
Abstract
Endoplasmic reticulum (ER) stress is associated with various cardiovascular diseases. However, its pathophysiological relevance and the underlying mechanisms in the context of hypoxia/reoxygenation (H/R) in endothelial cells are not fully understood. Previous findings have suggested that acetylcholine (ACh), the major vagal nerve neurotransmitter, protected against cardiomyocyte injury by activating AMP-activated protein kinase (AMPK). This study investigated the role of ER stress in endothelial cells during H/R and explored the beneficial effects of ACh. Our results showed that H/R triggered ER stress and apoptosis in endothelial cells, evidenced by the elevation of glucose-regulated protein 78, cleaved caspase-12 and C/EBP homologous protein expression. ACh significantly decreased ER stress and terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling positive cells and restored ER ultrastructural changes induced by H/R, possibly via protein kinase-like ER kinase and inositol-requiring kinase 1 pathways. Additionally, 4-diphenylacetoxy-N-methylpiperidine methiodide, a type-3 muscarinic ACh receptor (M3 AChR) inhibitor, abolished ACh-mediated increase in AMPK phosphorylation during H/R. Furthermore, M3 AChR or AMPK siRNA abrogated the ACh-elicited the attenuation of ER stress in endothelial cells, indicating that the salutary effects of ACh were likely mediated by M3 AChR-AMPK signaling. Overall, ACh activated AMPK through M3 AChR, thereby inhibited H/R-induced ER stress and apoptosis in endothelial cells. We have suggested for the first time that AMPK may function as an essential intermediate step between M3 AChR stimulation and inhibition of ER stress-associated apoptotic pathway during H/R, which may help to develop novel therapeutic approaches targeting ER stress to prevent or alleviate ischemia/reperfusion injury.
Abbreviations
| ACh | = | acetylcholine |
| AMPK | = | AMP-activated protein kinase |
| ATF6 | = | activating transcription factor 6 |
| CHOP | = | C/EBP homologous protein |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| ER | = | endoplasmic reticulum |
| GAPDH | = | glyceraldehyde 3-phospharte dehydrogenase |
| GRP78 | = | glucose-regulated protein 78 |
| H/R | = | hypoxia/reoxygenation |
| IRE1 | = | inositol-requiring kinase 1 |
| I/R | = | ischemia/reperfusion |
| MAChR | = | muscarinic acetylcholine receptor |
| PBS | = | phosphate-buffered saline |
| PERK | = | protein kinase-like ER kinase |
| TUNEL | = | terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling |
| 4-DAMP | = | 4-diphenylacetoxy-N-methylpiperidine methiodide |
| 4-PBA | = | 4-phenyl butyric acid |
Introduction
Ischemia/reperfusion (I/R) injury continues to be an important pathology of various cardiovascular diseases.Citation1,2 Endothelial cells locate the innermost layer of blood vessels, so damage to endothelial cells is usually considered as the initial step in the process of I/R.Citation3,4 The pathogenesis of I/R injury is complex, including excessive production of reactive oxygen species, inflammation and cell apoptosis.Citation5,6 Emerging evidence showed that endoplasmic reticulum (ER) stress occurred in the development of myocardial I/R,Citation7-9 and pharmacological inhibition of ER stress could decrease myocardial disorder in failing heart.Citation10,11 ER is abundant in endothelial cells, nevertheless, whether ER stress contributed to vascular endothelial injury during I/R and the underlying mechanism remains poorly defined. Therefore, it is conceivable that exploring novel therapeutics targeting ER stress may be a promising strategy to combat with I/R injury.
Recently, an increasing amount of attention has been focused on the importance of elevating vagal activity to cope with diverse cardiovascular diseases.Citation12 Previous studies have revealed that vagal nerve stimulation decreases infarct size, inflammatory markers and prevents vascular dysfunction induced by myocardial I/R.Citation13,14 Acetylcholine (ACh), the major neurotransmitter of the vagal nerve, elicits its anti-inflammatory and anti-oxidative effects through the activation of muscarinic ACh receptor (M AChR) in H9c2 cardiomyocytes.Citation15,16 Although the beneficial effects of ACh have been well-documented in cardiomyocytes, the impact of ACh on hypoxia/reoxygenation (H/R)-induced ER stress in endothelial cells have not been fully clarified. Improved understanding of the molecular mechanisms underlying ACh-mediated protection in endothelial cells will provide us novel therapeutic perspectives for prevention or treatment of I/R injury.
It should be noted that ACh could increase the activation of AMP-activated protein kinase (AMPK), leading to attenuation of H/R injury in cardiomyocytes.Citation17 AMPK, a threonine/serine kinase, is considered to be a conserved energy sensor.Citation18 There is growing evidence indicating that dysfunctional AMPK is related to the development and progression of many cardiovascular diseases including atherosclerosis, myocardial I/R injury and cardiac remodeling.Citation19-21 Notably, a recent report has demonstrated that AMPK activation ameliorates heart failure by suppressing ER stress.Citation22 However, the role of AMPK in ER stress in human umbilical vein endothelial cell (HUVEC) during H/R remains unknown. The present study was aimed to examine whether aberrant ER stress and apoptosis were involved in H/R-induced endothelial injury and whether ACh functioned as an AMPK activator to suppress ER stress and subsequent cell apoptosis. Furthermore, we explored the possible mechanism of ACh-afforded endothelial protection with a focus on M3 AChR-AMPK signaling cascades.
Results
ER stress and apoptosis occurred during H/R in endothelial cells
We first determined the time course of changes in ER markers after reoxygenation. HUVECs were subjected to hypoxia (1% O2, 8 h), followed by reoxygenation for varying durations (2, 4, 8, 16, 24 h). As shown in , compared with the control group, the expression of glucose-regulated protein 78 (GRP78), an ER-located molecular chaperones, was increased in response to H/R and reached the maximum at 16 h. The protein levels of cleaved caspase-12 and C/EBP homologous protein (CHOP), the indexes of ER stress-related apoptosis, were also increased significantly at 16 h in the context of H/R (). These results revealed that ER stress and apoptosis were triggered during H/R in endothelial cells. Based on the above results, reoxygenation for 16 h was chosen in the subsequent experiments.
Figure 1. ER stress-associated apoptosis was involved in H/R-induced endothelial injury. (A-C) Cell lysates from HUVECs were immunoblotted with antibodies of GRP78, caspase-12 and CHOP with different reoxygenation time course. (D-F) Protein expression of GRP78, caspase-12 and CHOP in endothelial cells treated with an ER stress inhibitor 4-PBA (10−4 M) in the context of H/R. GAPDH served as internal control. Open bar, normoxia; filled bar, H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. *P < 0.05 and **P < 0.01 vs Con; ##P < 0.01 vs H/R.
