation22-24
In this review we have summarized recent progress in the structural characterization of mitoribosomes, their biogenetic process and the factors involved during assembly. We also discuss human diseases related to disruption of mitoribosome assembly.
Lessons from High-Resolution cryo-EM Structures of Yeast and Mammalian Mitoribosomal Subunits
In 2009 the Nobel Prize in Chemistry was jointly awarded to Venkatraman Ramakrishnan (MRC Laboratory of Molecular Biology, Cambridge, UK), Thomas A. Steitz (Yale University, CT, USA) and Ada E. Yonath (Weizmann Institute of Science, Rehovot, Israel) “for studies of the structure and function of the bacterial ribosome.” However, determination of the mitoribosome structure was halted for many years as the low yield of mitoribosomes from most tissues made the crystallization process nearly impossible. Amidst these challenges, in 2003 Rajendra K. Agrawal was able to resolve the first 3-dimensional cryo-electron microscopic (cryo-EM) map of the bovine mitochondrial 55S ribosome carrying a tRNA at its P siteCitation5 at 13.5 Å resolution, and subsequently in 2009 the cryo-EM reconstruction of the mitochondrial ribosome with minimal RNA in the protist Leishmania tarentolae at 14.1 Å resolution.Citation4
Nearly a decade after the first structure, 2 groups - namely those of V. Ramakrishnan (Cambridge, UK) and Nenad Ban (ETH Zurich, Switzerland) - made a rapid advancement in the mitochondrial ribosome structure field by resolving the mtLSU of both yeast and mammals at a much higher resolution. Utilizing the advancements in EM and algorithms for computer-based analysis for “noisy” EM images of single particles, the Ramakrishnan group resolved the yeast mtLSU at a resolution of 3.2Å,Citation14 the human mtLSU with nascent polypeptide being folded in the exit tunnel at 3.4Å resolution.Citation12 The Ban group focused mainly on the mammalian mtLSU, where the porcine mtLSU (Sus scrofa) was resolved at both 4.9Å and 3.4Å resolution.Citation11,15 Contributing to the era of mitochondrial structural revolution, Agrawal's group resolved the cryo-EM structure of the mammalian mtSSU at 7Å resolution.Citation16 To finalize the impressive race between the Ramakrishnan and Ban groups, during the preparation of this manuscript, they have resolved at 3.5Å or 3.8Å resolution the complete structures of the 55S humanCitation25 or porcineCitation26 mitoribosomes, respectively.
The high-resolution structures have confirmed that the mammalian 55S mitoribosomes differ from bacterial (70S) and cytoplasmic ribosomes (80S) in their lower RNA:protein ratio, where mitochondrion-specific proteins have been recruited to prevent destabilization caused by rRNA reduction. In the yeast 74S mitoribosome, both protein and RNA moieties are substantially larger than their bacterial counterparts.Citation13 Almost all the mitochondrial ribosomal proteins with bacterial homologues have N- and/or C- terminal extensions, functions of some of which are unclearCitation6 and ∼25% of mito-specific proteins serve to directly compensate for the lost RNA secondary structures in mammalian mitoribosomes.Citation5,17,27-29 However, the early cryo-EM map of the bovine mitoribosome has already shown that many of additional or larger mitoribosomal proteins occupy new positions within the ribosome and that missing RNA segments are not always replaced by any protein mass.Citation5 In addition, high-resolution structure of human mitoribosome that was trapped in 3 different conformations, revealed a wider spectrum of mtSSU orientations than was observed for bacterial or cytoplasmic ribosomes.Citation17 Overall, the high-resolution reconstructions have provided a wealth of novel information, specifically noting the similarities and differences between yeast and mammalian mitoribosomes ().Citation4,5,13,15,16,25, 26
Figure 1. Structure of mitochondrial ribosomes. Top panel, human 55S; bottom panel, yeast LSU. LSU rRNA is shown in blue, SSU rRNA is yellow, tRNA-Val is red. Individual proteins are depicted in different colors, and those mentioned in the text are labeled.
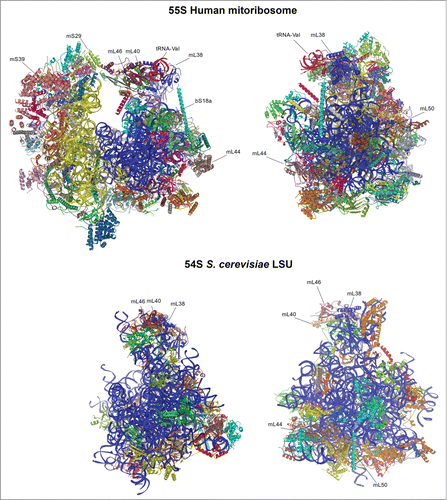
Yeast mtLSU has a core conserved with the bacterial LSU, while the mitochondrion-specific proteins and extensions of known bacterial homologues and rRNA expansion segments lie on the surface of the conserved core.Citation13 The rRNA expansion segments act as scaffolding for the additional new proteins. With regard to the rRNA, there is an expansion of 392 additional nucleotides spreading over 11 expansion segments (ES) compared to the bacterial LSU. However, there was no evidence of a 5S rRNA or putative 5S rRNA binding proteins L18 and L25, as had been proposed for the mammalian LSU. In its place, this missing area is occupied by mitochondrion-specific proteins (mL38, mL40 and mL46), 16S rRNA expansion clusters (82-ES and 84-ES1) and extensions from proteins homologous to bacteria. All these additions do not structurally mimic the 5S rRNA but they made the yeast mtLSU central protuberance (CP) into triple the volume of the bacterial CP. A new protuberance is observed adjacent to the mouth of the tunnel exit site consisting of both mitochondrion-specific proteins and rRNA expansion segments. Owing to its location, this bulged structure is proposed to be involved in tethering the mitoribosome to the membrane, which is independent of nascent peptide insertion. Furthermore, the yeast mtLSU has an additional contact with the E site tRNA that is not seen in bacteria. This contact is mediated through the C-terminal extension of bL33 with the tRNA acceptor stem, thus creating additional stability for the incoming tRNA. The polypeptide tunnel channel and its exit sites are highly conserved among previously studied ribosomes. However, when the structure of the yeast LSU is superimposed onto the bacterial ribosome, the region where the path is located appears blocked by a mitochondrion-specific extension of uL23. Notably, a new path that deviates from the bacterial path was observed, presenting a new exit site, surrounded by mitochondrion-specific proteins mL44 and mL50. Interestingly, these proteins are also involved in forming the mitochondrial inner membrane-facing protuberance, an orientation expected to promote mitoribosome interaction with the membrane to facilitate the co-translational membrane insertion of newly synthesized, highly hydrophobic OXPHOS subunits. Several inner membrane proteins are known to mediate membrane binding of ribosomes such as the Oxa1 machinery, which enables the insertion of nascent polypeptides into the inner membrane.Citation30-36 The binding of mitoribosomes to the inner membrane is discussed more extensively in section 5.2.
The high resolution cryo-EM reconstruction of the mammalian mitoribosome revealed extensive differences not only with bacterial cytoplasmic ribosomes but also with yeast mitoribosome.Citation11,12,15,25,26 The mammalian mitoribosome is highly protein-rich compared to both yeast and bacterial ribosomes. While the total rRNA content has increased in yeast with only minor deletions at the tunnel exit site, the mammalian rRNA content has nearly halved owing to numerous deletions evenly distributed when compared with the bacterial rRNA. The ribosomal proteins that are homologous to bacteria have extensions, which are shorter and not conserved with yeast. These extensions mainly interact with mitochondrion-specific proteins while only a few engage in filling the space of deleted rRNA. The mitochondrion-specific proteins are mainly peripherally distributed over the solvent-accessible surface forming clusters at the central protuberance, the L7/L12 stalk, and adjacent to the polypeptide exit site. Similar to the protein extensions, these proteins accommodate novel positions rather than compensate for the missing rRNA.
In addition to the mitochondrion-specific protein cluster at the CP, a second L-shaped RNA molecule was discovered in the mammalian mitoribosome at the same position where the 5S rRNA is found in the bacterial ribosome. However, the structure of the human mtLSUCitation12 showed that it is not a 5S rRNA molecule as it had been long speculatedCitation37 but rather tRNAVal, which plays an integral structural role at the CP. In the mtDNA, the tRNAVal gene is located in the middle of the genes encoding for the 12S and 16S rRNAs. These three RNAs are transcribed together as a polycistronic transcript similar to that of the bacterial rRNA operon. Owing to the presence of tRNAVal, the CP of the human mtLSU is substantially different from other ribosomes, including the yeast mitoribosome. Despite the major remodeling at the CP, the interactions it makes with the small subunit and the tRNAs bound to the ribosome are conserved. Nevertheless, in the yeast mitoribosome, this inter-subunit communication occurs via rRNA while in the mammalian mitoribosome it is coordinated entirely by mitochondrion-specific proteins,Citation5,11,12,14,16 a feature that is different from bacterial and cytoplasmic ribosomes that typically consist of only RNA-RNA intersubunit connections. Even though this suggests that, during evolution, mito-specific proteins have functionally replaced the RNA, it is more likely that much of the additional ribosomal proteins in mitochondria were recruited to stabilize the general structure of the ribosome.Citation7 Furthermore, the number of intersubunit bridges is lower than in the bacterial ribosome, probably to allow the 2 subunits to have flexibility in the conformation allowing them to tilt freely.Citation25,26 An additional major remodeling is observed at the A (aminoacyl) and P (peptidyl) tRNA binding sites, where some subunits present in bacterial ribosomes (e.g. uL5 at the P site, bL25 and the A site) have been lost in order to accommodate human mitochondrial tRNAs, which contain highly variable loops at the tRNA elbow. However the P-site finger, unique to the mammalian mitoribosome, compensates for these missing interactions.Citation25,26 A distinctive property of mitoribosomes is the acquisition of an intrinsic GTPase activity through the GTP–binding protein, mS29 of the 28S mtSSU subunit.Citation25,26 The GTPase activity is probably linked to subunit association, since mS29 locates at the subunit interface and is involved in coordinating 2 mitochondria-specific bridges.Citation25,26
The polypeptide exit tunnel is adapted to the transit of hydrophobic nascent peptides.Citation11,12 The tunnel exit site consists of conserved proteins from bacteria, namely bL23, bL29, bL22, bL24 and bL17, which create a ring around the exit site. Moreover, this conserved core is surrounded by a second layer of protein density consisting of bL33 and mL45, which promote anchoring of the mtLSU to the inner mitochondrial membrane.Citation11,12 The structure of the human mitoribosome with nascent polypeptide shows its extensive interactions with specific hydrophobic residues of the tunnel wall.Citation17 Membrane anchoring aligns the polypeptide exit site with the OXA1L translocon to facilitate co-translational membrane insertion of newly synthesized proteins. The overall tunnel path is similar to that of bacterial and cytoplasmic ribosomes but different from yeast mitoribosomes. Thus, the upper tunnel path does not show the constriction and the alternative exit tunnel observed in yeast.
The recent cryo-EM reconstructions of the 55S humanCitation25 and porcineCitation26 mitoribosome at high 3.5Å or 3.8Å resolution, have allowed to distinguish important features in the mammalian mtSSU. Similar to the mtLSU, several peripheral rRNA helices present in bacteria are either truncated or missing in the 12S rRNA, but the tertiary structure of the 12S rRNA core and the overall positions of the MRPs with bacteria homologs are preserved in the structure. A significant structural remodeling is observed at the mRNA entrance of the mtSSU compared to the bacterial SSU.Citation16,25,26 This remodeling serves to accommodate mammalian mitochondrial mRNAs, which have either null or very short 5′-untranslated regions (UTR),Citation38 since the structural data shows that in the proximity of the channel entrance a pentatricopeptide repeat (PPR) protein (mS39) is bound, it could be involved in recruiting the leaderless mRNA during translation initiation.Citation16,25,26
According to the mammalian mitoribosomal protein complement there are 3 isoforms for mammalian mtSSU protein bS18.Citation39,40 S18 isoforms are not uncommon. E. coli contains a single S18, but other prokaryotes such as Streptomyces coelicolor contain 2 S18 variants. A single bS18 form is present in yeast mitochondria, 2 in worms and 3 in flies and mammals.Citation39 Apparently two rounds of duplication of the gene encoding the mammalian bS18 occurred, giving rise to 3 very different variants termed bS18A, bS18B and bS18C. The primary sequences of these variants are very divergent and have conserved regions with prokaryotic S18s only in their central portion.Citation39 The existence of several bS18 variants led to the speculation that each mitoribosome would have a single variant leading to a heterogeneous population of mitochondrial ribosomesCitation9,39 for performance of specialized functions. However, the recent cryo-EM structures have identified bS18A to be in the mtLSU subunit (in porcine liver and human HEK293 cells),Citation11,12 but not in the human mtSSU. Instead, the human mtSSU contains bS18B at the position of S18 in bacterial ribosomes, and bS18C at a different position.Citation25,26 The precise role of these subunits remains to be disclosed.
Trans-acting factors Required for Mitoribosome Assembly from Yeast to Mammals
With the high-resolution mitoribosome structures resolved, the question still remains of how these macromolecular structures are assembled and what elements of the pathway are conserved across evolution. Bacterial ribosome assembly has been reconstituted in vitro and a large amount of information on the assembly pathway of bacterial and cytoplasmic ribosomes is already available. Ribosome assembly involves the coordinated processing and modification of rRNAs with the temporal association of ribosomal proteins. The entire process is regulated by several classes of ribosome biogenetic factors, including nucleases, rRNA modifying enzymes, DEAD-box helicases, GTPases and chaperones.Citation19,20,41 The current understanding of the mitoribosome assembly pathway and the factors involved (), which is still far from complete, will be summarized in this section.
Table 2. Role of mitoribosome assembly cofactors. Nucleases and RNA processing enzymes are not included
Factors required for rRNA processing and modification
Nucleases
The rRNA genes are often organized in operons that are transcribed into a single polycistronic transcript that requires processing to liberate the individual large and small rRNAs. Subsequently, rRNAs are further processed at both 5′ and 3′ end to yield mature rRNAs. The processing activities are performed by exo and/or endonucleases. In the mitochondrial genomes, rRNA organization depends on the species, as will be discussed below. A common trait among all species is the polycistronic nature of the transcript and the large number of RNA-processing events to liberate not only rRNAs but also mRNAs and tRNAs for translation.
In yeast S. cerevisiae the mtDNA contains 35 genes. Eight genes encode proteins; 7 subunits of OXPHOS enzymes and one SSU ribosomal protein (Var1). Twenty-seven other genes encode RNAs required for translation, specifically 24 tRNAs and 2 rRNAs (21S rRNA and 15S rRNA), and one gene for the RNA component of RNase P (RPM1). Only three genes contain either group I or group II introns,Citation42 namely one intron in 21S rRNA, 5 in COB, and 7 in COX1. Ten of these introns contain ORFs coding for homing endonuclease genes (HEG)Citation43, which in some cases have gained maturase (intron splicing) activity. All yeast mitochondrial genes are transcribed as polycistronic precursors from 11 distinct promotersCitation44. Regardless of the organization in the original transcript, all precursor tRNAs have 5′ and 3′ extensions that require further processing. The 5′-tRNA endonucleolytic processing is executed by an almost ubiquitous RNaseP, which contains the nucleus-encoded Rpm2 protein, and an RNA component encoded in the mtDNA RPM1 gene in yeast.Citation45 The 3′-tRNA processing is an endonucleolytic cleavage catalyzed by tRNase Z,Citation46 which is encoded by the TRZ1 gene in yeast.Citation47
The main 3′-5′-exoribonuclease in yeast mitochondria is the mitochondrial degradosome (mtEXO). It is composed of 2 large subunits: an RNase and an RNA helicase encoded by nuclear genes DSS1 and SUV3, respectively.Citation48 Lack of Suv3 or Dss1 attenuates RNA turnover, and also affects 5′- and 3′-processing of mitochondrial RNAs,Citation49 suggesting that turnover and processing of mitochondrial transcripts could be linked. All mRNAs of yeast mitochondria are processed at their 3′ ends within a conserved dodecamer sequence, 5′-AAUAAUAUUCUU-3′. It is relevant to mention that a dominant mutation in SUV3 gene was found to suppress a dodecamer deletion at the 3′ end of the VAR1 gene,Citation50 which is transcribed as part of a polycistronic transcript that contains tRNASer and ATP9.Citation44
In S. cerevisiae, the mtLSU 21S rRNA precursor is a ∼5.1–5.4 kb transcript containing a 1.2 kb intron (ρ1), tRNAThr2, tRNACys and tRNAHis.Citation44 The 3.1 Kb mature 21S rRNA transcript is formed by excision of the intron and removal of a ∼900bp extension from the 3′- end.Citation51 Processing of 21S rRNA transcripts containing the ρ1 intron requires the Suv3 helicase.Citation52,53 The 15S rRNA is also a part of a primary transcript containing tRNATrp. However, the single 15S rRNA precursor transcript is a 15.5S RNA with approximately 80 additional nucleotides at the 5′- end than the mature15S rRNA.Citation54 While it is not clear how the processing occurs, since no evidence for an endonucleolytic cleavage mechanism has been reported, it has been shown that the precursor is not processed in rho− mitochondria,Citation54 nor in isolated organelles,Citation48 suggesting that it may depend on functional mitochondrial translation and/or metabolism. The RNA binding PPR-protein called Dmr1Citation55 is required to maintain 15S rRNA integrity. Even though the 15.5S precursor was detected in dmr1Δ mutants, it is not clear whether Dmr1 is directly involved in 15.5S processing. The only known mitochondrial 5′–3′ exonuclease activity in S. cerevisiae is performed by Pet127Citation56,57 that processes the 5′ end of the 15S rRNA precursor. Interestingly, this processing is not essential for ribosome assembly and function, as pet127Δ mutants are respiratory-competent, even though they accumulate 15S rRNA with the unprocessed 5′ end.Citation56 Because pet127 mutants were isolated as partial suppressors of mutations affecting the COX3 translational activator Pet122,Citation58 it is possible that ribosomes with unprocessed 15S rRNA precursor exhibit slight changes in translation activation or specificity, yet these are not sufficient to significantly disrupt their function.