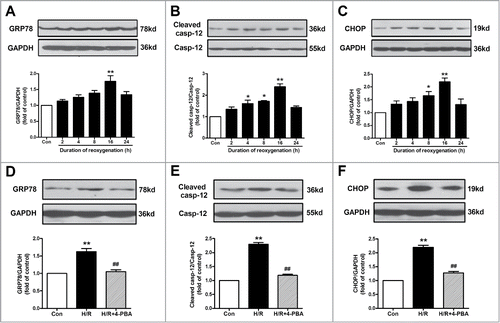
To determine whether ER stress was involved in H/R-induced cell apoptosis, 4-phenyl butyric acid (4-PBA; a widely-used ER stress inhibitor, 10−4 M) was applied in the present study. As shown in , 4-PBA inhibited H/R-induced ER stress, as evidenced by the decrease in the protein level of GRP78. Compared with the H/R group, the expression of cleaved caspase-12 as well as CHOP was also down-regulated in 4-PBA-treated H/R group (). These results suggested that H/R-induced cell apoptosis was attributed, at least in part, to the activation of ER stress-associated apoptotic pathway.
ACh decreased H/R-induced ER stress and apoptosis in a concentration-dependent manner
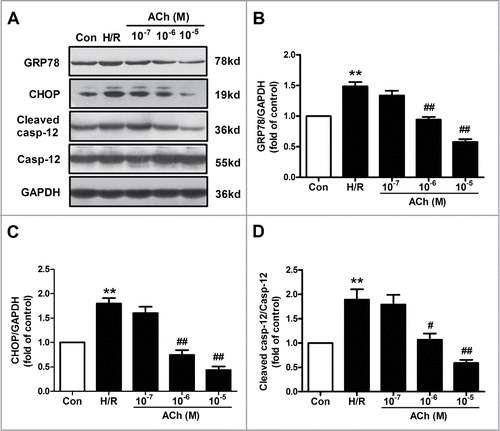
The effect of ACh on ER stress and apoptosis induced by H/R in endothelial cells was shown in . The increased expression in GRP78, cleaved caspase-12 and CHOP were ameliorated by the treatment of ACh (10−7–10−5 M) in a dose-dependent manner, indicating that ACh attenuated H/R-induced ER stress and cell apoptosis. According to the results, ACh was used at the concentration of 10−6 M in the remaining experiments.
Figure 2. ACh diminished H/R-induced ER stress and apoptosis in endothelial cells in a dose-dependent manner. (A) Representative immunoblots for GRP78, caspase-12, CHOP and GAPDH. (B–D) Quantitative analysis results of the expression of GRP78, caspase-12 and CHOP after ACh treatment in the context of H/R. Open bar, normoxia; filled bar, H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. **P <0.01 vs Con; #P < 0.05 and ##P < 0.01 vs H/R.
ACh inhibited ER stress-related apoptosis via M3 AChR
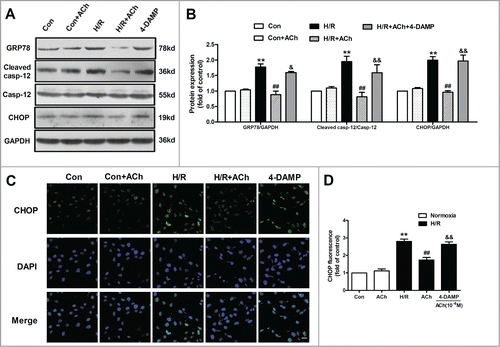
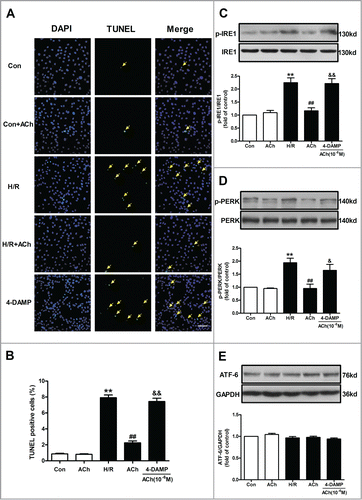
Previous studies have documented that M3 AChR exerts a major role in vasodilatory actions of ACh in vascular endothelium.Citation23 In order to examine which type of ACh receptor plays the predominant role in ACh-mediated suppression of cell apoptosis and ER stress, 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP; a M3 AChR inhibitor, 10−6 M) was used in the present study. The changes of protein expression were assessed by Western blot. As shown in , ACh significantly dampened the increase in GRP78, cleaved caspase-12 and CHOP expression induced by H/R in endothelial cells, and these protective effects of ACh were blunted by 4-DAMP. In order to further confirm abovementioned results, CHOP was selected to determine the immunofluorecense. The fluorescence intensity of CHOP was shown in , which was consistent with Western blot results. ACh significantly decreased H/R-induced elevation of CHOP immunofluorescence compared with the H/R group, and the beneficial effects of ACh were lost in HUVECs treated with the 4-DAMP.
Figure 3. ACh inhibited ER-related apoptosis through M3 AChR. (A) Representative immunoblots for GRP78, caspase-12 and CHOP. (B) ACh treatment decreased the upregulated expression of GRP78, cleaved caspase-12 and CHOP induced by H/R. The beneficial effects of ACh was abolished by M3 AChR antagonist 4-DAMP (10−6 M). (C and D) ACh inhibited CHOP immunofluorescence during H/R. Scale bar = 25 μm. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. **P < 0.01 vs Con; ##P < 0.01 vs H/R; &P < 0.05 and &&P < 0.01 vs ACh-treated H/R group.
ACh restored H/R-induced ultrastructural changes of ER
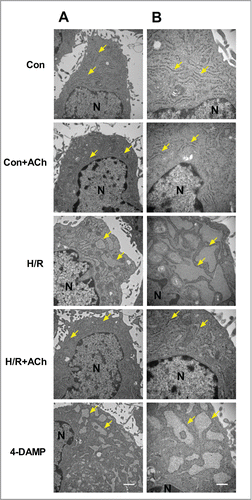
We attempted to evaluate the effect of ACh on ultrastructural changes in HUVECs using transmission electron microscopy. As depicted in , endothelial cells subjected to H/R showed the sack-like cisternae of ER. ACh treatment restored the ER morphology which appeared to be similar to that of the control endothelial cells. In 4-DAMP-treated cells, severe ER expansion was observed. The transmission electron microscopic observations indicated that ACh could preserve endothelial cellular ultrastructural changes triggered by H/R, in particular the changes of ER.