In humans, the mtDNA is a double-stranded molecule that encodes 13 proteins, 2 rRNAs (12S and 16S rRNAs) and 22 tRNAs. Mitochondrial transcription occurs on both DNA strands and gives rise to 3 polycistronic primary transcripts.Citation59 Two of these transcripts correspond to each whole strand. Most ORFs and rRNAs in these transcripts are punctuated by tRNAs.Citation60 Cleavage of tRNAs at their 5′ and 3′ ends by ribonuclease P (RNase P,Citation61 and the RNase Z-type enzyme ELAC2,Citation62,63 respectively, releases individual RNA species, which are subsequently matured.Citation38 The third transcript is smaller and contains, in consecutive order, tRNAPhe, 12S rRNA, tRNAVal and 16S rRNA, the last 3 being components of the mitoribosome. RNase P and ELAC2 are also involved in tRNAVal excision. It remains unknown whether and how tRNAVal excision is coordinated with assembly of 39S mtLSU.
rRNA-modifying enzymes
An important step in ribosome assembly is the modification of rRNA at conserved regions, frequently in the catalytic domains. These modifications can take place co-transcriptionally or immediately after transcription, while others occur when the rRNA is assembled in the pre-ribosomal particle.Citation64 rRNA modifications are of 3 major kinds, namely pseudouridylation, base methylation and 2′-O-ribose methylation. Pseudouridylation of rRNA is synthesized by pseudouridine synthases. Methylations are introduced by methyltransferases (MTases). Base methylations are carried out by specific MTases that recognize their targets directly.Citation65 Unlike in the eukaryotic system, where a small nucleolar RNAs (snoRNAs) is required to locate the target nucleotide, the prokaryotic rRNA modifications are performed by site-specific enzymes without the help of a guide RNA.Citation66 Similarly in mitochondria, MTases catalyze rRNA modifications without a guided small RNA.
Compared with the prokaryotic and the eukaryotic cytoplasmic ribosomes, which contain more than 30 and more than 200 modifications, respectively, the mitochondrial rRNA is scarcely modified. In S. cerevisiae, the mtSSU 15S rRNA lack methylated nucleotides and might have only one pseudouridylation site,Citation67 in which the position on the rRNA and the enzyme responsible remain unknown. S. cerevisiae mtLSU 21S rRNA has 3 modified nucleotides, a pseudouridine (ψ 2819) and 2 2′-O ribose methylations (Gm2270 and Um2791) located at the Peptidyl Transferase Center (PTC) domain. Much of the understanding of the yeast mtLSU 21S rRNA modifications was facilitated by the previous studies in E.coli 23S rRNA, in which 3 ribose methylations are found on universally conserved nucleotides within domain V located at PTC, G2251, C2498 and U2552. The corresponding methylation site of E.coli G2251 in yeast 21S rRNA is G2270. Gm2270 is methylated by a nuclear-encoded site-specific rRNA ribose MTase called Pet56 or Mrm1. Mrm1 does not co-sedement with the mtLSU but ribosome assembly is affected when Mrm1 expression is impaired.Citation68,69 Methylation by Mrm1 can proceed in naked RNA, indicating that methylation could occur early in the mitoribosome assembly pathway. The methylation site corresponding to E.coli U2552 in the yeast 21S rRNA is Um2791.Citation70 The MTase responsible for Um2791 is Mrm2, which methylates U2791 only when the 21S rRNA is assembled into the LSU, indicating that this modification probably occurs at a later stage of mitoribosome maturation. Mrm2 co-sediments with the mtLSU on sucrose gradientsCitation71; however, no data is currently available on the accumulation of ribosome assembly intermediates in the absence of Mrm2. The 21S rRNA has only one pseudouridylation at residue ψ2819 that is synthesized by the pseudouridine synthase Pus5. This residue, ψ2819, is conserved in the mtLSU rRNA from species such as humans and mouse and also in LSU rRNA from gram-negative bacteria (e.g., E. coli) but not in gram-positive bacteria (e.g. Bacillus subitilis) or in the 26S rRNA of eukaryotic cytoplasmic ribosomes. ψ2819 is located in the most highly conserved region of the PTC domain; hence, it is likely that the ψ2819 residue is playing a functionally important role during peptide bond formation.Citation72
Modifications in the mammalian mitochondrial rRNA were first mapped using hamster cells,Citation73 which include a total of 9 modifications. The mammalian mtSSU 12S rRNA contains 5 base methylations but does not have any methylated ribose residues. These modifications include 2 adenine di-methylations (A936 and A937), one uracil methylation U429 and 2 cytosine methylations (C839 and C841/C842), numbered according to human 12S rRNA numbering.
The two adenines (A937 and A938) at the stem loop structure of the 3′ end of the 12S rRNA are conserved in all domains of life, archaebacteria, eubacteria and eukaryotes.Citation74 In bacterial SSU rRNA these modifications are present in a region that contains the mRNA decoding center and the binding site for the large ribosome subunit,Citation75 suggesting the importance of this modification for translation regulation. In the mammalian mtSSU rRNA, the 2 adjacent adenines are methylated by mitochondrial transcription factor B (TFB1M), which is a functional homolog of bacterial MTase KsgA.Citation76 Studies in mice indicated that knocking out (KO) TFB1M produces embryonic lethality. A conditional TFB1M KO in the heart led to mitochondrial cardiomyopathy owing to complete loss of adenine dimethylation of the SSU rRNA, which led to impaired mitoribosome assembly and abolished mitochondrial translation.Citation76
The mammalian mitochondrial cytosine MTase NSUN4 is the homolog of E. coli YebU, known to methylate the cytosine at position 1407 of the SSU 16S rRNA. Remarkably, YebU cannot act on free rRNA, in vitro, but can exert its function only if the rRNA is assembled in the SSU. Studies in a conditional Nsun4 KO mice showed that NSUN4 is required for mitochondrial translation.Citation77 Deep sequencing bisulfate-treated RNA showed that NSUN4 methylates C911 in 12S rRNA of the SSU, a modification that is lost in Nsun4 KO mice. Further NSUN4 forms a stoichiometric complex with MTERF4, a member of the mitochondrial transcription termination factor family.Citation78 The 3D crystal structure of the human MTERF4-NSUN4 complex showed a very stable interaction between both subunits that occurs at the carboxy-terminus of MTERF4.Citation79 MTERF4 is composed of structurally repeated MTERF-motifs that form a nucleic acid binding domain, while NSUN4 lacks an N- or C-terminal extension commonly used by other related RNA MTases for RNA recognition. Instead, NSUN4 binds to the C-terminus of MTERF4 and a positively charged surface forms an RNA binding path across both MTERF4 and NSUN4, suggesting that both proteins in the complex together contribute to RNA recognition. Interestingly, NSUN4 is still capable of methylating its target (C911 in mice SSU rRNA) in Mterf4 KO mice.Citation77 However, deletion of Merf4 gene in the mice heart leads to inhibition of mitochondrial translation and prevents NSUN4 being targeted to the mtLSU. In the absence of MTERF4, the SSU and LSU levels are increased but mature mitoribosomes are not formed.Citation78 This implies that NSUN4 is a bi-functional protein, where NSUN4 alone methylates C911 in mtSSU 12S rRNA and also forms a complex with MTERF4 and targets mtLSUCitation77 to regulate mitoribosomal assembly.
Regarding uracil methylations in mtSSU rRNA, a single methylation at nucleotide 426 was identified in the 13S hamster mtSSU rRNA.Citation80 However, its presence has not been confirmed in other rodent or human mtSSU.
The human 16S rRNA has 3 2′-O-ribose methylations and one pseudouridylation. The three 2-O-ribose methylations are a Gm residue at G1145Citation81 and a UmGm at U1369G1370. These sites are conserved in both bacterial and cytoplasmic rRNA, in which they stabilize the RNA structure critical for the peptidyltransferase reaction and t-RNA recognition. The single pseudouridylation site is found at the U1397.
The three methyltransferases that modify the 16S rRNA are MRM1, MRM2 and RMTL1 (also called MRM3).Citation81 Specifically, MRM1 catalyzes Gm1145. However, MRM1 does not co-sediment with mitoribosomes in glycerol gradients but a small fraction of this protein was found at the bottom of the gradient where nucleoidsCitation81 and probably other large structures, such as the RNA granules, sediment. Both Um1369 and Gm1370 lie in the A-loop of the mtLSU rRNA. The A-loop is an essential RNA component of the ribosomal PTC that directly interacts with aminoacyl (A)-site tRNA. This structure is well conserved through evolution and the highly conserved nucleotides in the A-loop often contain 2′-O-ribose methylations. In human mitochondria, the enzyme responsible for the Um1369 is MRM2 protein, which is the ortholog of bacterial RrmJ and yeast Mrm2.Citation81,82 RNAi-mediated depletion of MRM2 decreases the stability of the mtLSU, impairs mitochondrial translation and consequently mitochondrial respiration.Citation82 Intriguingly, MRM2 was shown to interact not only with the mtLSU but also with the monosome,Citation81,82 suggesting a second function for this protein during mitoribosome maintenance or mitochondrial translation.
The Gm1370 is exclusive to the mammalian 16S rRNA and is not present in E. coli or yeast mitochondrial 21S rRNA. It is catalyzed by the MTase MRM3, which associates with the mtLSU, but not with the monosome.Citation81 RNAi-mediated depletion of MRM3 gave rise to mtLSU pre-ribosomal particles,Citation82 indicating that, if the pre-ribosomal particles were on pathway intermediates, yet to be demonstrated, MRM3 functions at a late stage of mitoribosome assembly.Citation82
Even though no pseudouridylated nucleotides were identified in the hamster mitochondrial LSU rRNA, the human 16S rRNA could have one pseudouridylated base at U1397 (according to human 16 rRNA numbering).Citation83 However, the enzyme responsible and the precise role of this modification are yet to be identified.
Factors required for formation of the ribonucleoprotein particle
In addition to modification and processing of the rRNAs, formation of the ribonucleoprotein particle also requires non-ribosomal proteins, largely known as ribosome assembly factors. In the prokaryotic and the eukaryotic systems where ribosome assembly is studied in depth, many of these assembly factors include GTPases and ATP-dependent RNA helicases. These enzymes consume energy by hydrolyzing nucleotide triphosphates and provide directionality and accuracy at specific stages during assembly. Concerning mitoribosome biogenesis only a handful of assembly factors have been identified so far, raising the need for more intensive investigation in this area of study.
GTPases
Members of the G-protein super family are key regulators of many cellular processes found in all domains of life. GTPases can be considered as molecular switch proteins as they alternate an active GTP-bound conformation, an inactive GDP bound form and an inactive intermediate apo-state.Citation84 These proteins are composed of a G-domain that has a characteristic fold and 4–5 highly conserved motifs (G1-G5) with additional N- and C- terminal domains.Citation43 G-proteins play important roles in ribosome assembly.Citation20 So far, 3 conserved G-proteins have been identified in mitoribosome assembly in yeast and humans, namely Mtg1, Mtg2 and Mtg3 (mitochondrial GTPases 1,2 and 3). In addition, the mammalian-specific GTPase named ERAL1 (Era-like 1) is also involved in mitoribosome biogenesis.
MTG1 or GTP-binding protein 7 (GTPBP7) is a conserved YiqF/YawG family protein. Prokaryotes have one MTG1 homolog, the RbgA protein, also termed YIqF.Citation85 RbgA homologs are widely present in gram-positive bacteria such as Bacillus subtilis, archaea, and in a few gram-negative bacteria (but not E. coli). RbgA proteins have a highly conserved GTP-binding domain and belong to a family of circularly permuted GTPases (cpGTPases) in which the highly conserved 5 GTPase motifs are aligned in an unusual order (G5-G4-G1-G2-G3).Citation86-88 In B. subtilis, deletion of the highly conserved N-terminal region inhibits cell growth even in the presence of wild-type RbgA, thus acting as a dominant-negative variant.Citation89 RbgA in B. subtilis is essential for cellular growth and participates in the late steps of 50S LSU ribosome assembly and maturation. It interacts with the 50S subunit in a GTP-dependent manner and its GTPase activity induces its dissociation from the ribosome.Citation90 According to in vitro studies the GTPase activity is stimulated more than 60-fold in the presence of the 50S subunit.Citation87,89-91 In RbgA depleted cells the mature 50S subunits are not formed; instead, a 45S complex (lacking ribosomal proteins –L16 and L36,Citation86,90-94 and reduced amounts of L27, L33, and L35Citation92) is accumulated. Spontaneous rbgA suppressors have been mapped to the gene encoding L6, and in vitro maturation assays have suggested that RbgA is necessary for correct positioning of L6 on the ribosome, prior to the incorporation of L16 and other late assembly proteins.Citation92
In eukaryotes, MTG1 was first identified in mitochondria in S. cerevisiae.Citation95 In yeast, an mtg1 null mutation induces a decrease in 21S rRNA and a severe defect in mitochondrial protein synthesis. This phenotype is partially suppressed by spontaneous mutations in the 21S rRNA conferring resistance to erythromycin,Citation95 thus linking the role of Mtg1 to mtLSU biogenesis. In B. subtilis, RbgA interacts with helix 38 (H38) of 23S rRNA and the mutual stabilization between the central protuberance and H38 ensures further maturation of the 23S rRNA.Citation89,96 H38 is conserved in the yeast 21S rRNA but studies exploring a possible interaction of MTG1 with H38 are still missing. In human, however, H38 has been lost and additional stabilizing interactions involve mitochondrion-specific MRPs and the tRNAVal.Citation12 Human MTG1 shares some common characteristics with its yeast homolog, such as: i) Mtg1 localizes to the mitochondria inner membrane facing the matrix side, where it associates with the mtLSU; ii) mtLSU stimulates the GTPase activity of Mtg1, while the intrinsic GTPase activity is undetectable; iii) silencing of MTG1 decreases mitochondrial translation activityCitation97; iv) heterologous expression of human MTG1 partially rescues the mitochondrial protein synthesis defect in yeast mtg1 null mutant cells,Citation95 suggesting a functional conservation of MTG1 from yeast to humans.
Mtg2 is an Obg family protein. The Escherichia coli Obg protein (ObgE) binds to the large and the small ribosomal subunit. Deletion of ObgE leads to a decrease in the 70S ribosome levels while increasing the 30S and 50S subunits, and accumulation of a 50S subunit assembly intermediates containing reduced amounts of L16, L33 and L34.Citation98 Yeast and human mitochondrial Mtg2 associate with the inner mitochondrial membrane facing the matrix, a location consistent with a role in mitoribosome assembly. Deletion of yeast mtg2 protein leads to a loss of mitochondrial DNA, which is often related to a defect in mitochondrial protein synthesisCitation99. Moreover, cells carrying temperature-sensitive mutant alleles are unable to carry out mitochondrial protein synthesis and contain lowered levels of mitochondrial ribosomal subunits. Sucrose gradient sedimentation patterns have shown co-sedimentation of Mtg2 with the mtLSU, indicating a role in the biogenesis of this subunit. Importantly, yeast Mtg2 was identified as a high copy suppressor of the thermosensitive respiratory defect of a 21S rRNA methyltransferase mutant, mrm2, suggesting that Mtg2 acts in the mtLSU assembly pathway at a step prior Mrm2. The accumulation of mtLSU assembly intermediates in yeast mtg2 mutants remains to be investigated.
The mammalian MTG2/ObgH1, also known as GTP-binding protein 5 (GTPBP5), specifically associates with the LSU. However, unlike the mammalian MTG1, only an intrinsic GTPase activity was detected in ObgH1. Interestingly, knockdown studies have not shown any clear indication that ObgH1 is involved in ribosome assembly.Citation97
Mtg3 belongs to the YqeH protein family. In Bacillus subtilis, the YqeH protein contains a circularly permutated GTP-binding domain (G4-G5-G1-G2-G3). YqeH binds to the 30S small ribosome subunit in a GTP/GDP-dependent fashion and is required for the efficient assembly of the 30S small subunit and 70S ribosomes. B. subtilis cells depleted of YqeH are greatly reduced in functional 70S ribosomes and 30S subunits.Citation55,100 In yeast, Mtg3 functions in the assembly of the mtSSU. A null mutation in mtg3 prevents the maturation of 15S rRNA and the consequent accumulation of a 15S precursor transcript.Citation101 Notably, the respiratory deficiency and 15S rRNA precursor processing defect in the mtg3 null mutant can be partially suppressed by overexpression of uL4, an mtLSU MRP located near the tunnel exit site. These data suggest that Mtg3, together with uL4, regulates assembly of the small subunit by modulating processing of the 15S rRNA precursor.Citation101
The human homolog of Mtg3 is C4orf14, a protein that interacts with the 28S mtSSU.Citation102 Sucrose gradient sedimentation analysis showed the protein to co-sediment with the mtSSU. Gene silencing of C4orf14 specifically affected components of the small subunit, leading to decreased mitochondrial protein synthesis. The GTPase activity of C4orf14 is critical for its interaction with the 28S mtSSU and other factors involved in mitochondrial translation. It was therefore proposed that C4orf14, with bound GTP, binds to components of the 28S subunit facilitating its assembly and GTP hydrolysis acts as the release mechanism. C4orf14 was also found to be associated with human mitochondrial nucleoids. C4orf14 is capable of binding to DNA in vitro and silencing of the C4orf14 gene in human cells led to mitochondrial DNA depletion. The association of C4orf14 with both mitochondrial translation factors and the mitochondrial nucleoid has suggested that the 28S mtSSU could be assembled at the mitochondrial nucleoid, enabling the direct transfer of mRNA from the nucleoid to the mitoribosome,Citation102 as it will be discussed in section 5.