Figure 4. ACh protected against H/R-induced destruction of ER ultrastructure. The transmission electron microscopic observations indicated that ACh could preserve cellular ultrastructural changes triggered by H/R in HUVECs, especially the changes of ER. (A) 20000×, Scale bar = 1 μm. (B) 40000×, Scale bar = 500 nm.
ACh inhibited PERK and IRE1 signaling pathways to protect HUVECs against H/R injury
Cell apoptosis was evaluated using terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling (TUNEL) staining. As shown in , ACh treatment significantly attenuated the H/R-induced TUNEL-positive HUVECs. Treating the cells with 4-DAMP inhibited anti-apoptotic effects of ACh, suggestive of an essential role of M3 AChR in ACh-induced endothelial protection from H/R injury. ER stress triggers protein kinase-like ER kinase (PERK)-, inositol-requiring kinase 1 (IRE1)- and activating transcription factor 6 (ATF6)-mediated apoptotic pathways to induce cell injury.Citation24 Therefore, we examined the role of 3 branches of ER stress signaling cascades and found that PERK and IRE1 phosphorylation were elevated in the H/R group compared with the control group. ACh markedly decreased IRE1 and PERK phosphorylation, and these effects were abolished by 4-DAMP (). Additionally, there is no significant difference in the ATF6 expression between any of the groups (), indicating that ATF6 pathway may be not involved in the H/R-induced endothelial cell apoptosis.
Figure 5. ACh inhibited PERK and IRE1 signaling pathways to protect HUVECs against H/R injury. (A and B) H/R-induced increase in TUNEL-positive cells was inhibited by ACh, 4-DAMP (10−6 M) abrogated the benefits of ACh in the context of H/R. Scale bar = 200 μm. The changes of p-IRE1/IRE1 (C), p-PERK/PERK (D) and ATF6 (E) expression were examined by Western blot. Open bar, normoxia; filled bar, H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. **P < 0.01 vs Con; ##P < 0.01 vs H/R; &P < 0.05 and &&P < 0.01 vs ACh-treated H/R group.
Knockdown of M3 AChR blocked the anti-ER apoptotic effects of ACh
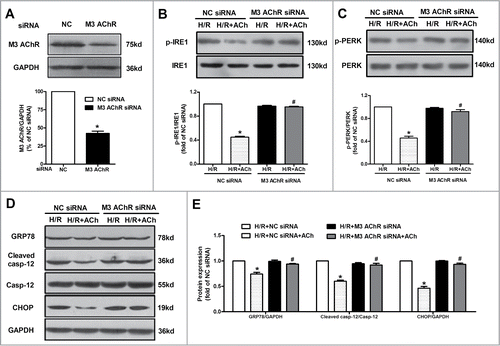
To further determine whether M3 AChR is responsible for the beneficial effects of ACh on anti-ER apoptotic pathway during H/R, we suppressed M3 AChR expression by transfecting siRNA in endothelial cells. As shown in , M3 AChR expression was downregulated by its corresponding siRNA. Next, we assessed the role of M3 AChR in suppression of ER stress and apoptosis by ACh in H/R-injured HUVECs. M3 AChR-depleted cells displayed higher levels of IRE1 and PERK phosphorylation compared with the negative control (NC) siRNA group in the presence of ACh (). Moreover, compared with NC siRNA group, M3 AChR knockdown efficiently blocked the attenuation of GRP78, cleaved caspase-12 and CHOP expression by ACh (). All these data clearly supported the roles of M3 AChR in the ACh-elicited inhibition of ER stress and cell apoptosis in H/R-induced endothelial injury.
Figure 6. Knockdown of M3 AChR blocked the effects of ACh during H/R. (A) The silencing efficiency of M3 AChR siRNA. Cells were transfected with siRNA followed by H/R. The expression changes of p-IRE1/IRE1 (B) and p-PERK/PERK (C) were determined after M3 AChR/NC siRNA with or without ACh. (D and E) Representative immunoblots and quantitative analysis of GRP78, cleaved caspase-12 and CHOP after M3 AChR/NC siRNA with or without ACh. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. *P < 0.01 vs NC siRNA group. #P < 0.01 vs ACh-treated NC siRNA group.
AMPK signaling was responsible for ACh-mediated inhibition of cellular injury
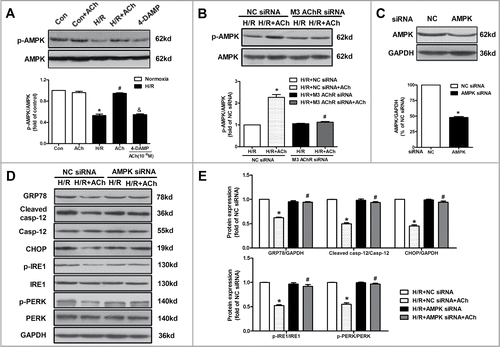
Activation of AMPK protects the heart from myocardial I/R injury via the inhibition of oxidative stress and cell apoptosis.Citation25,26 Thus we evaluated whether AMPK activation contributed to the favorable effects of ACh in endothelial cells. As shown in , H/R caused a reduction of AMPK phosphorylation compared with the control group. Treatment with ACh at reoxygenation increased phosphorylated AMPK in the context of H/R, which was abolished by 4-DAMP. Moreover, M3 AChR siRNA exhibited lower levels of AMPK phosphorylation compared with the NC siRNA group in the presence of ACh (). Based on the above results, AMPK seems to be a potential link between activation of M3 AChR and the inhibition of ER stress in ACh-mediated protection in endothelial cells. Thus we further suppressed AMPK expression by transfecting siRNA in endothelial cells. AMPK expression was down-regulated by its corresponding siRNA (). We assessed the effects of AMPK silence on cell apoptosis and ER stress in H/R-injured HUVECs. Compared with NC siRNA group, knockdown AMPK resulted in elevated protein levels of GRP78, cleaved caspase-12 and CHOP (). Furthermore, AMPK-depleted cells revealed higher levels of IRE1 and PERK phosphorylation compared with the NC siRNA group (). These results suggest that AMPK may function as an important link between inhibition of ER stress-associated apoptotic pathway and M3 AChR activation-induced endothelial protection.
Figure 7. AMPK activation was responsible for the protection of ACh in the context of H/R. (A) ACh increased the phosphorylation of AMPK after H/R, which was blocked by 4-DAMP (10−6 M). *P < 0.01 vs Con; #P < 0.01 vs H/R; &P < 0.01 vs ACh-treated H/R group. (B) M3 AChR-depleted cells reduced the AMPK phosphorylation in the context of H/R. (C) The silencing efficiency of AMPK siRNA. (D) Representative immunoblots and quantitative analysis of GRP78, cleaved caspase-12, CHOP, p-PERK, PERK, p-IRE1 and IRE1 after AMPK/NC siRNA with or without ACh in the context of H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. *P < 0.01 vs NC siRNA group. #P < 0.01 vs ACh-treated NC siRNA group.
Discussion
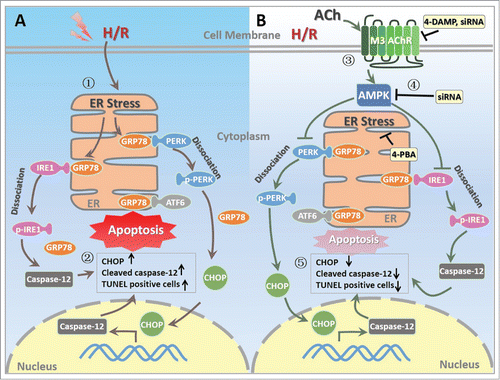
The current study provides novel insights into the mechanism underlying the suppression of ER stress-associated apoptotic pathways by ACh in endothelial cells subjected to H/R. The first intriguing phenomenon was that H/R triggered ER stress, evidenced by increase in the expression of GRP78, cleaved caspase-12 and CHOP, which induced subsequent apoptosis in endothelial cells. The second finding was that ACh treatment significantly decreased ER stress and TUNEL positive cells and restored ER ultrastructural changes induced by H/R, possibly via PERK and IRE1 signaling pathways, but not ATF6 cascade. Finally, M3 AChR or AMPK siRNA abrogated the ACh-elicited the attenuation of ER stress in endothelial cells, indicating that the salutary effects of ACh were likely mediated by M3 AChR-AMPK signaling cascades. We have suggested for the first time that AMPK may function as an essential intermediate step between M3 AChR stimulation and inhibition of ER stress-associated apoptotic pathway during H/R (), indicating M3 AChR-AMPK pathway may be a novel therapeutic target for modulation of ER stress in endothelial cells.
Figure 8. Proposed schematic illustration of the mechanism by which ACh exerts its endothelial protective effects against H/R injury in the present study. (1) H/R induces ER stress in endothelial cell; (2) H/R upregulates the expression of CHOP and cleaved caspase-12 as well as TUNEL positive HUVECs; (3) ACh activates M3 AChR to elicit its endothelial protection; (4) ACh enhances AMPK phosphorylation, thereby inhibits pro-apoptotic cascades; (5) ACh decreases ER stress and apoptosis during H/R in endothelial cells, presumably by M3 AChR signaling pathway. ACh, acetylcholine; AMPK, AMP-activated protein kinase; ATF6, activating transcription factor 6; CHOP, C/EBP homologous protein; ER, endoplasmic reticulum; GRP78, glucose-regulated protein 78; H/R, hypoxia/reoxygenation; IRE1, inositol-requiring kinase 1; M3 AChR, type-3 muscarinic acetylcholine receptor; PERK, protein kinase-like ER kinase; TUNEL, terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling; 4-DAMP, 4-diphenylacetoxy-N-methylpiperidine methiodide; 4-PBA, 4-phenyl butyric acid.
Substantial evidence has accumulated that ER stress plays an essential role in the pathological processes of myocardial I/R and heart failure.Citation22,27 Excessive or prolonged ER stress usually promotes apoptotic cascades by induction of transcription factor CHOP or activation of caspase-12.Citation28,29 Knockdown of CHOP or caspase-12 resulted in decreased ER stress and subsequent cell apoptosis in cardiomycytes.Citation16,27 In endothelial cells, ER is abundant and critical for governing calcium storage, lipid synthesis, protein folding and maturation.Citation30,31 Indeed, disruption of ER calcium homeostasis results in ER stress.Citation32 A previous study has demonstrated that increased Ca2+ flux from ER to mitochondria may trigger celastrol-induced paraptotic cell death in cancer cells.Citation33 Our recent findings have also shown that Ca2+ transfer from ER to mitochondria via the voltage-dependent anion channel-1/glucose-regulated protein 75/inositol 1,4,5-trisphosphate receptor 1 complex, induce ER-associated apoptosis during H/R in endothelial cells.Citation34 Thus, ER plays an important role in endothelial function. In other words, perturbations in ER homeostasis (e.g., ER stress) exert a maladaptive effect on physiological functions of endothelium. Our findings showed that, in response to H/R stimuli, the expression of CHOP and cleaved caspase-12 as well as TUNEL positive cells significantly increased in endothelial cells, which was in line with previous data in the cardiomyocytes.Citation16 These results indicated that ER stress occurred in endothelial cells subjected to H/R. Thus, it is conceivable that reversing ER stress could favorably affect endothelial function and represent a strategic target for I/R injury.
Several lines of evidence suggest that vagal nerve stimulation and ACh elicit favorable effects in various cardiovascular diseases, including myocardial I/R, hypertension and heart failure.Citation14,35,36 Previous study has demonstrated that ACh inhibits oxidative stress-mediated apoptosis in mouse embryonic stem cells and H9c2 cells.Citation37,38 A recent study from Li et al has suggested that ACh suppresses hypoxia-induced apoptosis by inhibiting inflammatory response in cardiomyocytes.Citation39 However, little information is available on the impact of ACh on the regulation of ER stress-associated apoptosis in endothelial cells during H/R. The present study showed that ACh reduced ER stress evidenced by decrease in GRP78, cleaved caspase-12 and CHOP expression, as well as TUNEL positive cells, indicating that inhibition of ER stress was responsible for ACh-afforded endothelial protection.
It should be noted that accumulation of unfolded proteins in the ER activates PERK-, IRE1- and ATF6-mediated pathways to induce cell apoptosis.Citation40,41 Consistently, we found that PERK and IRE1 dissociated from ER-localized GRP78 and initialed distinct pathway to induce apoptosis in endothelial cells during H/R, which was reversed by ACh treatment. Interestingly, in our current study, we did not observe changes of ATF6 in the context of H/R. This may be explained by the fact that the timing of activation of 3 ER stress signaling cascades could be different.Citation42 Previous study showed that the response of both IRE1 and PERK was induced by thapsigargin-triggered ER stress, whereas the response of ATF6 was markedly delayed.Citation43 Moreover, Wu et al manifested that ATF6 was activated by ischemia but inactivated on reperfusion, and this may be responsible for the absence of ATF6 potential role observed here.Citation44 Further studies are required to explore these issues.