Another mitochondrial member of the conserved GTP-binding proteins with RNA-binding activity is ERAL1, a homolog of Ras-like protein Era protein in E. coli, present in high eukaryotes. The bacterial Era binds near the 3′ terminus of the 16S rRNA within the 30S small ribosomal subunit and plays a crucial role in ribosome assemblyCitation103. Human mitochondrial ERAL1 was identified to coimmunoprecipitate with either mitochondrial transcription factor A (TFAM), a main component of mitochondrial nucleoids, or with the mtRRF (mitochondrial ribosome recycling factor). ERAL1 localizes in the mitochondrial matrix, associates with mitoribosomal proteins, binds to the mtSSU 12S rRNA and plays an important role in the formation of the 28S mtSSU.Citation104,105 Bacterial Era associates with a 3′ unstructured nonanucleotide immediately downstream of the terminal stem-loop (helix 45) of 16S rRNA. This site contains a highly conserved AUCA sequence directly upstream of the anti-Shine-Dalgarno sequence, which is conserved in bacteria. This has suggested that Era plays an essential role in ribosome maturation and quality control. In human mitochondria, the entire Era-binding region is absent from 12S rRNA and instead ERAL1 binds to a 33-nucleotide section on the 3′ terminal stem-loop region of 12S rRNACitation105 that contains 2 dimethylated adenine residues, as mentioned in the previous section. Furthermore, in contrast with the bacterial ortholog, loss of ERAL1 leads to rapid decay of nascent 12S rRNA,Citation104,105 consistent with a role as a mitochondrial RNA chaperone. It is worth noting that some phenotypes observed in human cells depleted of ERAL1 are similar to those in cells from mice devoid of TFB1M. They include dramatically reduced levels of the 12S rRNA, whereas mitochondrial mRNA levels remain largely unaffected or even increase. This has suggested that binding of ERAL1 to the 12S rRNA may require or have a greater affinity for the adenine dimethylated stem loop.Citation105
RNA helicases
RNA helicases bind and remodel RNA and RNP complexes in an ATP- (or NTP-) dependent manner. These proteins have a structurally conserved helicase core containing characteristic sequence elements and structural motifs. Additional domains confer specificity by recruitment to target RNPs, while the conserved helicase core interacts with RNA in a sequence-independent manner. One critical family of RNA helicases is the DEAD-box proteins, which received the name from the unique amino acid sequence Asp-Glu-Ala-Asp in one of their motifs (motif II). Members of the DExD/H-box family of ATP-dependent RNA helicases participate in virtually all aspects of cellular RNA metabolism, including pre-mRNA splicing, mRNA export, translation initiation, RNA turnover and ribosome biogenesis. One of the key differences of the DEAD box helicases from conventional helicases is their ability to unwind duplexes and/or remodel RNA-protein complexes locally within the strands in a non-processive way. The conventional helicases on the other hand act processively and translocate along single-stranded RNA in a defined polarity toward the duplex region.Citation106
The highly conserved helicase core in DEAD-box proteins consists of 2 virtually identical domains that resemble the bacterial recombination protein recombinase A (RecA) connected by a flexible linker. The two domains bend in such a way that a cleft is formed that harbors the binding sites for both ATP and RNA at the 2 opposite ends.Citation107-109 The cleft between the 2 domains must be closed to productively bind and hydrolyze ATP.Citation107-109 Frequently the helicase core is flanked by variable N- and C-terminal extensions that might be critical for the specific function of the individual protein. DEAD-box proteins acquire substrate specificity by interaction with co-factors that target the enzymes to their place of action.Citation110
The much-studied ribosomal assembly pathways of bacterial and eukaryotic systems have shown the involvement of many DExD/H-box proteins at various stages of the assembly process.Citation111 They often engage in unwinding of double-stranded RNA, assisting dissociation of RNA-binding proteins to favor recruitment of downstream factors or mediating local structural remodeling of pre-ribosomal complexes and providing accessibility for endo- or exonucleases and RNA modifying enzymes.Citation112
E.coli has 5 DExD/H-ATPases, namely DeaD/CsdA, RhlB, RhlE, DbpA and SrmB. Some of these proteins are multifunctional as they participate in ribosome assembly, RNA degradation and/or translational regulation.Citation20,113 DeaD/CsdA and SrmB participate in ribosome biogenesis, by associating with the 50S subunit, and the absence of either protein causes ribosome assembly defects that result in accumulation of 40S late assembly intermediates.Citation114,115 DbpA interacts with 23S rRNA (rRNA), specifically with helix 92, an interaction that stimulates the ATPase and helicase activities of the enzyme.Citation116,117 Although dbpa mutants have normal ribosome assembly, a dominant negative mutant induces the accumulation of a ribosome intermediate.Citation118 RhlE plays a role in the interconversion of rRNA-folding intermediates that are further processed by DeaD/CsdA or SrmB during ribosome maturation.Citation21 No obvious orthologues of these proteins are known in mitochondria.
With regard to mitoribosome assembly, participating DEAD-box helicases have been identified only recently. When our group performed an in silico screen in search of mitochondrial putative RNA helicases required for mitoribosome biogenesis and/or translation in S. cerevisiae, our focus was directed to Mrh4 (4th putative mitochondrial DEAD box RNA helicase). Mrh4 is essential for mtLSU biogenesis. Mrh4 interacts with the 21S rRNA, mitoribosome subassemblies and fully assembled mitoribosomes. In the absence of Mrh4, the 21S rRNA is matured and forms part of a large on-pathway assembly of intermediate missing proteins bL9, uL16 and bL33. In E. coli, L16 and L33 play important structural and perhaps catalytic roles.Citation119 Whereas L33 does not seem to play a major role in ribosome assembly,Citation120 L16 accelerates the late steps of in vitro assemblyCitation121 and induces a conformational change in the 50S, which may in turn affect the peptidyltransferase activity and subunit association of the ribosome.Citation122 The stable incorporation of these proteins is essential for ribosomal subunit association. Several bacterial ribosomal mutants accumulate 40S–45S particles, each lacking small sets of proteins that overlap with those missing in the Δmrh4 pre-54S particle. Particularly, the dominant-negative mutant of dbpa, when overexpressed in E. coli, induced a deficit in 50S subunits giving rise to a 45S particle containing reduced levels of L16, L25, L27, L28, L33, L34, and L35.Citation118 Several of the missing MRPs bind near the PTC, suggesting the requirement of a DbpA-mediated conformational change for the binding of these MRPs at this site.
For Mrh4 on the other hand, we have proposed that it plays an essential role during the late stages of mitoribosome assembly by promoting remodeling of the 21S rRNA-protein interactions.Citation123 Because Mrh4 was found to interact with the monosome, the possibility exists that it could perform additional roles during translation.
Looking for putative mammalian homologues of Mrh4, we performed cluster analysis of all identified DEAD/H helicases across species, which grouped yeast Mrh4 with mouse/rat/human DDX28. Heterologous expression of DDX28 in a yeast mrh4 null mutant, however, did not rescue its mitoribosome assembly defect.Citation123 DDX28 displays RNA-sensitive Mg2+-dependent ATPase activity in vitro.Citation124 It was shown that DDX28 has dual subcellular localization in monkey COS1 cells; whereas the majority of the protein permanently resides in mitochondria, a small portion was found in the nucleolus.Citation124,125 GFP-fused DDX28 was detected in punctate structures in COS1 cell mitochondria, about half of which co-localized with, or were adjacent to the mitochondrial nucleoids.Citation126 These results were confirmed in cultured human cells although biochemical studies localized most of the protein in mitochondria.Citation127 DDX28 interacts with the 16S rRNA and the mtLSU,Citation127 most probably with a helix in domain V.Citation128 RNAi-mediated DDX28 silencing in HEK293T cells does not affect mitochondrial mRNA stability, 16S rRNA processing or modification. However, it leads to reduced levels of 16S rRNA and mtLSU proteins, impaired mtLSU assembly and deeply attenuated mitochondrial protein synthesis.Citation127 Our findings have identified DDX28 essential for 16S rRNA stability, perhaps during the early stages of mitoribosome mtLSU biogenesis.
The DHX30 helicase also participates in mtLSU assembly, although its precise role still remains to be investigated. This protein localizes to foci in the vicinity of nucleoids,Citation129 the RNA granules.Citation128 Silencing of DHX30 in human cells produces a very severe translation defect that correlates with a profound loss of monosomes. DHX30 seems to interact with the mtLSU, mtSSU and the monosome, suggesting that it remains closely associated with the ribosome at all stages of assembly.
Other mitoribosome assembly factors
In mammalian cells, 2 mitochondrial transcription factor family proteins play roles in mtLSU assembly, perhaps coordinating transcription and ribosome biogenesis. The precise role of mTERFD1 (or mTERF3) in promoting mtLSU biogenesis is not understood,Citation130 but mTERF4 forms a complex with the rRNA methyltransferase NSUN4 to promote its recruitment to the mtLSUCitation78 in order to facilitate monosome assembly,Citation77 as mentioned earlier.
Moreover, C7orf30, a member of the DUF143 family of ribosomal silencing factors, participates in mtLSU assemblyCitation131-133 by interacting with uL14 and promoting its incorporation into the mtLSU.Citation132 Knockdown of C7orf30 by short hairpin RNA (shRNA) does not alter the sedimentation profile of the mtLSU, but results in mtLSU depletion and consequent decreased monosome formation that leads to a mitochondrial translation defect.
The matrix AAA-protease system is involved in the processing of the S. cerevisiae 54S mtLSU protein bL32, allowing its association with pre-ribosomal particles in the late stages of mtLSU assembly.Citation134 bL32 folding depends on its mitochondrial targeting sequence. The post-translocational processing by m-AAA protease is required as opposed to co-translocational cleavage by the general mitochondrial processing peptidase, as bL32 requires presequence-assisted folding for complete import.Citation134
The pre-54S intermediate that accumulates in the absence of Mrh4 contains bL32, which supports the late-stage nature of the intermediate and suggests the requirement of Mrh4 subsequent to the incorporation of bL32. The role of the AAA-protease system in mitoribosome maturation is conserved in mammals, since maturation of bL32 and mitochondrial protein synthesis are also impaired in a hereditary spastic paraplegia (HSP) mouse model lacking the m-AAA protease subunit paraplegin.Citation134 Expression of the human homolog proteins AFG3L2/SPG7 in a yta12/yta10 mutant yeast strain complements the lack of the yeast m-AAA protease,Citation135 further suggesting functional conservation.
GRSF1 (G-rich sequence binding factor 1) is the core component of mitochondrial RNA granules, where newly synthesized RNAs accumulate and where their processing, storage, sorting or translation is regulated.Citation136,137 Furthermore, a small portion of GRSF1 co-sediments with the mtSSUCitation127 and GRSF1 depletion induces a defect in the assembly of the mtSSU and leads to a marked attenuation of mitochondrial protein synthesis.Citation136,137 These observations indicate a role for GRSF1 in mtSSU biogenesis.
FASTKD2, a protein of the Fas-activated serine-threonine kinase (FASTK)- domain-containing family, also localizes to RNA granules, binds to the 16S rRNA and is required for mtLSU assembly.Citation128
Defining the Assembly Pathway for Biogenesis of Mitoribosomes in Yeast and Mammals
Lessons from studies in vitro and on bacterial ribosomes
A wealth of information is already available on the assembly of the bacterial ribosome and these studies can provide valuable hints as to how this process could occur in mitochondria, as well as to what approaches can be used for that purpose. Readers interested in bacterial ribosome biogenesis are referred to several recent excellent reviews.Citation20,88,111 In general, the assembly of ribosomes involves the coordinated processing and modification of rRNAs with the temporal association of ribosomal proteins. Both studies in vitro and in vivo have identified precursors and assembly factors that lead to the formation of the bacterial ribosome.
Despite the complex composition of ribosomal subunits, earlier pioneering work in the 1960s led by NomuraCitation138,139 and NierhausCitation140,141 demonstrated that active bacterial subunits could be reconstituted in vitro from individual unmodified rRNAs and proteins. The process did not require any additional components (i.e., assembly factors), but has a marked dependence on non-physiological temperature and ionic conditions. Through numerous measurements of binding interdependences among different proteins, they generated general in vitro assembly maps for the 30S and 50S subunits.Citation138-142 Their data revealed that ribosomal proteins are incorporated in different orders, displaying both hierarchical and parallel manners. For example, in the early stages of 30S assembly, primary ribosomal proteins bind directly to the 5′-, central and 3′-domains of 16S rRNA, inducing changes in the rRNA structure, and facilitating subsequent binding of secondary and tertiary ribosomal proteins.Citation139,143
More recent studies using time-resolved hydroxyl radical footprinting,Citation144 matrix-assisted laser description/ionization mass spectrometry (MS),Citation145 time-resolved electron microscopyCitation146 and in vivo studies of E. coli cells treated with neomycin,Citation147,148 have revealed that even in the presence of ribosomal protein binding dependencies, assembly can proceed through multiple parallel pathways generating heterogeneous populations of assembly intermediates.
Inside the cell, subunit assembly is highly efficient, as the process is facilitated by a variety of cofactors with diverse functions (nucleases, GTPases, RNA helicases, chaperones) that prevent the formation of kinetic traps during the assembly process. Disruption of cofactors by genetic deletion or mutation leads to growth defects and accumulation of subunit precursors, whose analyses by quantitative MS (qMS) have provided hints regarding the step in which they intervene and of rate-limiting steps in the assembly pathway (reviewed inCitation20). In the same line, studies based on systematic chromosomal deletion of ribosomal protein genes in E. coli have shown that although most ribosomal proteins are essential, several proteins (L9, L15, L21, L24, L27, L29, L30, L34, S9, and S17) appear to be dispensable for ribosome assembly and translation. However, these proteins can be important for efficient and accurate assembly and protein synthesis capacity under some conditions.Citation41 These studies, combined in some cases with Cryo-EM structures of usually late-stage assembly intermediates, have provided an idea about conformational maturation of the rRNA and of the growing ribonucleoprotein particle.Citation93,96 Until recently the identification and characterization of in vivo assembly intermediates in wild-type bacterial cells have been challenging owing to the fewer number of ribosomes undergoing biogenesis (2–5%), the rapid kinetics and asynchronous transcription from rDNA loci.Citation149 Nevertheless, with the use of high-resolution high-throughput qMS methodologyCitation19,150 the Williamson group was able to characterize in vivo ribosome assembly intermediates and associated assembly factors in wild-type E. coli cells. They analyzed the composition of sucrose gradient fractions prepared using an in vivo stable isotope pulse-labeling (SILAC) approach to distinguish degradation products and dead-end assembly intermediates from on-pathway intermediates.Citation19,150 This methodology provided snapshots of the assembly intermediates that confirmed 4 subassemblies for the 30S subunit and 6 50S precursors that correlated well with the in vitro reconstitution data, although they were not identical to the ones found in vitro.Citation19
For the 30S subunit, analyses of sucrose gradient fractions revealed the existence of 5 subassemblies, whose composition is summarized in .Citation19,146 In one of the reports, sucrose gradient fractions were analyzed in parallel using single-particle negative stain EM to elucidate the structures of assembly intermediates and mature ribosomes present in each sample, which revealed the heterogeneity of the 5 assembly intermediates.Citation146 Another profitable approach to study the 30S assembly pathway, applied by Culver's group, consists of aptamer-based RNA-tagging and affinity-purification systems to isolate SSU or LSU assembly intermediates from wild-type cells.Citation151,152 This strategy elucidated one predominant specie of pre-16S rRNA (17S rRNA) to be associated with all purified assembly intermediates in the SSU assembly pathway.Citation152 As in Williamson's experiments, Culver's group observed several independent pathways for maturation of 17S rRNA in SSU subassemblies, results that further suggest that flux through SSU biogenesis involves multiple pathways.Citation152
Figure 2. Mitoribosome assembly lines. Assembly pathway of the (A) mitochondrial large subunit (SSU) and (B) mitochondrial small subunit (SSU). The assembly pathways for the bacterial subunits are presented as a reference. Only some ribosome biogenetic factors are included. The assembly stage at which they act is depicted only approximately.