The present study demonstrated that ACh inhibited endothelial cell apoptosis through IRE1 and PERK signaling pathways under H/R condition. ACh acts on either muscarinic or nicotinic AChR to exert its effect.Citation45 Previous studies in our lab have shown that ACh benefits to the heart and endothelium by acting on M AChR.Citation14-16,46 M3 AChR siRNA as well as 4-DAMP, significantly abolished the protective effect elicited by ACh, suggesting that the action of ACh on ER stress was mediated mainly by M3 AChR. Intriguingly, our results also showed that M3 AChR siRNA abolished ACh-elicited restoration of AMPK during H/R. Furthermore, AMPK-depleted cells exhibited higher levels of ER stress markers and cell apoptosis, as compared to NC siRNA-treated H/R group in the presence of ACh, indicating that M3 AChR-AMPK pathway plays a role in the beneficial effects of ACh. Consistent with our present study, Terai et al. have showed that activation of AMPK inhibited ER stress and protected cardiomyocyte against hypoxic injury.Citation27 In a rat model of isoproterenol-induced heart failure, metformin administration activated AMPK and remarkably attenuated ER stress, thus exerting a prominent cardioprotection.Citation22 These results indicate that AMPK might be important for maintaining ER homeostasis and that aberrant ER stress might lead to the initiation and progression of endothelial cell injury following H/R. Therefore, we speculate that ACh mediates endothelial protection in this cellular model may attributed, at least in part, to the activation of AMPK.
In the present study, we focused on the role of ER stress in ACh-induced endothelial protection at the cellular level. A recent report has shown that attenuation of the ER stress exerts cardioprotective effects in the rats suffered from myocardial I/R.Citation47 Cell injury from ER stress is emerging as a critical pathophysiology of a wide range of prevalent human diseases, including cardiovascular diseases, diabetes and stroke.Citation11,28 Thus, inhibition of ER stress will hopefully lead to new strategies to combat these ER stress-associated diseases in clinics. Interestingly, our recent study demonstrated that activation of M3 AChR by choline ameliorated I/R-induced vascular dysfunction in a rat model, showing the in vivo functional significance of increasing the ACh level and its mediated signaling cascades.Citation48 The effects of ACh or enhanced vagal activity on ER stress in vivo in experimental animals required further investigation. Additionally, previous study has shown that ACh level in the rabbit left ventricle induced by vagal nerve stimulation is at the nM level.Citation49 However, the amount of ACh in the synaptic space can reach the millimolar range after quantal release.Citation50 Moreover, the therapeutic concentration of ACh under the pathological conditions is generally higher than the physiological level.Citation37,38 In our study, ACh (10−6 M) effectively protected against ER stress and showed prominent endothelial protection. Therefore, we chose 10−6 M ACh for our experiments, which is consistent with previous studies.Citation16,51
In summary, the salient findings of this study are that ACh mediates inhibition of ER stress and related apoptotic signaling through M3 AChR-AMPK pathway in endothelial cells, providing new insights into the benefits of ACh in the context of I/R (). AMPK may function as an important upstream regulator of ER stress in endothelial cells, thus activation of AMPK may be a novel target for vascular endothelial protection. These results also represent a significant addition to the understanding of ACh-mediated pathways and the potential roles they play in regulating ER stress, which may help to develop novel therapeutic approaches targeting ER stress to prevent or alleviate I/R injury.
Materials and Methods
Cell line, cell culture
The HUVEC line (CRL-1730) was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and was incubated in Ham's F12K medium (Macgene Biotech Co., Ltd, Beijing, China) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 0.03 mg/ml endothelial cell growth supplement (Macgene Biotech Co., Ltd), 100 U/ml penicillin (Sigma, St. Louis, MO, USA) and 100 μg/ml streptomycin (Sigma) in a humidified incubator with 95% air and 5% CO2 at 37°C. Cells were fed every 2–3 d and split 1 to 3 at 80% confluence. The 4th to 8th passages of HUVECs were used in the present study.
Cell treatment
The medium was replaced by a fresh medium prior to each experiment. After grown to 80% confluence, cells were starved in serum-free Ham's F12K (FBS was eliminated in all the following experiments) for 12 h and then were transferred into a hypoxic incubator containing 1% O2, 5% CO2, and 94% N2 with a modified ischemia-mimetic solution (in mM: NaCl, 135; KCl, 8; NaH2PO4, 0.33; MgCl2, 0.5; Hepes, 5.0; CaCl2, 1.8; and lactate, 20, pH 6.80). The endothelial cells were subjected to hypoxia for 8 h to induce simulated ischemia as previously described.Citation46 For reoxygenation, the medium was replaced with fresh serum-free F12K medium and rapidly transferred into a normoxic incubator (5% CO2 and 95% air) for 16 h. Then the cells were divided randomly into the following groups for treatment: (1) Con: culture normally with F12K; (2) ACh: administration of ACh (10−6 M) alone; (3) H/R: 8 h of hypoxia followed by reoxygenation for 16 h; (4) H/R + 4-PBA: 4-PBA (10−4 M) was added at the onset of reoxygenation; (5) H/R + 4-DAMP + ACh: M3 AChR inhibitor 4-DAMP (10−6 M) was used in combination with ACh (10−6 M) at the onset of reoxygenation. These agents were all purchased from Sigma.
Western blot analysis
HUVECs were rinsed twice with ice-cold phosphate-buffered saline (PBS) and lysed in RIPA buffer (Beyotime Biotech, Haimen, China) containing 1 mM phenylmethylsulfonyl fluoride. The lysates were further centrifuged at 13000 rpm for 15 min at 4°C, then the supernatants were collected and stored at –80°C. Total protein concentrations were determined using Bicinchoninic acid protein assay kit (Beyotime Biotech). Equal amount of protein (30 μg) with loading buffer were separated run on the SDS-poly-acrylamide gels, electrotransferred to nitrocellulose membranes (Millipore, Billerica, MA, USA). The membranes were blocked for 1 h in Tris-buffered saline containing 5% nonfat milk and 0.1% Tween 20 at room temperature, and then incubated at 4°C overnight with primary antibody against GAPDH (diluted 1:5000; Sinopept, Beijing, China), CHOP (diluted 1:500; Signalway Antibody, Pearland, TX, USA), caspase-12 (diluted 1:1000; Abnova, Walnut, CA, USA), GRP78 (diluted 1:1000; Cell Signaling Technology, Beverly, MA, USA), PERK (diluted 1:1000; Cell Signaling Technology), phospho-PERK(Thr981) (diluted 1:200; Santa Cruz Biotechnology, CA, USA), IRE1 (diluted 1:1000; Cell Signaling Technology); phospho-IRE1(Ser724) (diluted 1:1000; Novus Biologicals, Littleton, CO, USA), ATF6 (diluted 1:1000; Bioworld Technology, MN, USA); AMPK (diluted 1:1000; Cell Signaling Technology); phospho-AMPK(Thr172) (diluted 1:1000; Cell Signaling Technology); M3 AChR (diluted 1:1000; Millipore). After incubation with appropriate horseradish peroxidase-labeled secondary antibodies (diluted 1:5000; Signalway Antibody) for 40 min at room temperature, the proteins were detected by ECL-Plus reagent (Millipore), followed by grayscale analysis using the Gel-Pro Analyzer (Media Cybernetics, Bethesda, MD, USA).
Transmission electron microscopy
HUVECs were harvested and fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2–7.4) for 2 h at 4°C. After washing in phosphate buffer, the endothelial cells were post-fixed with 1% osmium tetroxide in 0.1 M phosphate buffer for 2 h. The samples were dehydrated in a graded ethanol series, infiltrated with propylene oxide, embedded in epoxy resin and sliced into ultrathin sections with glass knives on a LKB-V ultramicrotome. After double-stained with uranyl acetate and lead citrate, electron microphotographs were obtained by a transmission electron microscope (H-7650; Hitachi, Tokyo, Japan).Citation52
Immunofluorescence and confocal microscopy
Immunofluorescence staining was performed according to the manufacturer's instructions. In brief, HUVECs were washed twice by PBS and fixed with 4% paraformaldehyde (Sigma) for 30 min, followed by blocking with normal goat serum (Boster; Wuhan, China) for 1 h at room temperature. Then the samples were incubated with primary antibody against CHOP (diluted 1:100) overnight at 4°C. Corresponding fluorescein isothiocyanate (FITC)-conjugated secondary antibody (1:200, Zhongshan Goldenbridge Biotechnology; Beijing, China) was added for 30 min at 37°C and then cells were rinsed twice in PBS. 4′,6-diamidino-2-phenylindole (DAPI) was used to visualize nucleus. The staining was observed under a laser confocal microscope (Nikon C2 system; Nikon, Tokyo, Japan).
TUNEL staining
HUVECs were stained using a DeadEnd Fluorometric TUNEL system (Promega, Madison, WI, USA) for apoptosis detection according to the manufacturer's instructions.Citation15 Briefly, cells were grown on collagen-coated coverslips and washed with PBS after treatment. Then the cells were fixed with 4% paraformaldehyde (Sigma) and incubated with 50 µl of TUNEL reaction mixture added to each sample for 1 h at 37°C. After incubation with the fluorescent label DAPI, the staining was observed under a fluorescence microscope (TE-2000U, Nikon, Japan). At least 10 randomly chosen fields with 400 DAPI-positive cells were scored, and the number of TUNEL-positive cells was presented as a percentage of positive nuclei/total nuclei counted 100 %.
Small interfering RNA of M3 AChR and AMPK
Cells were seeded into 6-well plates. After the cells reached 70–80% confluence, they were transfected with M3 AChR siRNA, AMPK siRNA or NC siRNA, which were synthesized and purchased from Shanghai GenePharma Co. Ltd. (Shanghai, China). Transfections were performed using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions with 100 nM of each siRNA.Citation38 The efficiency of siRNA-mediated AMPK, M3 AChR knockdown were verified by Western blot 48 h after transfection.
Statistical analysis
Data were expressed as mean ± SEM. All results were performed using student's t-test or one-way analysis of variance followed by a Tukey post hoc test for multiple comparisons. The package GraphPad Prism Version 5.01 (GraphPad Software; San Diego, CA, USA) was used to perform all the statistical analysis. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1060383Supplements.docx
Download MS Word (315.3 KB)Acknowledgments
We appreciate the technical support and materials from the electron microscope center of Xi'an Jiaotong University.
Funding
This work is supported by National Natural Science Foundation of China (Major International Joint Research Project, No. 81120108002; General Project, No. 81473203), Specialized Research Fund for the Doctoral Program of Higher Education (No. 20130201130008). We are grateful to Dr. Wendy W. Zang for English editing.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 2012; 298:229–317; PMID:22878108; http://dx.doi.org/10.1016/B978-0-12-394309-5.00006-7
- Bi XY, He X, Zhao M, Yu XJ, Zang WJ. Role of endothelial nitric oxide synthase and vagal activity in the endothelial protection of atorvastatin in ischemia/reperfusion injury. J Cardiovasc Pharmacol 2013; 61:391–400; PMID:23364605; http://dx.doi.org/10.1097/FJC.0b013e318286baf3
- Xie P, Duan Y, Guo X, Hu L, Yu M. SalA attenuates hypoxia-induced endothelial endoplasmic reticulum stress and apoptosis via down-regulation of VLDL receptor expression. Cell Physiol Biochem 2015; 35:17–28; PMID:25547648; http://dx.doi.org/10.1159/000369671
- Wang J, Chen S, Ma X, Cheng C, Xiao X, Chen J, Liu S, Zhao B, Chen Y. Effects of endothelial progenitor cell-derived microvesicles on hypoxia/reoxygenation-induced endothelial dysfunction and apoptosis. Oxid Med Cell Longev 2013; 2013:572729; PMID:24288585; http://dx.doi.org/10.1155/2013/572729
- Vemula P, Gautam B, Abela GS, Wang DH. Myocardial ischemia/reperfusion injury: potential of TRPV1 agonists as cardioprotective agents. Cardiovasc Hematol Disord Drug Targets 2014; 14:71–8; PMID:24304232; http://dx.doi.org/10.2174/1871529´13666131129103759
- Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol 2010; 106:360–8; PMID:20643246; http://dx.doi.org/10.1016/j.amjcard.2010.03.032
- Liu XH, Zhang ZY, Sun S, Wu XD. Ischemic postconditioning protects myocardium from ischemia/reperfusion injury through attenuating endoplasmic reticulum stress. Shock 2008; 30:422–27; PMID:18323739; http://dx.doi.org/10.1097/SHK.0b013e318164ca29
- Wang Z, Wang Y, Ye J, Lu X, Cheng Y, Xiang L, Chen L, Feng W, Shi H, Yu X, et al. bFGF attenuates endoplasmic reticulum stress and mitochondrial injury on myocardial ischaemia/reperfusion via activation of PI3K/Akt/ERK1/2 pathway. J Cell Mol Med 2015; 19:595–607; PMID:25533999; http://dx.doi.org/10.1111/jcmm.12346