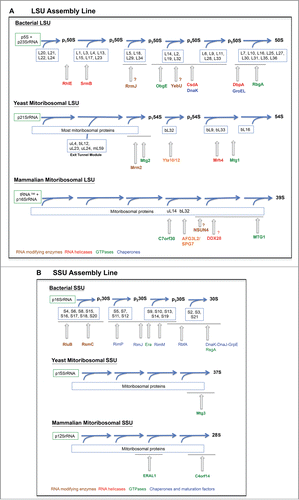
For the 50S subunit, the qMS approach identified 6 distinct assembly groups of ribosomal proteins,Citation19 whose composition is also depicted in . These studies have shown that the central protuberance assembles last.Citation19 This has suggested that key functional sites of the 50S subunit, such as the tRNA binding sites, remain unstructured until the late stages of maturation, preventing the incomplete subunit from prematurely engaging in translation.Citation93
Current knowledge on mitoribosome assembly pathways
Unlike the bacterial ribosome, the mitochondrial ribosome is permanently bound to the inner mitochondrial membrane in order to facilitate protein synthesis with membrane insertion,Citation153 and the assembly process has been proposed to occur in contact with the inner membrane.Citation123,154 Even though during the last decade information on mitoribosome assembly factors and the complement of MRPs was greatly enhanced, a detailed map of the assembly pathway has yet to be determined (). The delay in information could be due to 2 major causes. First, yeast ribosome assembly mutants tend to lose their mitochondrial DNA, therefore complicating their analyses. Second, the mammalian ribosome assembly mutants described so far apparently do not accumulate assembly intermediates that could be characterized.
The challenge of yeast has been bypassed by the use of thermo-sensitive mutants that allow for controlled induction of the mutant phenotype.Citation95,101 A better approach has resulted from the identification of genetic suppressors that maintain mtDNA stability in the absence of mitoribosome assembly, thus allowing the study of assembly intermediates.Citation123 Also, expression of some truncated ribosome proteins have resulted in ribosome assembly lesions without affecting mtDNA stability.Citation154 Three studies have demonstrated the existence of late-assembly intermediates, which are tethered to the inner mitochondrial membrane. First, the LSU r-protein bL32 is imported into the mitochondrial matrix as a precursor that needs to be proteolytically processed by m-AAA proteases Yta10/Yta12 prior to bL32 insertion into the mitoribosome.Citation134 In the absence of the protease, LSU assembly is perturbed at a late step prior to bL32 incorporation.Citation134 As a result a membrane-bound large pre-54S assembly intermediate is accumulated in yta10/yta12 mutants. Second, deletion of the C-terminal mitochondria-specific domain of uL23 (mrp20ΔC) results in accumulation of a membrane-bound pre-54S assembly intermediate of approximately 150kDa.Citation154 Interestingly, this intermediate is composed of uL23, uL24, uL4, and mL41,Citation154 all located at the LSU tunnel exit siteCitation13,35 as well as mL59. Hence, it has been proposed that different modules of the 54S ribosome could assemble independently before completion of the full ribosomal subunit. And third, a recent report by our group on the characterization of Mrh4 described the accumulation of a membrane-bound on-pathway pre-54S late-stage assembly intermediate (containing bL32), missing only 3 late assembly MRPs, namely bL33, uL16 and bL9 in the absence of the DEAD-box helicase.Citation123 Putting together this data, the assembly map that can be drawn for the 54S LSU particle is extremely incomplete () and thus further in-depth investigation in this area of research is much needed. Currently, no assembly intermediates for the yeast 37S SSU have been identified.
Studies on human mitoribosome assembly have adapted the proteomics approach developed to study the in vivo assembly of bacterial ribosomes.Citation155 The only available SILAC pulse-chase experiments have discriminated the newly synthesized proteins enriched in the nucleoid fraction (and its vicinity) in comparison to the ribosome-enriched fraction. Higher resolution is required to identify mitoribosome assembly intermediates.
Compartmentalization of Mitoribosome Assembly
Nucleoids, RNA granules and mitoribosome assembly
A recent breakthrough in mitoribosome assembly relates to the mitochondrial compartmentalization of this process. At least 3 distinct types of foci relevant to mtDNA expression have been identified and visualized within the mitochondrial matrix of human cells. Those are the mitochondrial nucleoids, RNA granules and the RNA degradosome.Citation156 Here we will discuss the available data suggesting that the nucleoid and the RNA granule could be the factories where mitoribosomes are assembled.
The mammalian mitochondrial nucleoid is a nucleoprotein complex that contains mtDNA and proteins involved in mtDNA maintenance, replication and transcription.Citation129,157-160 Within the nucleoid, which can contain up to 5–6 mtDNAs, each genome replicates and transcribes independently of the others.Citation161 Early studies aiming to identify proteins associated with mt-nucleoids were based on affinity purification of known nucleoid components (mitochondrial transcription factor A (mtTFA or TFAM) and mitochondrial single strand binding protein (mtSSB)) followed by mass spectrometry. They identified proteins involved in mtDNA metabolism and additionally many mitoribosome proteins and proteins required for mitochondrial translation such as ATAD3,Citation162 suggesting an intimate association between nucleoids, components of the mitochondrial translation machinery and foci containing ribonucleoprotein complexes. For example, the human GTPase C4orf14 that functions in the assembly of the 28S mtSSU, associates with mitochondrial translation factors and the mitochondrial nucleoid.Citation102 Furthermore, the absence of MPV17L2, a protein proposed to be required for the assembly of the monosome and whose stability depends on mtDNA, induces the trapping of 28S mtSSU proteins in enlarged nucleoids.Citation163 This has suggested the hypothesis that the 28S subunit is assembled at the mitochondrial nucleoid, enabling the direct transfer of mRNA from the nucleoid to the mitoribosome.Citation102 Similar observations were recently reported in a proteomics study that found that mitochondrial RNA processing enzymes involved in tRNA excision - ribonuclease P (RNase P),Citation61 and the RNase Z-type enzyme ELAC2,Citation62,63 as well as a subset of mitochondrial ribosomal proteins (MRPs) - associate with nucleoids or foci located near the nucleoids to initiate RNA processing and ribosome assembly.Citation155 In an important experiment, formaldehyde cross-linking was performed to determine the nucleoid proteins that are in close contact with the mtDNA.Citation129 A set of core nucleoid proteins was found in both native and cross-linked nucleoids, including 13 proteins with known roles in mtDNA transactions. Several other metabolic proteins and chaperones identified in native nucleoids, including ATAD3 and ribosomal proteins, were not observed to cross-link to mtDNA.Citation129 These results suggested a model for a layered structure of mtDNA nucleoids in which replication and transcription occur in the central core, whereas RNA processing and translation may occur in the peripheral region.Citation129
However, analysis of bromouridine (BrU) labeling of newly transcribed RNA in human mitochondria has identified distinct BrU-positive RNA foci in close proximity to the nucleoids.Citation161 These foci, recently termed RNA granules, contain the protein GRSF1 (G-rich sequence binding factor 1) and RNase P, and accumulate mtRNAs to regulate their processing, storage, sorting or translation.Citation136,137 These foci also contain ribosomal proteinsCitation137 and rRNA modifying enzymes.Citation164 Furthermore, GRSF1 depletion induces a defect in the assembly of the mitoribosomal SSU and a marked attenuation in mitochondrial protein synthesis.Citation136,137 Consistently, our sucrose gradient fractionation data has shown co-sedimentation of GRSF1 with mtSSU markers.Citation127 This has raised the possibility that ribosome biogenesis could occur near or within the RNA granules. Affinity purification of tagged GRSF1 identified ribosomal proteinsCitation137 and the 16S rRNA methyltransferases MRM1, MRM2 and MRM3/RMTL1.Citation164 Affinity purification of tagged MRM3 co-eluted ribosomal proteins and ribosome assembly factors that co-localize to the RNA granules, such as GRSF-1 and the DEAD-box helicase DDX28.Citation164,165 Recent comprehensive affinity purification studies of GRSF1 and DDX28 followed by mass spectrometry analysis by 2 independent groups have yielded similar results.Citation127,128 In the study performed by our group, DDX28 was found associated with 4 major groups of relevant proteins.Citation165 The first group included most mitoribosomal proteins (45 mtLSU and 29 mtSSU MRPs). The second group was comprised of several previously identified factors involved in the biogenesis of the mtLSU (MRM3, MTG1, C7orf30, FASTKD2 and MTERF-D1), the mtSSU (ERAL1) or both (DHX30), together with a set of potential mitoribosomal assembly/maintenance factors including proteases, GTPases and chaperones. The third group involved mitochondrial RNA metabolism proteins, including GRSF1, LRPPRC, and SLIRP involved in the stability, polyadenylation and coordination of translation of mitochondrial mRNAsCitation166,167; RNase P; the 16S methyltransferase RMTL1/MRM3; the pentatricopeptide repeat domain protein 1 (PTCD1) involved in the processing of mitochondrial polycistronic transcripts that contain leucine tRNACitation168; and the translational regulator PTCD3.Citation150 And the fourth consisted of a set of translation factors and most aminoacyl tRNA synthetases. These studies also identified other proteins previously associated with mitochondrial nucleoids, e.g., the helicase DHX30 that acts as a transcriptional regulator, and ATAD3 mentioned earlier.
The overlapping pool of proteins associated with either mitochondrial nucleoids or RNA granules reflects their proximity and the lack of membranous boundaries sealing these compartments. Their functional and physical dynamics is highlighted by the observation by fluorescence microscopy that approximately 10–15% of nucleoids and RNA granules do actually co-localize in most studies, probably reflecting sites of active transcription.Citation136,137,156,161 A quantitative assessment of these dynamics showed that after an initial short pulse of 20 min, most BrU-positive RNA foci colocalize within 200 nm of mtDNA, but after longer periods (or a pulse-chase) the BrU-positive RNA foci become distributed randomly.Citation137 Thus, mitoribosome assembly could be initiated within or near the nucleoids,Citation155,163 possibly in a co-transcriptional manner as it occurs in bacteria.Citation20 SILAC pulse-chase experiments showed that most newly synthesized mitoribosome proteins from the 2 subunits were initially recruited to the nucleoid-containing fraction and the authors concluded that the first steps in ribosome biosynthesis occurred at the nucleoids.Citation155 However, it is more likely that RNA granules co-sedimented with the nucleoids in sucrose gradients and therefore the SILAC results cannot exactly discriminate where mitoribosome assembly occurs. Studies of the composition of human DDX28Citation165 and GRSF interactomesCitation128 mentioned earlier point toward the conclusion that most steps of mitoribosome biogenesis could actually occur in the RNA granules. It is necessary, however, to stress the concept of RNA granules as dynamic structures. As portrayed in , newly transcribed rRNAs and/or early mitoribosome assembly intermediates are probably transferred from nucleoids to the RNA granules, where mitoribosome assembly is completed. These mitochondrial matrix subcompartments are therefore reminiscent of the nucleolus. Within the nucleus, the membrane-less nucleolus is organized around the chromosomal regions that contain the genes for the rRNAs, and is the site of rRNA transcription and processing, and of ribosome assembly. Equivalent features pertain to the mitochondrial RNA granule, which we have proposed to term “the mitochondriolus” ().Citation127
Figure 3. Compartmentalization of mitoribosome biogenesis. The processes of mtDNA replication and expression occur in submitochondrial matrix compartments. The mtDNA and proteins involved in mtDNA metabolism form a nucleoprotein complex called the mitochondrial nucleoid. Upon transcription, RNAs are sorted at the mitochondriolus (RNA granule), where ribosome assembly largely occurs.
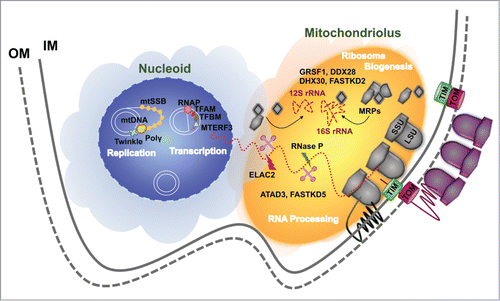
Furthermore, the nucleoids are tethered directly or indirectly through both mitochondrial membranes to kinesin, and to microtubules in the surrounding cytoplasm.Citation161,169 On the other hand, the RNA granules colocalize with components of the cytoplasmic machinery that imports nuclear-encoded proteins such as the translocase in the outer membrane (TOM22). Therefore, it has been hypothesized that the mitochondrial nucleoids and RNA granules could organize the mitochondrial and cytoplasmic translation machineries on both sides of mitochondrial membranes to facilitate efficient assembly of proteins synthesized by the 2 distinct translation machineries into mitochondrial complexes.Citation161
In yeast, the mitochondrial gene expression system is organized in distinct ribosome-containing clusters.Citation170 Similar to what has been described in mammalian cells, a subset of these clusters is engaged in a large complex with or near the nucleoid,Citation170 reminiscent of the mammalian mitochondrial RNA granules. Studies of BrU-RNA staining in yeast mitochondria, currently missing, will help clarify whether the “mitochondriolus” appeared early in eukaryotic evolution and whether this is the universal mitoribosome factory.
Membrane localization of mitoribosomes and mitoribosome assembly
Yeast and mammalian mitochondrial ribosomes are bound to the inner membrane, presumably as a result of their specialization in the synthesis of hydrophobic membrane proteins, which are co-translationally inserted into the inner membrane (2). Even synthesis of the yeast ribosomal subunit Var1 is membrane-localized.Citation171
In yeast, several inner membrane proteins mediate membrane binding of ribosomes such as those in the Oxa1 machinery, which facilitates the insertion of nascent polypeptides into the inner membrane.Citation30-36 Oxa1 interacts with the mitoribosomal LSU via its C-terminal α-helix ribosome-binding domain.Citation30,32 Also Mba1 and Mdm38 tether the mitoribosome to the membrane in both translationally active and inactive statesCitation34,36 and additionally exhibit overlapping regulatory functions in translation of selected mitochondrial mRNAs.Citation34 Studies of the proteins located at the polypeptide tunnel exit of the yeast mitochondrial ribosome showed that Mba1 is positioned in proximity to the ribosomal proteins uL29 and uL22.Citation35 This sub-ribosomal localization of Mba1 differs from that of Oxa1, which is located near uL24 and uL23.Citation30,31 Furthermore, yeast mitoribosomes remain docked to the membrane even in the absence of Oxa1 machinery associated with the tunnel exit, indicating that additional and as yet uncharacterized anchors must exist.Citation172 The yeast LSU structure has shown that the LSU central protuberance has a membrane-facing location, which makes it a candidate to be involved in the tethering of mitoribosomes to the membrane.Citation13
On the other hand, the 4.9 Å Cryo-EM reconstruction of the mitoribosomal 39S LSU subunit from porcine (Sus scrofa) liver tissue has indicated that the mammalian mitochondrion-specific mL45 (MRPL45) subunit, localized at the polypeptide tunnel exit, anchors the mitoribosome to the inner mitochondrial membrane.Citation15 mL45 displays homology to the C-terminal domain, membrane-binding segment of TIM44, a subunit of the inner membrane translocase essential for protein import.Citation173 Notably, mL45 also presents homology to the yeast mitochondrial protein Mba1,Citation6 which tethers the mitoribosome to the inner membrane as discussed earlier. Thus, mL45 may bind the mitoribosome to the inner membrane in a manner that would expose the translated nascent polypeptide to the solvent matrix milieu,Citation15 making it accessible to specific maturation and assembly factors involved in the biogenesis of OXPHOS complexes. As in yeast, mitoribosomal membrane-anchoring would also facilitate alignment of the nascent polypeptide tunnel exit with mammalian OXA1L (homolog of yeast Oxa1) to facilitate co-translational membrane insertion of newly synthesized proteinsCitation174,175
Recent data has suggested that the inner mitochondrial membrane could act as a platform for the ribosome assembly process.Citation123,154 The expression of a truncated form of uL23 (Mrp20) in yeast Δmrp20 cells resulted in the accumulation of a ribosomal assembly intermediate composed of tunnel-exit-site proteins, which was found associated with the inner membrane.Citation154 Likewise, the late pre-54S particle that accumulates in the absence of the DEAD-box helicase Mrh4 is associated with the inner membrane. Strikingly, similar membrane-binding behavior occurs for non-assembled ribosomal proteins in cells devoid of mtDNA (rho zero cells) and thus of mtrRNA, which suggests the possibility of specialized membrane-associated compartments to which ribosomal proteins become targeted after their import into mitochondria,Citation123,154 prior to being recruited for binding to mtrRNA or perhaps where ribosome assembly actually occurs.
Defective Ribosome Assembly and Human Disease
Defects in mitochondrial translation are responsible for devastating human disorders (). They constitute a specific class of mitochondrial diseases, biochemically characterized by alterations in the 4 OXPHOS enzymes that contain mtDNA-encoded subunits. In this group of disorders, mutations in most mtDNA-encoded tRNAs, as well as in nuclear genes encoding mitoribosomal proteins, translation initiation and elongation factors, are responsible for clinically and genetically heterogeneous infantile multisystemic diseases, such as Leigh's syndrome, sensorineural hearing loss, encephalomyopathy and hypertrophic cardiomyopathy.Citation22-24,176 The existence of many patients afflicted by mitochondrial disorders with multienzymatic OXPHOS defects of unknown genetic cause predicts that numerous genes involved in the biogenesis and function of the mitochondrial translation machinery remain to be identified.
Table 3. Mitoribosome assembly and human diseases. CPEO, chronic progressive external ophthalmoplegia; MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes; MNGIE, Myo-, neuro-, gastrointestinal encephalopathy. For most mutations, only one or a few references were included. A complete set of references can be found in MitoCartaCitation221
Focusing on changes in ribosomal components associated with human diseases, only a few mutations have been so far described in rRNAs and mitoribosomal proteins. On the contrary, several mutations have been identified in the MT-TV gene encoding for tRNAVal (see ).