- Guo XF, Yang XJ. Endoplasmic reticulum stress response in spontaneously hypertensive rats is affected by myocardial ischemia reperfusion injury. Exp Ther Med 2015; 9:319–326; PMID:25574192; http://dx.doi.org/10.3892/etm.2014.2094
- Zhang GG, Teng X, Liu Y, Cai Y, Zhou YB, Duan XH, Song JQ, Shi Y, Tang CS, Yin XH, et al. Inhibition of endoplasm reticulum stress by ghrelin protects against ischemia/reperfusion injury in rat heart. Peptides 2009; 30:1109–1116; PMID:19406177; http://dx.doi.org/10.1016/j.peptides.2009.03.024
- Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol 2013; 75:49–67; PMID:23020580; http://dx.doi.org/10.1146/annurev-physiol-030212-183707
- He X, Zhao M, Bi X, Sun L, Yu X, Zhao M, Zang W. Novel strategies and underlying protective mechanisms of modulation of vagal activity in cardiovascular diseases. Br J Pharmacol 2015; PMID:25378088; http://dx.doi.org/10.1111/bph.13010 [Epub ahead of print]
- Calvillo 1, Vanoli E, Andreoli E, Besana A, Omodeo E, Gnecchi M, Zerbi P, Vago G, Busca G, Schwartz PJ. Vagal stimulation, through its nicotinic action, limits infarct size and the inflammatory response to myocardial ischemia and reperfusion. J Cardiovasc Pharmacol 2011; 58:500–7; PMID:21765369; http://dx.doi.org/10.1097/FJC.0b013e31822b7204
- Zhao M, He X, Bi XY, Yu XJ, Gil Wier W, Zang WJ. Vagal stimulation triggers peripheral vascular protection through the cholinergic anti-inflammatory pathway in a rat model of myocardial ischemia/reperfusion. Basic Res Cardiol 2013; 108:345; PMID:23519622; http://dx.doi.org/10.1007/s00395-013-0345-1
- Liu JJ, Li DL, Zhou J, Sun L, Zhao M, Kong SS, Wang YH, Yu XJ, Zhou J, Zang WJ. Acetylcholine prevents angiotensin II-induced oxidative stress and apoptosis in H9c2 cells. Apoptosis 2011; 16:94–103; PMID:20963497; http://dx.doi.org/10.1007/s10495-010-0549-x
- Miao Y, Bi XY, Zhao M, Jiang HK, Liu JJ, Li DL, Yu XJ, Yang YH, Huang N, Zang WJ. Acetylcholine inhibits tumor necrosis factor α activated endoplasmic reticulum apoptotic pathway via EGFR-PI3K signaling in cardiomyocytes. J Cell Physiol 2015; 230:767–74; PMID:25201632; http://dx.doi.org/10.1002/jcp.24800
- Zhao M, Sun L, Yu XJ, Miao Y, Liu JJ, Wang H, Ren J, Zang WJ. Acetylcholine mediates AMPK-dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cell Physiol Biochem 2013; 32:601–13; PMID:24021916; http://dx.doi.org/10.1159/000354464
- Song P, Zou MH. Regulation of NAD(P)H oxidases by AMPK in cardiovascular systems. Free Radic Biol Med 2012; 52:1607–19; PMID:22357101; http://dx.doi.org/10.1016/j.freeradbiomed.2012.01.025
- Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, Choi HC, Zou MH. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010; 121:792–803; PMID:20124121; http://dx.doi.org/10.1161/CIRCULATIONAHA.109.900928
- Kataoka Y, Shibata R, Ohashi K, Kambara T, Enomoto T, Uemura Y, Ogura Y, Yuasa D, Matsuo K, Nagata T, et al. Omentin prevents myocardial ischemic injury through AMP-activated protein kinase- and Akt-dependent mechanisms. J Am Coll Cardiol 2014; 63:2722–33; PMID:24768874; http://dx.doi.org/10.1016/j.jacc.2014.03.032
- Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol 2010; 45:276–95; PMID:20522000; http://dx.doi.org/10.3109/10409238.2010.488215
- Zhuo XZ, Wu Y, Ni YJ, Liu JH, Gong M, Wang XH, Wei F, Wang TZ, Yuan Z, Ma AQ, et al. Isoproterenol instigates cardiomyocyte apoptosis and heart failure via AMPK inactivation-mediated endoplasmic reticulum stress. Apoptosis 2013; 18:800–10; PMID:23620435; http://dx.doi.org/10.1007/s10495-013-0843-5
- Khurana S, Chacon I, Xie G, Yamada M, Wess J, Raufman JP Kennedy RH. Vasodilatory effects of cholinergic agonists are greatly diminished in aorta from M3R−/− mice. Eur J Pharmacol 2004; 493:127–32; PMID:15189773; http://dx.doi.org/10.1016/j.ejphar.2004.04.012
- Groenendyk J, Sreenivasaiah PK, Kim do H, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res 2010; 107:1185–97; PMID:21071716; http://dx.doi.org/10.1161/CIRCRESAHA.110.227033
- Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ, Lopez BL, Christopher TA, Tian R, Koch W, et al. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation 2009; 119:835–44; PMID:19188503; http://dx.doi.org/10.1161/CIRCULATIONAHA.108.815043
- Chen K, Li G, Geng F, Zhang Z, Li J, Yang M, Dong L, Gao F. Berberine reduces ischemia/reperfusion-induced myocardial apoptosis via activating AMPK and PI3K-Akt signaling in diabetic rats. Apoptosis 2014; 19:946–57; PMID:24664781; http://dx.doi.org/10.1007/s10495-014-0977-0
- Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol 2005; 25:9554–75; PMID:16227605; http://dx.doi.org/10.1128/MCB.25.21.9554–9575.2005
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 2015; 10:173–94; PMID:25387057; http://dx.doi.org/10.1146/annurev-pathol-012513-104649
- Macaluso M, Caracciolo V, Rizzo V, Sun A, Montanari M, Russo G, Bellipanni G, Khalili K, Giordano A. Integrating role of T antigen, Rb2/p130, CTCF and BORIS in mediating non-canonical endoplasmic reticulum-dependent death pathways triggered by chronic ER stress in mouse medulloblastoma. Cell Cycle 2012; 11:1841–50; PMID:22544282; http://dx.doi.org/10.4161/cc.20242
- Estrada IA, Donthamsetty R, Debski P, Zhou MH, Zhang SL, Yuan JX, Han W, Makino A. STIM1 restores coronary endothelial function in type 1 diabetic mice. Circ Res 2012; 111:1166–75; PMID:22896585; http://dx.doi.org/10.1161/CIRCRESAHA.112.275743