Mutations in the 12S rRNA are an important cause of hearing loss. Specifically, the homoplasmic A1555G and C1494T mutations at the highly conserved decoding site of the 12S rRNA gene are well documented as being associated with either aminoglycoside-induced or nonsyndromic hearing loss in many families of various ethnic backgrounds.Citation177-180 Aminoglycosides (such as gentamicin, kanamycin, and streptomycin) are used as antibacterial therapeutic agents that inhibit protein synthesis by interacting with the A-site of the 16S rRNA in the 30S ribosomal subunit.Citation181,182 They exert pleiotropic actions during the 3 phases of protein synthesis. They affect initiation, perturb peptide elongation at the 30S ribosomal subunit and impair translational proofreading, leading to misreading of the RNA message, premature termination, or both.Citation183,184 In the human mitochondrial 12S rRNA A-site, C1494 forms a non-canonical RNA base pair with A1555.Citation185 The bacterial sensitivity to aminoglycosides involves their direct binding to a 16S rRNA C–G base pair at positions 1409–1491.Citation186 The A1555G transition results in a decrease in flexibility of the decoding center and makes the secondary structure of rRNA more similar to the corresponding region of E. coli 16S rRNA therefore contributing to create a binding site for aminoglycoside drugs.Citation25,26,187 The effect of creating an A-T pair by the C1494T transition is apparently similar.Citation25,26 As a consequence, exposure to aminoglycosides can induce or aggravate hearing loss in individuals carrying one of these mutations. Phenotypic expression of the deafness-associated homoplasmic A1555G mutation varies from profound congenital hearing loss to normal hearing.Citation177,188 This variability in clinical expression in most patients is due to the complex inheritance of multiple nuclear-encoded modifier genes. Such modifier genes include the mitochondrial transcription factor B1 (TFB1M),Citation189 which methylates adenine residues in the adjacent loop of the A1555G mutation in the 12S rRNA gene.Citation74 Other modifiers are the genes MTO1Citation190,191 and GTPBP3Citation192 - human homologs of yeast MTO1 and MSS1 genes, respectively. Yeast Mto1 and Mss1 form a heterodimer complex, which interacts with the small mitochondrial ribosomal subunit at a site in the small rRNA close to or inclusive of paromomycin-resistance mutations, and may play a role in optimizing mitochondrial protein synthesis by a proof-reading mechanism.Citation193 Each human gene complements the respective yeast mutant, what indicates functional conservation during evolutionCitation190,192 and explains their roles as disease modifier genes.
While numerous patients carrying mutations in the 12S rRNA have been described, only 2 mutations in the 16S rRNA associated with a primary mitochondrial disorder have been reported. The first case, a young woman with severe myopathy, presented ragged red fibers with enlarged abnormal mitochondria, profound and combined deficiency of OXPHOS complexes, with the exception of complex II, not accompanied by mtDNA depletion.Citation194 An almost homoplasmic G>A substitution at the 3090 conserved position in the 16S rRNA was detected by sequencing in muscle, the more affected tissue, but was absent in fibroblasts, follicles and blood cells. The mutation was not detected in the blood and urine cells of the patient's mother, suggesting a de novo origin during embryonic development.Citation194 More recently, the mutation T2336C has been associated with hypertrophic cardiomyopathy in the 4 affected members of a Chinese family.Citation195 The mutation is homoplasmic in blood, urine, hair follicles and oral epithelium and it is predicted to disturb the T2336-A2438 base pairing in the first position of a stem loop in the 16S rRNA domain III. In lymphoblastoid cell lines derived from these patients, mitochondrial morphology is altered and oxygen consumption is reduced by approximately 37%.Citation195 However, data on the status of mitoribosome assembly or mitochondrial translation efficiency are not available for either one of the 2 pathogenic mutations reported until now in the 16S rRNA.
Mitochondrial tRNAVal is a recurrent target for mutations causing mitochondrial disorders. Reported nucleotide variants in the MT-TV gene include: (i) Heteroplasmic G1606A, affecting the acceptor stem of tRNAVal, which is associated with adult onset complex neurologic syndrome that included hearing loss, cataracts, ataxia, myoclonus, and dementiaCitation196,197; (ii) Homoplasmic C1624T affecting a base pair in the dihydrouridine loop produced multiple neonatal deaths and one surviving child with Leigh syndrome.Citation198 In one of the deceased patients, a selective and severe reduction of the steady-state mttRNAVal level was found in cardiac and skeletal muscle; (iii) Homoplasmic C1628T in a child presenting mitochondrial encephalocardiomyopathy.Citation199 Intriguingly, the patient had isolated complex I deficiency in skeletal muscle,Citation199 although complex V was not measured and the biochemical defect could be deeper in affected tissues. In all other patients reported and mentioned below, tRNAVal mutations caused a combined respiratory chain deficiency; (iv) Heteroplasmic A1630G in a patient with MNGIE-like gastrointestinal dysmotility and cachexiaCitation200; (v) Heteroplasmic G1642A associated with MELASCitation201,202; (vi) G1644A has been found in several patients. It results in progressive encephalocardiomyopathy with adult onset when present in homoplasmyCitation199 or when present in heteroplasmy, with adult onset Leigh syndromeCitation203 or with MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodesCitation204). In the homoplasmic case, it was reported that the levels of mutant tRNAVal were ∼20% of controls, suggesting that it was probably degraded. And (vii) heteroplasmic T1658C mutation was identified in a patient with chronic progressive external ophthalmoplegia (CPEO) in which the T loop structure of mitochondrial tRNAValCitation205 was changed. From all these studies, only 2 reported the levels of mutant tRNAVal, which were found markedly decreased. At present, there are no reports analyzing the effect of these mutations on mitoribosomal assembly.
Reported mutations in genes coding for mitochondrial ribosomal proteins affect 3 SSU subunits: uS7 (MRPS7), bS16 (MRPS16) and uS22 (MRPS22), and 3 LSU subunits, uL3 (MRPL3), bL12 (MRPL12) and mL44 (MRPL44). A homozygous mutation in MRPS7 was described in 2 affected siblings from one family suffering from congenital sensorineural deafness and progressive hepatic and renal failure.Citation206 A homozygous mutation in MRPS16 was described in one family with agenesis of corpus callosum, dysmorphism and fatal neonatal lactic acidosis.Citation207 MRPS22 mutations lead to cardiomyopathy, hypotonia and tubulopathy in a first familyCitation208 and Cornelia de Lange-like dysmorphic features, brain abnormalities and hypertrophic cardiomyopathy in a second family.Citation209 MRPL3 mutations were identified in 4 siblings of the same family presenting cardiomyopathy and psychomotor retardation.Citation210 A homozygous MRPL12 mutation was associated with growth retardation and neurological deterioration in a single patient born to consanguineous parents.Citation211 Finally, a homozygous MRPL44 mutation was identified in 2 siblings suffering from recessive hypertrophic cardiomyopathy.Citation212 Since the mammalian mitoribosome (55S) contains more than 80 protein components that make up the 28S SSU and 39S LSU, it is likely that more pathogenic mutations in the constituent polypeptides will be uncovered in the near future. Notably, the A181V amino acid substitution in bL12 affects a highly conserved residue in the C-terminal domain of the protein, whose bacterial homolog has been involved in the interaction of the ribosome with translation elongation factors,Citation213 suggesting a potential detrimental effect of the mutation on mitochondrial translation efficiency and/or accuracy.Citation211 The information gained through the study of prokaryotic ribosomes can be highly valuable in the analysis of conserved features. However, uS22 and mL44 are mitochondrion-specific subunits, and the uL3 reported missense mutation is located in a mitochondrial C-terminal extension of the protein absent in the E. coli homolog subunit, hampering homology modeling.Citation210
Remarkably, mutations in SSU and LSU proteins lead to decreased steady state levels of 12S rRNA and 16S rRNA, respectivelyCitation206,207,209-212 and apparent failure to accumulate assembly intermediates. Indeed, MRPL12 and MRPL44 mutant fibroblasts present with a reduction in fully assembled LSU without accumulation of subassembly particles at least containing the few subunits tested by immunoblotting (uL3, bL12, mL44 and uL13).Citation211,212 Unfortunately, no information on the mitochondrial ribosome assembly status is available for SSU pathogenic mutants. In cells and/or tissues from patients carrying mutations in mitochondrial ribosomal subunits, 12S and 16S rRNA amounts tightly correlate with the levels of residual mitochondrial translation and OXPHOS complexes, as well as with the severity of the clinical phenotype. In the case of the homozygous MRPS16 mutation, the patient died at 3 days of age of untreatable acidosis. The decrease of 12S rRNA steady-state level to 12% of control was accompanied by a dramatic defect in mitochondrial protein synthesis and a combined deficiency of OXPHOS complexes, with the exception of complex II, in muscle and liver homogenate.Citation207 The known pathogenic mutations in LSU subunits are instead associated with a partial reduction in 16S rRNA level, which results only in a moderate decrease of mitochondrial translation.Citation210-212 It is intriguing that this mildly reduced efficiency in mitochondrial gene expression does not affect equally the biogenesis of all OXPHOS complexes. In most cases, patients' muscle and fibroblasts present with a combined complex I – complex IV or isolated complex IV deficiency, while activities and steady state levels of complexes II and III are unaffected.Citation211,212 Complex V is additionally reduced in MRPL3 mutant cells.Citation210 Taken together, these observations suggest that the expression of mitochondrial-encoded complex IV subunits is more susceptible to partial deficiencies of the mitochondrial translational apparatus. Moreover, 2 siblings have been reported carrying the same homozygous mutation in MRPL44.Citation212 While the first patient died at 6 months of age of cardiac failure, the second one was an asymptomatic teenager at the time of publication. Fibroblasts from the latter showed a partial reduction of the fully assembled LSU and a dramatic complex IV defect despite an overall unaffected rate of mitochondrial translation, suggesting the existence of additional genetic and environmental factors responsible for the phenotypic variability.Citation212
Until present, no mutations have been found in rRNA modification enzymes but there are patients carrying mutations in mitoribosome assembly factors. A mutation in FASTKD2 was found in a patient with mitochondrial encephalopathy associated with an isolated complex IV deficiency,Citation214 although the molecular mechanism involved was not investigated. Mutations have been also identified in the 2 components of the heterodimeric AFG3L2/SPG7 mitochondrial m-AAA protease, also called paraplegin. The yeast Yta12/Yta10 m-AAA homolog protease is involved in protein quality control and additionally regulates ribosome assembly by controlling proteolytic maturation of the LSU protein bL32,Citation134 as explained earlier. Loss of function mutations in AFG3L2 result in either spinocerebellar ataxia type 28Citation215,216 or spastic ataxia.Citation217 SPG7 mutations are instead a common cause of hereditary spastic paraplegia,Citation218 although they have been also identified in cases of optic neuropathy and progressive external ophthalmoplegia.Citation219,220
Concluding Remarks and Perspectives
The high-resolution cryo-EM data has revealed unexpected differences between bacterial and mitochondrial ribosomes. The evolution of the mitoribosome is reflected by the differences observed between the yeast and mammalian counterparts. The structural knowledge is expected to speed up our understanding of how mitoribosomes are assembled by facilitating the generation of hypotheses of how specific stages of the process may occur. Mitoribosome biogenesis is complicated by the dual genetic origin of their components. The synthesis of rRNAs within the mitochondrial matrix should ensure co-transcriptional ribosome assembly, perhaps initiating near the nucleoid and finishing at the mitochondriolus. The precise compartmentalization of the different ribosome biogenesis stages needs further investigation. The list of mitoribosome assembly factors, although incomplete, keeps growing. We are still facing the necessity to expand the list, but importantly also the challenge to determine their precise molecular functions, which is most cases remain to be elucidated. New approaches, imported from the bacterial field and adapted to mitochondrial experimental needs, seem necessary. For example, SILAC-based approaches should allow characterizing the mitoribosome assembly landscape by quantitative mass spectrometry, and high-resolution cryo-EM will facilitate determining the structure of pre-ribosomal intermediates. Classical genetic and biochemical approaches will be always instrumental to analyze the proteins and/or RNA domains that interact with specific assembly factors. New developments in gene-editing in mammalian cells will allow the creation of cell line KOs for ribosome proteins or assembly factors, thus opening a new world of possible investigations. Overall, the next few years are anticipated to witness a further expansion of the mitoribosome assembly field, expected to crystallize on the discovery of intricate molecular details and new biogenetic factors occurring along the maturation pathway of yeast and mammalian mitoribosomes. Furthermore, this knowledge is expected to be instrumental for a better understanding of the pathogenic mechanisms underlying mitochondrial diseases associated with mitochondrial translation defects
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Brant Watson for critical reading of the manuscript and Dr. Golik Pawel (University of Warsaw, Poland) for his suggestions on the mitochondrial nucleases section. We apologize to those not cited owing to space limitations.
Funding
AB is supported by grants from the National Institutes of Health (NIH) RO1 GM071775, GM105781 and GM112179. FF is supported by an American Heart Association (AHA) development grant and by a Stanley Glaser Award. DD and YT are recipients of predoctoral AHA fellowships.
References
- O'Brien TW. The general occurrence of 55 S ribosomes in mammalian liver mitochondria. J Biol Chem 1971; 246:3409-3417; PMID:4930061
- O'Brien TW, Kalf GF. Ribosomes from rat liver mitochondria. I. Isolation procedure and contamination studies. J Biol Chem 1967; 242:2172-2179; PMID:6022863
- Grivell LA, Reijnders L, Borst P. Isolation of yeast mitochondrial ribosomes highly active in protein synthesis. Biochim Biophys Acta 1971; 247:91-103; PMID:4946284
- Sharma MR, Booth TM, Simpson L, Maslov DA, Agrawal RK. Structure of a mitochondrial ribosome with minimal RNA. Proc Natl Acad Sci U S A 2009; 106:9637-9642; PMID:19497863
- Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell 2003; 115:97-108; PMID:14532006; http://dx.doi.org/10.1016/S0092-8674(03)00762-1
- Smits P, Smeitink JA, van den Heuvel LP, Huynen MA, Ettema TJ. Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res 2007; 35:4686-4703; PMID:17604309
- Mears JA, Sharma MR, Gutell RR, McCook AS, Richardson PE, Caulfield TR, Agrawal RK, Harvey SC. A structural model for the large subunit of the mammalian mitochondrial ribosome. J Mol Biol 2006; 358:193-212; PMID:16510155
- Herrmann JM, Woellhaf MW, Bonnefoy N. Control of protein synthesis in yeast mitochondria: The concept of translational activators. Biochim Biophys Acta 2012; 1833:286-294; PMID:22450032
- O'Brien TW, O'Brien BJ, Norman RA. Nuclear MRP genes and mitochondrial disease. Gene 2005; 354:147-151; PMID:15908146; http://dx.doi.org/10.1016/j.gene.2005.03.026
- Christian BE, Spremulli LL. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta 2012; 1819:1035-1054; PMID:22172991
- Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R, Ban N. The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 2014; 515:283-6; PMID:25271403
- Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science 2014; 2:1258026; PMID:25278503
- Amunts A, Brown A, Bai XC, Llácer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science 2014; 343:1485-1489; PMID:24675956; http://dx.doi.org/10.1126/science.1249410
- Amunts A, Brown A, Bai XC, Llácer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science 2014; 343:1485-1489; PMID:24675956; http://dx.doi.org/10.1126/science.1249410
- Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 2014; 505:515-519; PMID:24362565; http://dx.doi.org/10.1038/nature12890
- Kaushal PS, Sharma MR, Booth TM, Haque EM, Tung CS, Sanbonmatsu KY, Spremulli LL, Agrawal RK. Cryo-EM structure of the small subunit of the mammalian mitochondrial ribosome. Proc Natl Acad Sci U S A 2014; 111:7284-7289; PMID:24799711; http://dx.doi.org/10.1073/pnas.1401657111
- Amunts A, Brown A, Toots J, Scheres SH, Ramakrishnan V. The structure of the human mitochondrial ribosome. Science 2015; 348:95-8; PMID:25838379
- Woolford JL, Jr., Baserga SJ. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013; 195:643-681; PMID:24190922; http://dx.doi.org/10.1534/genetics.113.153197
- Chen SS, Williamson JR. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J Mol Biol 2013; 425:767-779; PMID:23228329; http://dx.doi.org/10.1016/j.jmb.2012.11.040
- Shajani Z, Sykes MT, Williamson JR. Assembly of bacterial ribosomes. Annu Rev Biochem 2011; 80:501-526; PMID:21529161; http://dx.doi.org/10.1146/annurev-biochem-062608-160432
- Nierhaus KH. The assembly of prokaryotic ribosomes. Biochimie 1991; 73:739-755; PMID:1764520; http://dx.doi.org/10.1016/0300-9084(91)90054-5
- Rotig A. Human diseases with impaired mitochondrial protein synthesis. Biochim Biophys Acta 2011; 1807:1198-1205; PMID:21708121; http://dx.doi.org/10.1016/j.bbabio.2011.06.010
- Jacobs HT, Turnbull DM. Nuclear genes and mitochondrial translation: a new class of genetic disease. Trends Genet 2005; 21:312-314; PMID:15922826; http://dx.doi.org/10.1016/j.tig.2005.04.003
- Perez-Martinez X, Funes S, Camacho-Villasana Y, Marjavaara S, Tavares-Carreón F, Shingú-Vázquez M. Protein synthesis and assembly in mitochondrial disorders. Curr Top Med Chem 2008; 8:1335-1350; PMID:18991722; http://dx.doi.org/10.2174/156802608786141124
- Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal 2012; 16:1323-1367; PMID:22146081; http://dx.doi.org/10.1089/ars.2011.4123
- Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol 2014; 2:123-139; PMID:24455476; http://dx.doi.org/10.1016/j.redox.2013.12.011
- Koc EC, Cimen H, Kumcuoglu B, Abu N, Akpinar G, Haque ME, Spremulli LL, Koc H. Identification and characterization of CHCHD1, AURKAIP1, and CRIF1 as new members of the mammalian mitochondrial ribosome. Front. Physiol 2013; 4:183; PMID:23908630; http://dx.doi.org/10.3389/fphys.2013.00183
- Suzuki T, Terasaki M, Takemoto-Hori C, Hanada T, Ueda T, Wada A, Watanabe K. Proteomic analysis of the mammalian mitochondrial ribosome. Identification of protein components in the 28 S small subunit. J Biol Chem 2001; 276:33181-33195; PMID:11402041; http://dx.doi.org/10.1074/jbc.M103236200
- Koc EC, Burkhart W, Blackburn K, Moyer MB, Schlatzer DM, Moseley A, Spremulli LL. The large subunit of the mammalian mitochondrial ribosome. Analysis of the complement of ribosomal proteins present. J Biol Chem 2001; 276:43958-43969; PMID:11551941; http://dx.doi.org/10.1074/jbc.M106510200
- Jia L, Dienhart M, Schramp M, McCauley M, Hell K, Stuart RA. Yeast Oxa1 interacts with mitochondrial ribosomes: the importance of the C-terminal region of Oxa1. EMBO J 2003; 22:6438-6447; PMID:14657017; http://dx.doi.org/10.1093/emboj/cdg624
- Jia L, Kaur J, Stuart RA. Mapping of the Saccharomyces cerevisiae Oxa1-mitochondrial ribosome interface and identification of MrpL40, a ribosomal protein in close proximity to Oxa1 and critical for oxidative phosphorylation complex assembly. Eukaryot Cell 2009; 8:1792-1802; PMID:19783770; http://dx.doi.org/10.1128/EC.00219-09
- Szyrach G, Ott M, Bonnefoy N, Neupert W, Herrmann JM. Ribosome binding to the Oxa1 complex facilitates co-translational protein insertion in mitochondria. EMBO J 2003; 22:6448-6457; PMID:14657018; http://dx.doi.org/10.1093/emboj/cdg623
- Kohler R, Boehringer D, Greber B, Bingel-Erlenmeyer R, Collinson I, Schaffitzel C, Ban N. YidC and Oxa1 form dimeric insertion pores on the translating ribosome. Mol Cell 2009; 34:344-353; PMID:19450532; http://dx.doi.org/10.1016/j.molcel.2009.04.019
- Bauerschmitt H, Mick DU, Deckers M, Vollmer C, Funes S, Kehrein K, Ott M, Rehling P, Herrmann JM. Ribosome-binding proteins Mdm38 and Mba1 display overlapping functions for regulation of mitochondrial translation. Mol Biol Cell 2010; 21:1937-1944; PMID:20427570; http://dx.doi.org/10.1091/mbc.E10-02-0101
- Gruschke S, Gröne K, Heublein M, Hölz S, Israel L, Imhof A, Herrmann JM, Ott M. Proteins at the polypeptide tunnel exit of the yeast mitochondrial ribosome. J Biol Chem 2010; 285:19022-19028; PMID:20404317; http://dx.doi.org/10.1074/jbc.M110.113837
- Ott M, Prestele M, Bauerschmitt H, Funes S, Bonnefoy N, Herrmann JM. Mba1, a membrane-associated ribosome receptor in mitochondria. EMBO J 2006; 25, 1603-1610; PMID:16601683; http://dx.doi.org/10.1038/sj.emboj.7601070
- Smirnov A, Entelis N, Martin RP, Tarassov I. Biological significance of 5S rRNA import into human mitochondria: role of ribosomal protein MRP-L18. Genes Dev 2011; 25:1289-1305; PMID:21685364; http://dx.doi.org/10.1101/gad.624711
- Temperley RJ, Wydro M, Lightowlers RN, Chrzanowska-Lightowlers ZM. Human mitochondrial mRNAs–like members of all families, similar but different. Biochim Biophys Acta 2010; 1797:1081-1085; PMID:20211597; http://dx.doi.org/10.1016/j.bbabio.2010.02.036
- Cavdar Koc E, Burkhart W, Blackburn K, Moseley A, Spremulli LL. The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J Biol Chem 2001; 276:19363-19374; PMID:11279123; http://dx.doi.org/10.1074/jbc.M100727200
- O'Brien TW. Properties of human mitochondrial ribosomes. IUBMB Life 2003; 55:505-513; PMID:14658756; http://dx.doi.org/10.1080/15216540310001626610
- Shoji S, Dambacher CM, Shajani Z, Williamson JR, Schultz PG. Systematic chromosomal deletion of bacterial ribosomal protein genes. J Mol Biol 2011; 413:751-761; PMID:21945294; http://dx.doi.org/10.1016/j.jmb.2011.09.004
- Lang BF, Laforest MJ, Burger G. Mitochondrial introns: a critical view. Trends Genet 2007; 23:119-125; PMID:17280737; http://dx.doi.org/10.1016/j.tig.2007.01.006
- Belfort M, Roberts RJ. Homing endonucleases: keeping the house in order. Nucleic Acids Res 1997; 25:3379-3388; PMID:9254693; http://dx.doi.org/10.1093/nar/25.17.3379
- Turk EM, Das V, Seibert RD, Andrulis ED. The mitochondrial RNA landscape of Saccharomyces cerevisiae. PLoS One 2013; 8:e78105; PMID:24143261; http://dx.doi.org/10.1371/journal.pone.0078105
- Morales MJ, Dang YL, Lou YC, Sulo P, Martin NC. A 105-kDa protein is required for yeast mitochondrial RNase P activity. Proc Natl Acad Sci U S A 1992; 89:9875-9879; PMID:1409716; http://dx.doi.org/10.1073/pnas.89.20.9875
- Chen JY, Martin NC. Biosynthesis of tRNA in yeast mitochondria. An endonuclease is responsible for the 3′-processing of tRNA precursors. J Biol Chem 1988; 263:13677-13682; PMID:2843529
- Zhao Z, Su W, Yuan S, Huang Y. Functional conservation of tRNase ZL among Saccharomyces cerevisiae, Schizosaccharomyces pombe and humans. Biochem J 2009; 422:483-492; PMID:19555350; http://dx.doi.org/10.1042/BJ20090743
- Dziembowski A, Piwowarski J, Hoser R, Minczuk M, Dmochowska A, Siep M, van der Spek H, Grivell L, Stepien PP. The yeast mitochondrial degradosome. Its composition, interplay between RNA helicase and RNase activities and the role in mitochondrial RNA metabolism. J Biol Chem 2003; 278:1603-1611; PMID:12426313; http://dx.doi.org/10.1074/jbc.M208287200
- Dziembowski A, Stepien PP. Genetic and biochemical approaches for analysis of mitochondrial degradosome from Saccharomyces cerevisiae. Methods Enzymol 2001; 342:367-378; PMID:11586909
- Butow RA, Zhu H, Perlman P, Conrad-Webb H. The role of a conserved dodecamer sequence in yeast mitochondrial gene expression. Genome 1989; 31:757-760; PMID:2698840; http://dx.doi.org/10.1139/g89-134
- Merten S, Synenki RM, Locker J, Christianson T, Rabinowitz M. Processing of precursors of 21S ribosomal RNA from yeast mitochondria. Proc Natl Acad Sci U S A 1980; 77:1417-1421; PMID:6990410; http://dx.doi.org/10.1073/pnas.77.3.1417
- Stepien PP, Kokot L, Leski T, Bartnik E. The suv3 nuclear gene product is required for the in vivo processing of the yeast mitochondrial 21s rRNA transcripts containing the r1 intron. Curr Genet 1995; 27:234-238; PMID:7736607; http://dx.doi.org/10.1007/BF00326154
- Margossian SP, Li H, Zassenhaus HP, Butow RA. The DExH box protein Suv3p is a component of a yeast mitochondrial 3′-to-5′ exoribonuclease that suppresses group I intron toxicity. Cell 1996; 84:199-209; PMID:8565066; http://dx.doi.org/10.1016/S0092-8674(00)80975-7
- Osinga KA, Evers RF, Van der Laan JC, Tabak HF. A putative precursor for the small ribosomal RNA from mitochondria of Saccharomyces cerevisiae. Nucleic Acids Res 1981; 9:1351-1364; PMID:6262728; http://dx.doi.org/10.1093/nar/9.6.1351
- Puchta O, Lubas M, Lipinski KA, Piatkowski J, Malecki M, Golik P. DMR1 (CCM1/YGR150C) of Saccharomyces cerevisiae encodes an RNA-binding protein from the pentatricopeptide repeat family required for the maintenance of the mitochondrial 15S ribosomal RNA. Genetics 2010; 184:959-973; PMID:20124025; http://dx.doi.org/10.1534/genetics.110.113969
- Wiesenberger G, Fox TD. Pet127p, a membrane-associated protein involved in stability and processing of Saccharomyces cerevisiae mitochondrial RNAs. Mol Cell Biol 1997; 17:2816-2824; PMID:9111353
- Fekete Z, Ellis TP, Schonauer MS, Dieckmann CL. Pet127 governs a 5′ -> 3′-exonuclease important in maturation of apocytochrome b mRNA in Saccharomyces cerevisiae. J Biol Chem 2008; 283:3767-3772; PMID:18086665; http://dx.doi.org/10.1074/jbc.M709617200
- Haffter P, Fox TD. Suppression of carboxy-terminal truncations of the yeast mitochondrial mRNA-specific translational activator PET122 by mutations in two new genes, MRP17 and PET127. Mol Gen Genet 1992; 235:64-73; PMID:1279374; http://dx.doi.org/10.1007/BF00286182
- Montoya J, Gaines GL, Attardi G. The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 1983; 34:151-159; PMID:6883508; http://dx.doi.org/10.1016/0092-8674(83)90145-9
- Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981; 290:470-474; PMID:7219536; http://dx.doi.org/10.1038/290470a0
- Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 2008; 135:462-474; PMID:18984158; http://dx.doi.org/10.1016/j.cell.2008.09.013
- Brzezniak LK, Bijata M, Szczesny RJ, Stepien PP. Involvement of human ELAC2 gene product in 3′ end processing of mitochondrial tRNAs. RNA Biol 2011; 8:616-626; PMID:21593607; http://dx.doi.org/10.4161/rna.8.4.15393
- Sanchez MI, Mercer TR, Davies SM, Shearwood AM, Nygård KK, Richman TR, Mattick JS, Rackham O, Filipovska A. RNA processing in human mitochondria. Cell Cycle 2011; 10:2904-2916; PMID:21857155; http://dx.doi.org/10.4161/cc.10.17.17060
- Venema J, Tollervey D. Ribosome synthesis in Saccharomyces cerevisiae. Annu Rev Genet 1999; 33, 261-311; PMID:10690410; http://dx.doi.org/10.1146/annurev.genet.33.1.261
- Motorin Y, Helm M. RNA nucleotide methylation. Wiley Interdiscip Rev RNA 2011; 2:611-631; PMID:21823225
- Ofengand J. Ribosomal RNA pseudouridines and pseudouridine synthases. FEBS Lett 2002; 514:17-25; PMID:11904174; http://dx.doi.org/10.1016/S0014-5793(02)02305-0
- Klootwijk J, Klein I, Grivell LA. Minimal post-transcriptional modification of yeast mitochondrial ribosomal RNA. J Mol Biol 1975; 97:337-350; PMID:1102710; http://dx.doi.org/10.1016/S0022-2836(75)80044-1
- Sirum-Connolly K, Mason TL. Functional requirement of a site-specific ribose methylation in ribosomal RNA. Science 1993; 262:1886-1889; PMID:8266080; http://dx.doi.org/10.1126/science.8266080
- Sirum-Connolly K, Mason TL. The role of nucleotide modifications in the yeast mitochondrial ribosome. Nucleic Acids Symp Ser 1995:73-75; PMID:8643404
- Sirum-Connolly K, Peltier JM, Crain PF, McCloskey JA, Mason TL. Implications of a functional large ribosomal RNA with only three modified nucleotides. Biochimie 1995; 77:30-39; PMID:7541254; http://dx.doi.org/10.1016/0300-9084(96)88101-6
- Pintard L, Bujnicki JM, Lapeyre B, Bonnerot C. MRM2 encodes a novel yeast mitochondrial 21S rRNA methyltransferase. EMBO J 2002; 21:1139-1147; PMID:11867542; http://dx.doi.org/10.1093/emboj/21.5.1139
- Ansmant I, Massenet S, Grosjean H, Motorin Y, Branlant C. Identification of the Saccharomyces cerevisiae RNA:pseudouridine synthase responsible for formation of psi(2819) in 21S mitochondrial ribosomal RNA. Nucleic Acids Res 2000; 28:1941-1946; PMID:10756195; http://dx.doi.org/10.1093/nar/28.9.1941
- Dubin DT, Taylor RH. Modification of mitochondrial ribosomal RNA from hamster cells: the presence of GmG and late-methylated UmGmU in the large subunit (17S) RNA. J Mol Biol 1978; 121:523-540; PMID:671547; http://dx.doi.org/10.1016/0022-2836(78)90398-4
- Seidel-Rogol BL, McCulloch V, Shadel GS. Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat Genet 2003; 33:23-24; PMID:12496758; http://dx.doi.org/10.1038/ng1064
- Xu Z, O'Farrell HC, Rife JP, Culver GM. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat Struct Mol Biol 2008; 15:534-536; PMID:18391965; http://dx.doi.org/10.1038/nsmb.1408
- Metodiev MD, Lesko N, Park CB, Cámara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson NG. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab 2009; 9:386-397; PMID:19356719; http://dx.doi.org/10.1016/j.cmet.2009.03.001
- Metodiev MD, Spåhr H, Loguercio Polosa P, Meharg C, Becker C, Altmueller J, Habermann B, Larsson NG, Ruzzenente B. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet 2014; 10:e1004110; PMID:24516400; http://dx.doi.org/10.1371/journal.pgen.1004110
- Camara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, et al. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab 2011; 13:527-539; PMID:21531335; http://dx.doi.org/10.1016/j.cmet.2011.04.002