- Park IJ, Kim MJ, Park OJ, Choe W, Kang I, Kim SS, Ha J. Cryptotanshinone induces ER stress-mediated apoptosis in HepG2 and MCF7 cells. Apoptosis 2012; 17:248–57; PMID:22113823; http://dx.doi.org/10.1007/s10495-011-0680-3
- Dong S, Teng Z, Lu FH, Zhao YJ, Li H, Ren H, Chen H, Pan ZW, Lv YJ, Yang BF, et al. Post-conditioning protects cardiomyocytes from apoptosis via PKC(epsilon)-interacting with calcium-sensing receptors to inhibit endo(sarco)plasmic reticulum-mitochondria crosstalk. Mol Cell Biochem 2010; 341:195–206; PMID:20383739; http://dx.doi.org/10.1007/s11010-010-0450-5
- Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, Choi KS. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget 2014; 5:6816–31; PMID:25149175
- He X, Bi XY, Lu XZ, Zhao M, Yu XJ, Sun L, Xu M, Wier WG, Zang WJ. Reduction of mitochondria-endoplasmic reticulum interactions by acetylcholine protects human umbilical vein endothelial cells from hypoxia/reoxygenation injury. Arterioscler Thromb Vasc Biol 2015; PMID:25977565; http://dx.doi.org/10.1161/ATVBAHA.115.305469 [Epub ahead of print]
- Petkovich BW, Vega J, Thomas S. Vagal modulation of hypertension. Curr Hypertens Rep 2015; 17:532; PMID:25794951; http://dx.doi.org/10.1007/s11906-015-0532-6
- Schwartz PJ, De Ferrari GM, Sanzo A, Landolina M, Rordorf R, Raineri C, Campana C, Revera M, Ajmone-Marsan N, Tavazzi L, et al. Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Fail 2008; 10:884–91; PMID:18760668; http://dx.doi.org/10.1016/j.ejheart.2008.07.016
- Kim MH, Kim MO, Heo JS, Kim JS, Han HJ. Acetylcholine inhibits long-term hypoxia-induced apoptosis by suppressing the oxidative stress-mediated MAPKs activation as well as regulation of Bcl-2, c-IAPs, and caspase-3 in mouse embryonic stem cells. Apoptosis 2008; 13:295–304; PMID:18049903; http://dx.doi.org/10.1016/10.1007/s10495-007-0160-y
- Miao Y, Zhou J, Zhao M, Liu J, Sun L, Yu X, He X, Pan X, Zang W. Acetylcholine attenuates hypoxia/reoxygenation-induced mitochondrial and cytosolic ROS formation in H9c2 cells via M2 acetylcholine receptor. Cell Physiol Biochem 2013; 31:189–98; PMID:23407103; http://dx.doi.org/10.1159/000343360
- Li DL, Liu JJ, Liu BH, Hu H, Sun L, Xu HF, Yu XJ, Ma X, Ren J, et al. Acetylcholine inhibits hypoxia-induced tumor necrosis factor-α production via regulation of MAPKs phosphorylation in cardiomyocytes. J Cell Physiol 2011; 226:1052–9; PMID:20857413; http://dx.doi.org/10.1002/jcp.22424
- Millott R, Dudek E, Michalak M. The endoplasmic reticulum in cardiovascular health and disease. Can J Physiol Pharmacol 2012; 90:1209–17; PMID:22897133; http://dx.doi.org/10.1139/y2012-058
- Zhou B, Li H, Liu J, Xu L, Zang W, Wu S, Sun H. Intermittent injections of osteocalcin reverse autophagic dysfunction and endoplasmic reticulum stress resulting from diet-induced obesity in the vascular tissue via the NFκB-p65-dependent mechanism. Cell Cycle 2013; 12:1901–13; PMID:23708521; http://dx.doi.org/10.4161/cc.24929
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011; 13:184–90; PMID:21364565; http://dx.doi.org/10.1038/ncb0311-184
- DuRose JB, Tam AB, Niwa M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol Biol Cell. 2006; 17:3095–107; PMID:16672378; http://dx.doi.org/10.1091/mbc.E06-01-0055
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell 2007; 13:351–64; PMID:17765679; http://dx.doi.org/10.1016/j.devcel.2007.07.005
- Ofek K, Soreq H. Cholinergic involvement and manipulation approaches in multiple system disorders. Chem Biol Interact 2013; 203:113–9; PMID:22898318; http://dx.doi.org/10.1016/j.cbi.2012.07.007
- Sun L, Zang WJ, Wang H, Zhao M, Yu XJ, He X, Miao Y, Zhou J. Acetylcholine promotes ROS detoxification against hypoxia/reoxygenation-induced oxidative stress through FoxO3a/PGC-1α dependent superoxide dismutase. Cell Physiol Biochem 2014; 34:1614–25; PMID:25402826; http://dx.doi.org/10.1159/000366364
- Wei K, Liu L, Xie F, Hao X, Luo J, Min S. Nerve growth factor protects the ischemic heart via attenuation of the endoplasmic reticulum stress induced apoptosis by activation of phosphatidylinositol 3-kinase. Int J Med Sci 2015; 12:83–91; PMID:25552923; http://dx.doi.org/10.7150/ijms.10101
- Lu XZ, Bi XY, He X, Zhao M, Xu M, Yu XJ, Zhao ZH, Zang WJ. Activation of M3 muscarinic acetylcholine receptors attenuates ischaemia/reperfusion-induced vascular injury via inhibition of the ROS-mediated CaMKII pathway. Br J Pharmacol 2015; PMID:25953628; http://dx.doi.org/10.1111/bph.13183 [Epub ahead of print]
- Kawada T, Akiyama T, Shimizu S, Kamiya A, Uemura K, Li M, Shirai M, Sugimachi M. Detection of endogenous acetylcholine release during brief ischemia in the rabbit ventricle: a possible trigger for ischemic preconditioning. Life Sci 2009; 85:597–601; PMID:19733187; http://dx.doi.org/10.1016/j.lfs.2009.08.015
- Engel AG, Shen XM, Selcen D, Sine S. New horizons for congenital myasthenic syndromes. Ann N Y Acad Sci 2012; 1275:54–62; PMID:23278578; http://dx.doi.org/10.1111/j.1749-6632.2012.06803.x
- Sugimoto K, Fujii S, Takemasa T, Yamashita K. Detection of intracellular nitric oxide using a combination of aldehyde fixatives with 4,5-diaminofluorescein diacetate. Histochem Cell Biol 2000; 113:341–7; PMID:0883393; http://dx.doi.org/10.1007/s004180000151
- He X, Zhao M, Bi XY, Yu XJ, Zang WJ. Delayed preconditioning prevents ischemia/reperfusion-induced endothelial injury in rats: role of ROS and eNOS. Lab Invest 2013; 93:168–80; PMID:23147223; http://dx.doi.org/10.1038/labinvest.2012.160