- Spahr H, Habermann B, Gustafsson CM, Larsson NG, Hallberg BM. Structure of the human MTERF4-NSUN4 protein complex that regulates mitochondrial ribosome biogenesis. Proc Natl Acad Sci U S A 2012; 109:15253-15258; PMID:22949673; http://dx.doi.org/10.1073/pnas.1210688109
- Baer RJ, Dubin DT. Methylated regions of hamster mitochondrial ribosomal RNA: structural and functional correlates. Nucleic Acids Res 1981; 9:323-337; PMID:6782552; http://dx.doi.org/10.1093/nar/9.2.323
- Lee KW, Bogenhagen DF. Assignment of 2′-O-methyltransferases to Modification Sites on the Mammalian Mitochondrial Large Subunit 16S rRNA. J Biol Chem 2014; 289:24936-42, epub ahead of print; PMID:25074936
- Rorbach J, Boesch P, Gammage PA, Nicholls TJ, Pearce SF, Patel D, Hauser A, Perocchi F, Minczuk M. MRM2 and MRM3 are involved in biogenesis of the large subunit of the mitochondrial ribosome. Mol Biol Cell 2014; 9:01-0014; PMID:25009282
- Ofengand J, Bakin A. Mapping to nucleotide resolution of pseudouridine residues in large subunit ribosomal RNAs from representative eukaryotes, prokaryotes, archaebacteria, mitochondria and chloroplasts. J Mol Biol 1997; 266:246-268; PMID:9047361; http://dx.doi.org/10.1006/jmbi.1996.0737
- Clementi N, Polacek N. Ribosome-associated GTPases: the role of RNA for GTPase activation. RNA Biol 2010; 7:521-527; PMID:20657179; http://dx.doi.org/10.4161/rna.7.5.12467
- Goto S, Muto A, Himeno H. GTPases involved in bacterial ribosome maturation. J Biochem 2013; 153:403-414; PMID:23509007; http://dx.doi.org/10.1093/jb/mvt022
- Kim do J, Jang JY, Yoon HJ, Suh SW. Crystal structure of YlqF, a circularly permuted GTPase: implications for its GTPase activation in 50 S ribosomal subunit assembly. Proteins 2008; 72:1363-1370; PMID:18536017; http://dx.doi.org/10.1002/prot.22112
- Gulati M, Jain N, Anand B, Prakash B, Britton RA. Mutational analysis of the ribosome assembly GTPase RbgA provides insight into ribosome interaction and ribosome-stimulated GTPase activation. Nuc Acids Res 2013; 41:3217-3227; PMID:23325847; http://dx.doi.org/10.1093/nar/gks1475
- Britton RA. Role of GTPases in bacterial ribosome assembly. Annu Rev Microbiol 2009; 63:155-176; PMID:19575570; http://dx.doi.org/10.1146/annurev.micro.091208.073225
- Matsuo Y, Morimoto T, Kuwano M, Loh PC, Oshima T, Ogasawara N. The GTP-binding protein YlqF participates in the late step of 50 S ribosomal subunit assembly in Bacillus subtilis. J Biol Chem 2006; 281:8110-8117; PMID:16431913; http://dx.doi.org/10.1074/jbc.M512556200
- Achila D, Gulati M, Jain N, Britton RA. Biochemical characterization of ribosome assembly GTPase RbgA in Bacillus subtilis. J Biol Chem 2012; 287:8417-23; PMID:22267738; http://dx.doi.org/10.1074/jbc.M111.331322
- Matsuo Y, Oshima T, Loh PC, Morimoto T, Ogasawara N. Isolation and characterization of a dominant negative mutant of Bacillus subtilis GTP-binding protein, YlqF, essential for biogenesis and maintenance of the 50 S ribosomal subunit. J Biol Chem 2007; 282:25270-25277; PMID:17613524
- Gulati M, Jain N, Davis JH, Williamson JR, Britton RA. Functional interaction between ribosomal protein L6 and RbgA during ribosome assembly. PLoS Genet 2014; 10:e1004694; PMID:25330043
- Jomaa A, Jain N, Davis JH, Williamson JR, Britton RA, Ortega J. Functional domains of the 50S subunit mature late in the assembly process. Nucleic Acids Res 2014; 42:3419-3435; PMID:24335279
- Uicker WC, Schaefer L, Britton RA. The essential GTPase RbgA (YlqF) is required for 50S ribosome assembly in Bacillus subtilis. Mol Microbiol 2006; 59:528-40; PMID:16390447
- Barrientos A, Korr D, Barwell KJ, Sjulsen C, Gajewski CD, Manfredi G, Ackerman S, Tzagoloff A. MTG1 codes for a conserved protein required for mitochondrial translation. Mol Biol Cell 2003; 14:2292-2302; PMID:12808030
- Li N, Chen Y, Guo Q, Zhang Y, Yuan Y, Ma C, Deng H, Lei J, Gao N. Cryo-EM structures of the late-stage assembly intermediates of the bacterial 50S ribosomal subunit. Nucleic Acids Res 2013; 41:7073-7083; PMID:23700310
- Kotani T, Akabane S, Takeyasu K, Ueda T, Takeuchi N. Human G-proteins, ObgH1 and Mtg1, associate with the large mitochondrial ribosome subunit and are involved in translation and assembly of respiratory complexes. Nucleic Acids Res 2013; 41:3713-3722; PMID:23396448
- Sato A, Kobayashi G, Hayashi H, Yoshida H, Wada A, Maeda M, Hiraga S, Takeyasu K, Wada C. The GTP binding protein Obg homolog ObgE is involved in ribosome maturation. Genes Cells 2005; 10:393-408; PMID:15836769
- Datta K, Fuentes JL, Maddock JR. The yeast GTPase Mtg2p is required for mitochondrial translation and partially suppresses an rRNA methyltransferase mutant, mrm2. Mol Biol Cell 2005; 16:954-963; PMID:15591131
- Anand B, Surana P, Prakash B. Deciphering the catalytic machinery in 30S ribosome assembly GTPase YqeH. PLoS One 2010; 5:e9944; PMID:20376346; http://dx.doi.org/10.1371/journal.pone.0009944
- Paul MF, Alushin GM, Barros MH, Rak M, Tzagoloff A. The putative GTPase encoded by MTG3 functions in a novel pathway for regulating assembly of the small subunit of yeast mitochondrial ribosomes. J Biol Chem 2012; 287:24346-24355; PMID:22621929
- He J, Cooper HM, Reyes A, Di Re M, Kazak L, Wood SR, Mao CC, Fearnley IM, Walker JE, Holt IJ. Human C4orf14 interacts with the mitochondrial nucleoid and is involved in the biogenesis of the small mitochondrial ribosomal subunit. Nucleic Acids Res 2012; 40:6097-6108; PMID:22447445
- Sharma MR, Barat C, Wilson DN, Booth TM, Kawazoe M, Hori-Takemoto C, Shirouzu M, Yokoyama S, Fucini P, Agrawal RK. Interaction of Era with the 30S ribosomal subunit implications for 30S subunit assembly. Mol Cell 2005; 18:319-329; PMID:15866174
- Dennerlein S, Rozanska A, Wydro M, Chrzanowska-Lightowlers ZM, Lightowlers RN. Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem J 2010; 430:551-558; PMID:20604745
- Uchiumi T, Ohgaki K, Yagi M, Aoki Y, Sakai A, Matsumoto S, Kang D. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res 2010; 38:5554-5568; PMID:20430825
- Pan C, Russell R. Roles of DEAD-box proteins in RNA and RNP Folding. RNA Biol 2010; 7:667-676; PMID:21045543
- Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 2011; 12:505-516; PMID:21779027
- Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene 2006; 367:17-37; PMID:16337753; http://dx.doi.org/10.1016/j.gene.2005.10.019
- Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Struct Biol 2010; 20:313-324; PMID:20456941
- Young CL, Khoshnevis S, Karbstein K. Cofactor-dependent specificity of a DEAD-box protein. Proc Natl Acad Sci U S A 2013; 110:E2668-2676; PMID:23630256
- Martin R, Straub AU, Doebele C, Bohnsack MT. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol 2013; 10:4-18; PMID:22922795
- Parsyan A, Svitkin Y, Shahbazian D, Gkogkas C, Lasko P, Merrick WC, Sonenberg N. mRNA helicases: the tacticians of translational control. Nat Mol Cell Biol 2011; 12:235-245; PMID:21427765
- Kaberdin VR, Blasi U. Bacterial helicases in post-transcriptional control. Biochim Biophys Acta 2013; 1829:878-883; PMID:23291566
- Peil L, Virumäe K, Remme J. Ribosome assembly in Escherichia coli strains lacking the RNA helicase DeaD/CsdA or DbpA. FEBS J 2008 275:3772-3782. Epub 02008 Jun 06528; PMID:18565105
- Charollais J, Pflieger D, Vinh J, Dreyfus M, Iost I. The DEAD-box RNA helicase SrmB is involved in the assembly of 50S ribosomal subunits in Escherichia coli. Mol Microbiol 2003; 48:1253-1265; PMID:12787353
- Fuller-Pace FV, Nicol SM, Reid AD, Lane DP. DbpA: a DEAD box protein specifically activated by 23s rRNA. EMBO J 1993; 12:3619-3626; PMID:8253085
- Diges CM, Uhlenbeck OC. Escherichia coli DbpA is an RNA helicase that requires hairpin 92 of 23S rRNA. EMBO J 2001; 20:5503-5512; PMID:11574482
- Sharpe Elles LM, Sykes MT, Williamson JR, Uhlenbeck OC. A dominant negative mutant of the E. coli RNA helicase DbpA blocks assembly of the 50S ribosomal subunit. Nucleic Acids Res 2009; 37:6503-6514; PMID:19734347
- Osswald M, Döring T, Brimacombe R. The ribosomal neighbourhood of the central fold of tRNA: cross-links from position 47 of tRNA located at the A, P or E site. Nucleic Acids Res 1995; 23:4635-4641; PMID:8524654
- Maguire BA, Wild DG. The roles of proteins L28 and L33 in the assembly and function of Escherichia coli ribosomes in vivo. Mol Microbiol 1997; 23:237-245; PMID:9044258
- Franceschi FJ, Nierhaus KH. Ribosomal proteins L15 and L16 are mere late assembly proteins of the large ribosomal subunit. Analysis of an Escherichia coli mutant lacking L15. J Biol Chem 1990; 265:16676-16682; PMID:2204629
- Martin-Marcos P, Hinnebusch AG, Tamame M. Ribosomal protein L33 is required for ribosome biogenesis, subunit joining, and repression of GCN4 translation. Mol Cell Biol 2007; 27:5968-5985; PMID:17548477; http://dx.doi.org/10.1128/MCB.00019-07
- De Silva D, Fontanesi F, Barrientos A. The DEAD-Box protein Mrh4 functions in the assembly of the mitochondrial large ribosomal subunit. Cell Metab 2013; 18:712-725; PMID:24206665; http://dx.doi.org/10.1016/j.cmet.2013.10.007
- Valgardsdottir R, Brede G, Eide LG, Frengen E, Prydz H. Cloning and characterization of MDDX28, a putative dead-box helicase with mitochondrial and nuclear localization. J Biol Chem 2001; 276:32056-32063; PMID:11350955; http://dx.doi.org/10.1074/jbc.M011629200
- Valgardsdottir R, Prydz H. Transport signals and transcription-dependent nuclear localization of the putative DEAD-box helicase MDDX28. J Biol Chem 2003; 278:21146-21154; PMID:12663657; http://dx.doi.org/10.1074/jbc.M300888200
- Valgardsdottir R, Ottersen OP, Prydz H. Regulated compartmentalization of the putative DEAD-box helicase MDDX28 within the mitochondria in COS-1 cells. Exp Cell Res 2004; 299:294-302; PMID:15350529; http://dx.doi.org/10.1016/j.yexcr.2004.05.019
- Tu YT, Barrientos A. The Human Mitochondrial DEAD-Box Protein DDX28 Resides in RNA Granules and Functions in Mitoribosome Assembly. Cell Rep 2015; 12:00058-00053; PMID:25683708
- Antonicka H, Shoubridge EA. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell Rep 2015; 12:00055-00058; PMID:25683715
- Bogenhagen DF, Rousseau D, Burke S. The layered structure of human mitochondrial DNA nucleoids. J Biol Chem 2008; 283:3665-3675; PMID:18063578; http://dx.doi.org/10.1074/jbc.M708444200
- Wredenberg A, Lagouge M, Bratic A, Metodiev MD, Spåhr H, Mourier A, Freyer C, Ruzzenente B, Tain L, Grönke S, et al. MTERF3 regulates mitochondrial ribosome biogenesis in invertebrates and mammals. PLoS Genet 2013; 9:e1003178; PMID:23300484; http://dx.doi.org/10.1371/journal.pgen.1003178
- Rorbach J, Gammage PA, Minczuk M. C7orf30 is necessary for biogenesis of the large subunit of the mitochondrial ribosome. Nucleic Acids Res 2012; 40:4097-4109; PMID:22238376; http://dx.doi.org/10.1093/nar/gkr1282
- Fung S, Nishimura T, Sasarman F, Shoubridge EA. The conserved interaction of C7orf30 with MRPL14 promotes biogenesis of the mitochondrial large ribosomal subunit and mitochondrial translation. Mol Biol Cell 2013; 24:184-193; PMID:23171548; http://dx.doi.org/10.1091/mbc.E12-09-0651
- Wanschers BF, Szklarczyk R, Pajak A, van den Brand MA, Gloerich J, Rodenburg RJ, Lightowlers RN, Nijtmans LG, Huynen MA. C7orf30 specifically associates with the large subunit of the mitochondrial ribosome and is involved in translation. Nucleic Acids Res 2012; 40:4040-4051; PMID:22238375; http://dx.doi.org/10.1093/nar/gkr1271
- Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 2005; 123:277-289; PMID:16239145; http://dx.doi.org/10.1016/j.cell.2005.08.003
- Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol 2007; 27:758-767; PMID:17101804; http://dx.doi.org/10.1128/MCB.01470-06
- Antonicka H, Sasarman F, Nishimura T, Paupe V, Shoubridge EA. The Mitochondrial RNA-Binding Protein GRSF1 Localizes to RNA Granules and Is Required for Posttranscriptional Mitochondrial Gene Expression. Cell Metab 2013 17:386-398; PMID:23473033; http://dx.doi.org/10.1016/j.cmet.2013.02.006
- Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska-Lightowlers ZM, Martinou JC. GRSF1 Regulates RNA Processing in Mitochondrial RNA Granules. Cell Metab 2013; 17:399-410; PMID:23473034; http://dx.doi.org/10.1016/j.cmet.2013.02.005
- Traub P, Nomura M. Structure and function of E. coli ribosomes. V. Reconstitution of functionally active 30S ribosomal particles from RNA and proteins. Proc Natl Acad Sci U S A 1968; 59:777-784; PMID:4868216; http://dx.doi.org/10.1073/pnas.59.3.777
- Held WA, Mizushima S, Nomura M. Reconstitution of Escherichia coli 30 S ribosomal subunits from purified molecular components. J Biol Chem 1973; 248:5720-5730; PMID:4579428
- Nierhaus KH, Dohme F. Total reconstitution of functionally active 50S ribosomal subunits from Escherichia coli. Proc Natl Acad Sci U S A 1974; 71:4713-4717; PMID:4612527; http://dx.doi.org/10.1073/pnas.71.12.4713
- Dohme F, Nierhaus KH. Total reconstitution and assembly of 50 S subunits from Escherichia coli ribosomes in vitro. J Mol Biol 1976; 107:585-599; PMID:794489; http://dx.doi.org/10.1016/S0022-2836(76)80085-X
- Sieber G, Nierhaus KH. Kinetic and thermodynamic parameters of the assembly in vitro of the large subunit from Escherichia coli ribosomes. Biochemistry 1978; 17:3505-3511; PMID:356881; http://dx.doi.org/10.1021/bi00610a013
- Stern S, Powers T, Changchien LM, Noller HF. RNA-protein interactions in 30S ribosomal subunits: folding and function of 16S rRNA. Science 1989; 244:783-790; PMID:2658053; http://dx.doi.org/10.1126/science.2658053
- Adilakshmi T, Bellur DL, Woodson SA. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature 2008; 455:1268-1272; PMID:18784650; http://dx.doi.org/10.1038/nature07298
- Talkington MW, Siuzdak G, Williamson JR. An assembly landscape for the 30S ribosomal subunit. Nature 2005; 438:628-632; PMID:16319883; http://dx.doi.org/10.1038/nature04261
- Mulder AM, Yoshioka C, Beck AH, Bunner AE, Milligan RA, Potter CS, Carragher B, Williamson JR. Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science 2010; 330:673-677; PMID:21030658; http://dx.doi.org/10.1126/science.1193220
- Bubunenko M, Korepanov A, Court DL, Jagannathan I, Dickinson D, Chaudhuri BR, Garber MB, Culver GM. 30S ribosomal subunits can be assembled in vivo without primary binding ribosomal protein S15. RNA 2006; 12:1229-1239; PMID:16682557; http://dx.doi.org/10.1261/rna.2262106
- Sykes MT, Shajani Z, Sperling E, Beck AH, Williamson JR. Quantitative proteomic analysis of ribosome assembly and turnover in vivo. J Mol Biol 2010; 403:331-345; PMID:20709079; http://dx.doi.org/10.1016/j.jmb.2010.08.005
- Lindahl L. Intermediates and time kinetics of the in vivo assembly of Escherichia coli ribosomes. J Mol Biol 1975; 92:15-37; PMID:1097701; http://dx.doi.org/10.1016/0022-2836(75)90089-3
- Sashital DG, Greeman CA, Lyumkis D, Potter CS, Carragher B, Williamson JR. A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30S subunit assembly in E. coli. Elife 2014; 3; PMID:25313868; http://dx.doi.org/10.7554/eLife.04491
- Youngman EM, Green R. Affinity purification of in vivo-assembled ribosomes for in vitro biochemical analysis. Methods 2005; 36:305-312; PMID:16076457; http://dx.doi.org/10.1016/j.ymeth.2005.04.007
- Gupta N, Culver GM. Multiple in vivo pathways for Escherichia coli small ribosomal subunit assembly occur on one pre-rRNA. Nat Struct Mol Biol 2014; 21:937-943; PMID:25195050; http://dx.doi.org/10.1038/nsmb.2887
- Liu M, Spremulli L. Interaction of mammalian mitochondrial ribosomes with the inner membrane. J Biol Chem 2000; 275:29400-29406; PMID:10887179; http://dx.doi.org/10.1074/jbc.M002173200
- Kaur J, Stuart RA. Truncation of the Mrp20 protein reveals new ribosome-assembly subcomplex in mitochondria. EMBO Rep 2011; 12:950-955; PMID:21779004; http://dx.doi.org/10.1038/embor.2011.133
- Bogenhagen DF, Martin DW, Koller A. Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab 2014; 19:618-629; PMID:24703694; http://dx.doi.org/10.1016/j.cmet.2014.03.013
- Borowski LS, Dziembowski A, Hejnowicz MS, Stepien PP, Szczesny RJ. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res 2013; 41:1223-1240; PMID:23221631; http://dx.doi.org/10.1093/nar/gks1130
- Holt IJ, He J, Mao CC, Boyd-Kirkup JD, Martinsson P, Sembongi H, Reyes A, Spelbrink JN. Mammalian mitochondrial nucleoids: organizing an independently minded genome. Mitochondrion 2007; 7:311-321; PMID:17698423; http://dx.doi.org/10.1016/j.mito.2007.06.004
- Brown TA, Tkachuk AN, Shtengel G, Kopek BG, Bogenhagen DF, Hess HF, Clayton DA. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol Cell Biol 2011; 31:4994-5010; PMID:22006021; http://dx.doi.org/10.1128/MCB.05694-11
- Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson NG, Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A 2011; 108:13534-13539; PMID:21808029; http://dx.doi.org/10.1073/pnas.1109263108
- Kucej M, Butow RA. Evolutionary tinkering with mitochondrial nucleoids. Trends Cell Biol 2007; 17:586-592; PMID:17981466; http://dx.doi.org/10.1016/j.tcb.2007.08.007
- Iborra FJ, Kimura H, Cook PR. The functional organization of mitochondrial genomes in human cells. BMC Biol 2004; 2:9; PMID:15157274; http://dx.doi.org/10.1186/1741-7007-2-9
- He J, Cooper HM, Reyes A, Di Re M, Sembongi H, Litwin TR, Gao J, Neuman KC, Fearnley IM, Spinazzola A, et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res 2012; 40:6109-6121; PMID:22453275; http://dx.doi.org/10.1093/nar/gks266
- Dalla Rosa I, Durigon R, Pearce SF, Rorbach J, Hirst EM, Vidoni S, Reyes A, Brea-Calvo G, Minczuk M, Woellhaf MW, et al. MPV17L2 is required for ribosome assembly in mitochondria. Nucleic Acids Res 2014; 42:8500-15; PMID:24948607
- Lee KW, Okot-Kotber C, LaComb JF, Bogenhagen DF. Mitochondrial rRNA Methyltransferase Family Members are Positioned to Modify Nascent rRNA in Foci Near the mtDNA Nucleoid. J Biol Chem 2013; 288:31386-31399; PMID:24036117; http://dx.doi.org/10.1074/jbc.M113.515692
- Hess KC, Liu J, Manfredi G, Mühlschlegel FA, Buck J, Levin LR, Barrientos A. A mitochondrial CO2-adenylyl cyclase-cAMP signalosome controls yeast normoxic cytochrome c oxidase activity. FASEB J 2014; 28:4369-4380; PMID:25002117; http://dx.doi.org/10.1096/fj.14-252890
- Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Cámara Y, Milenkovic D, Zickermann V, Wibom R, Hultenby K, et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J 2012; 31:443-456; PMID:22045337; http://dx.doi.org/10.1038/emboj.2011.392
- Chujo T, Ohira T, Sakaguchi Y, Goshima N, Nomura N, Nagao A, Suzuki T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res 2012; 40:8033-8047; PMID:22661577; http://dx.doi.org/10.1093/nar/gks506
- Rackham O, Davies SM, Shearwood AM, Hamilton KL, Whelan J, Filipovska A. Pentatricopeptide repeat domain protein 1 lowers the levels of mitochondrial leucine tRNAs in cells. Nucleic Acids Res 2009; 37:5859-5867; PMID:19651879; http://dx.doi.org/10.1093/nar/gkp627
- Prachar J. Mouse and human mitochondrial nucleoid–detailed structure in relation to function. Gen Physiol Biophys 2010; 29:160-174; PMID:20577028; http://dx.doi.org/10.4149/gpb_2010_02_160
- Kehrein K, Schilling R, Möller-Hergt BV, Wurm CA, Jakobs S, Lamkemeyer T, Langer T, Ott M. Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep 2015; 12:S2211-1247; PMID:25683707
- Fiori A, Mason TL, Fox TD. Evidence that synthesis of the Saccharomyces cerevisiae mitochondrially encoded ribosomal protein Var1p may be membrane localized. Eukaryot Cell 2003; 2:651-653; PMID:12796311
- Keil M, Bareth B, Woellhaf MW, Peleh V, Prestele M, Rehling P, Herrmann JM. Oxa1-ribosome complexes coordinate the assembly of cytochrome c oxidase in mitochondria. J Biol Chem 2012; 287:34484-34493; PMID:22904327; http://dx.doi.org/10.1074/jbc.M112.382630
- Schneider HC, Berthold J, Bauer MF, Dietmeier K, Guiard B, Brunner M, Neupert W. Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature 1994; 371:768-774; PMID:7935837; http://dx.doi.org/10.1038/371768a0
- Stiburek L, Fornuskova D, Wenchich L, Pejznochova M, Hansikova H, Zeman J. Knockdown of human Oxa1l impairs the biogenesis of F1Fo-ATP synthase and NADH:ubiquinone oxidoreductase. J Mol Biol 2007; 374:506-516; PMID:17936786; http://dx.doi.org/10.1016/j.jmb.2007.09.044
- Bonnefoy N, Kermorgant M, Groudinsky O, Minet M, Slonimski PP, Dujardin G. Cloning of a human gene involved in cytochrome oxidase assembly by functional complementation of an oxa1- mutation in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 1994; 91:11978-11982; PMID:7991568; http://dx.doi.org/10.1073/pnas.91.25.11978
- Jacobs HT. Disorders of mitochondrial protein synthesis. Hum Mol Genet 2003; 12:R293-301; PMID:12928485; http://dx.doi.org/10.1093/hmg/ddg285
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet 1993; 4:289-294; PMID:7689389; http://dx.doi.org/10.1038/ng0793-289
- Zhao H, Li R, Wang Q, Yan Q, Deng JH, Han D, Bai Y, Young WY, Guan MX. Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am J Hum Genet 2004; 74:139-152; PMID:14681830; http://dx.doi.org/10.1086/381133
- Ding Y, Leng J, Fan F, Xia B, Xu P. The role of mitochondrial DNA mutations in hearing loss. Biochem Genet 2013; 51:588-602. Epub 12013 Apr 10521; PMID:23605717; http://dx.doi.org/10.1007/s10528-013-9589-6
- Estivill X, Govea N, Barceló E, Badenas C, Romero E, Moral L, Scozzri R, D'Urbano L, Zeviani M, Torroni A. Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am J Hum Genet 1998; 62:27-35; PMID:9490575; http://dx.doi.org/10.1086/301676
- Recht MI, Fourmy D, Blanchard SC, Dahlquist KD, Puglisi JD. RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J Mol Biol 1996; 262:421-436; PMID:8893854; http://dx.doi.org/10.1006/jmbi.1996.0526
- Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 1996; 274:1367-1371; PMID:8910275; http://dx.doi.org/10.1126/science.274.5291.1367
- Lambert T. Antibiotics that affect the ribosome. Rev Sci Tech 2012; 31:57-64; PMID:22849268
- Davies J, Davis BD. Misreading of ribonucleic acid code words induced by aminoglycoside antibiotics. The effect of drug concentration. J Biol Chem 1968; 243:3312-3316; PMID:5656371
- Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu Rev Biochem 2005; 74:129-177; PMID:15952884; http://dx.doi.org/10.1146/annurev.biochem.74.061903.155440
- Purohit P, Stern S. Interactions of a small RNA with antibiotic and RNA ligands of the 30S subunit. Nature 1994; 370:659-662; PMID:8065453; http://dx.doi.org/10.1038/370659a0
- Hamasaki K, Rando RR. Specific binding of aminoglycosides to a human rRNA construct based on a DNA polymorphism which causes aminoglycoside-induced deafness. Biochemistry 1997; 36:12323-12328; PMID:9315872; http://dx.doi.org/10.1021/bi970962r
- Xing G, Chen Z, Cao X. Mitochondrial rRNA and tRNA and hearing function. Cell Res 2007; 17:227-239; PMID:17199108
- Bykhovskaya Y, Mengesha E, Wang D, Yang H, Estivill X, Shohat M, Fischel-Ghodsian N. Human mitochondrial transcription factor B1 as a modifier gene for hearing loss associated with the mitochondrial A1555G mutation. Mol Genet Metab 2004; 82:27-32; PMID:15110318; http://dx.doi.org/10.1016/j.ymgme.2004.01.020
- Li X, Li R, Lin X, Guan MX. Isolation and characterization of the putative nuclear modifier gene MTO1 involved in the pathogenesis of deafness-associated mitochondrial 12 S rRNA A1555G mutation. J Biol Chem 2002; 277:27256-27264; PMID:12011058; http://dx.doi.org/10.1074/jbc.M203267200
- Bykhovskaya Y, Mengesha E, Wang D, Yang H, Estivill X, Shohat M, Fischel-Ghodsian N. Phenotype of non-syndromic deafness associated with the mitochondrial A1555G mutation is modulated by mitochondrial RNA modifying enzymes MTO1 and GTPBP3. Mol Genet Metab 2004; 83:199-206; PMID:15542390; http://dx.doi.org/10.1016/j.ymgme.2004.07.009
- Li X, Guan MX. A human mitochondrial GTP binding protein related to tRNA modification may modulate phenotypic expression of the deafness-associated mitochondrial 12S rRNA mutation. Mol Cell Biol 2002; 22:7701-7711; PMID:12370316; http://dx.doi.org/10.1128/MCB.22.21.7701-7711.2002
- Colby G, Wu M, Tzagoloff A. MTO1 codes for a mitochondrial protein required for respiration in paromomycin-resistant mutants of Saccharomyces cerevisiae. J Biol Chem 1998; 273:27945-27952; PMID:9774408; http://dx.doi.org/10.1074/jbc.273.43.27945
- Coulbault L, Deslandes B, Herlicoviez D, Read MH, Leporrier N, Schaeffer S, Mouadil A, Lombès A, Chapon F, Jauzac P, et al. A novel mutation 3090 G>A of the mitochondrial 16S ribosomal RNA associated with myopathy. Biochem Biophys Res Commun 2007; 362:601-605; PMID:17761147; http://dx.doi.org/10.1016/j.bbrc.2007.08.040
- Liu Z, Song Y, Li D, He X, Li S, Wu B, Wang W, Gu S, Zhu X, Wang X, et al. The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy. J Med Genet 2014; 51:176-184; PMID:24367055; http://dx.doi.org/10.1136/jmedgenet-2013-101818
- Sacconi S, Salviati L, Gooch C, Bonilla E, Shanske S, DiMauro S. Complex neurologic syndrome associated with the G1606A mutation of mitochondrial DNA. Arch Neurol 2002 59:1013-1015; PMID:12056939; http://dx.doi.org/10.1001/archneur.59.6.1013
- Tiranti V, D'Agruma L, Pareyson D, Mora M, Carrara F, Zelante L, Gasparini P, Zeviani M. A novel mutation in the mitochondrial tRNA(Val) gene associated with a complex neurological presentation. Ann Neurol 1998; 43:98-101; PMID:9450773; http://dx.doi.org/10.1002/ana.410430116
- McFarland R, Clark KM, Morris AA, Taylor RW, Macphail S, Lightowlers RN, Turnbull DM. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet 2002; 30:145-146; PMID:11799391; http://dx.doi.org/10.1038/ng819
- Arredondo JJ, Gallardo ME, García-Pavía P, Domingo V, Bretón B, García-Silva MT, Sedano MJ, Martín MA, Arenas J, Cervera M, et al. Mitochondrial tRNA valine as a recurrent target for mutations involved in mitochondrial cardiomyopathies. Mitochondrion 2012; 12:357-362; PMID:21986556; http://dx.doi.org/10.1016/j.mito.2011.09.010
- Horvath R, Bender A, Abicht A, Holinski-Feder E, Czermin B, Trips T, Schneiderat P, Lochmüller H, Klopstock T. Heteroplasmic mutation in the anticodon-stem of mitochondrial tRNA(Val) causing MNGIE-like gastrointestinal dysmotility and cachexia. J Neurol 2009; 256:810-815; PMID:19252805; http://dx.doi.org/10.1007/s00415-009-5023-8
- de Coo IF, Sistermans EA, de Wijs IJ, Catsman-Berrevoets C, Busch HF, Scholte HR, de Klerk JB, van Oost BA, Smeets HJ. A mitochondrial tRNA(Val) gene mutation (G1642A) in a patient with mitochondrial myopathy, lactic acidosis, and stroke-like episodes. Neurology 1998; 50:293-295; PMID:9443499; http://dx.doi.org/10.1212/WNL.50.1.293
- Taylor RW, Chinnery PF, Haldane F, Morris AA, Bindoff LA, Wilson J, Turnbull DM. MELAS associated with a mutation in the valine transfer RNA gene of mitochondrial DNA. Ann Neurol 1996; 40:459-462; PMID:8797538; http://dx.doi.org/10.1002/ana.410400318
- Chalmers RM, Lamont PJ, Nelson I, Ellison DW, Thomas NH, Harding AE, Hammans SR. A mitochondrial DNA tRNA(Val) point mutation associated with adult-onset Leigh syndrome. Neurology 1997; 49:589-592; PMID:9270602; http://dx.doi.org/10.1212/WNL.49.2.589
- Tanji K, Kaufmann P, Naini AB, Lu J, Parsons TC, Wang D, Willey JZ, Shanske S, Hirano M, Bonilla E, et al. A novel tRNA(Val) mitochondrial DNA mutation causing MELAS. J Neurol Sci 2008; 270:23-27; PMID:18314141; http://dx.doi.org/10.1016/j.jns.2008.01.016
- Yan N, Cai S, Guo B, Mou Y, Zhu J, Chen J, Zhang T, Li R, Liu X. A novel mitochondrial tRNA(Val) T1658C mutation identified in a CPEO family. Mol Vis 2010; 16:1736-1742; PMID:20806033
- Menezes MJ, Guo Y, Zhang J, Riley LG, Cooper ST, Thorburn DR, Li J, Dong D, Li Z, Glessner J, et al. Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure, and lactic acidemia. Hum Mol Genet 2015; 24:2297-307; PMID:25556185
- Miller C, Saada A, Shaul N, Shabtai N, Ben-Shalom E, Shaag A, Hershkovitz E, Elpeleg O. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol 2004; 56:734-738; PMID:15505824; http://dx.doi.org/10.1002/ana.20282
- Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, Lombes A, Elpeleg O. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet 2007; 44:784-786; PMID:17873122; http://dx.doi.org/10.1136/jmg.2007.053116
- Smits P, Saada A, Wortmann SB, Heister AJ, Brink M, Pfundt R, Miller C, Haas D, Hantschmann R, Rodenburg RJ, et al. Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur J Hum Genet 2011; 19:394-399; PMID:21189481; http://dx.doi.org/10.1038/ejhg.2010.214
- Galmiche L, Serre V, Beinat M, Assouline Z, Lebre AS, Chretien D, Nietschke P, Benes V, Boddaert N, Sidi D, et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat 2011; 32:1225-1231; PMID:21786366; http://dx.doi.org/10.1002/humu.21562
- Serre V, Rozanska A, Beinat M, Chretien D, Boddaert N, Munnich A, Rötig A, Chrzanowska-Lightowlers ZM. Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim Biophys Acta 2013; 1832:1304-1312; PMID:23603806; http://dx.doi.org/10.1016/j.bbadis.2013.04.014
- Carroll CJ, Isohanni P, Pöyhönen R, Euro L, Richter U, Brilhante V, Götz A, Lahtinen T, Paetau A, Pihko H, et al. Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J Med Genet 2013; 50:151-159; PMID:23315540; http://dx.doi.org/10.1136/jmedgenet-2012-101375
- Diaconu M, Kothe U, Schlünzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 2005; 121, 991-1004; PMID:15989950; http://dx.doi.org/10.1016/j.cell.2005.04.015
- Ghezzi D, Saada A, D'Adamo P, Fernandez-Vizarra E, Gasparini P, Tiranti V, Elpeleg O, Zeviani M. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am J Hum Genet 2008; 83:415-423. Epub 2008 Sep 1014; PMID:18771761; http://dx.doi.org/10.1016/j.ajhg.2008.08.009
- Maltecca F, Magnoni R, Cerri F, Cox GA, Quattrini A, Casari G. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J Neurosci 2009; 29:9244-9254; PMID:19625515; http://dx.doi.org/10.1523/JNEUROSCI.1532-09.2009
- Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, Finardi A, Cagnoli C, Tempia F, Frontali M, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet 2010; 42:313-321; PMID:20208537; http://dx.doi.org/10.1038/ng.544
- Pierson TM, Adams D, Bonn F, Martinelli P, Cherukuri PF, Teer JK, Hansen NF, Cruz P, Mullikin For The Nisc Comparative Sequencing Program JC, Blakesley RW, et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet 2011; 7:e1002325; PMID:22022284; http://dx.doi.org/10.1371/journal.pgen.1002325
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998; 93:973-983; PMID:9635427; http://dx.doi.org/10.1016/S0092-8674(00)81203-9
- Pfeffer G, Gorman GS, Griffin H, Kurzawa-Akanbi M, Blakely EL, Wilson I, Sitarz K, Moore D, Murphy JL, Alston CL, et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014; 137:1323-1336; PMID:24727571; http://dx.doi.org/10.1093/brain/awu060
- Wedding IM, Koht J, Tran GT, Misceo D, Selmer KK, Holmgren A, Frengen E, Bindoff L, Tallaksen CM, Tzoulis C. Spastic paraplegia type 7 is associated with multiple mitochondrial DNA deletions. PLoS One 2014; 9:e86340; PMID:24466038; http://dx.doi.org/10.1371/journal.pone.0086340
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008; 134:112-123; PMID:18614015; http://dx.doi.org/10.1016/j.cell.2008.06.016
- Fischel-Ghodsian N, Prezant TR, Bu X, Oztas S. Mitochondrial ribosomal RNA gene mutation in a patient with sporadic aminoglycoside ototoxicity. Am J Otolaryngol 1993; 14:399-403; PMID:8285309; http://dx.doi.org/10.1016/0196-0709(93)90113-L
- Li R, Xing G, Yan M, Cao X, Liu XZ, Bu X, Guan MX, et al. Cosegregation of C-insertion at position 961 with the A1555G mutation of the mitochondrial 12S rRNA gene in a large Chinese family with maternally inherited hearing loss. Am J Med Genet A 2004; 124A:113-117; PMID:14699607; http://dx.doi.org/10.1002/ajmg.a.20305
- Distelmaier F, Haack TB, Catarino CB, Gallenmüller C, Rodenburg RJ, Strom TM, Baertling F, Meitinger T, Mayatepek E, Prokisch H, et al. MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy. Neurogenetics 2015; 24; PMID:25797485
- Klebe S, Depienne C, Gerber S, Challe G, Anheim M, Charles P, Fedirko E, Lejeune E, Cottineau J, Brusco A, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012; 135:2980-2993; PMID:23065789; http://dx.doi.org/10.1093/brain/aws240