
It can be argued that the development of effective anti-bacterial agents have shaped the history of the twentieth century. The success of these drugs is largely driven by the availability of unique bacterial proteins that are absent in human cells. Drugs that inhibit these proteins induce bacterial killing without significant toxicity to the patient. In contrast, cancer is a disease that is indelibly and undeniably human. With few exceptions,Citation1 there are no unique proteins in human cancer cells that are absent in normal cells and would trigger irreversible cancer death upon inhibition.
The discovery of oncogenic “driver” mutations led to the hypothesis that cancer cells may be uniquely dependent on onco-proteins encoded by these mutated genes.Citation2 However, the clinical experiences with drugs that selectively inhibit these proteins have been largely disappointing.Citation3 Efforts devoted to better understanding these “targeted” agents have taught us the following. Most oncogenes are members of large families of proto-oncogenes, and there are significant degrees of functional degeneracy between oncogenes and cellular proto-oncogenes. Consequently, cancer cells can circumvent the inhibition of onco-proteins through activation of alternative proto-oncogenes.Citation3
It is in this context that the work reported by Ying et al is of importance.Citation4 The authors studied glioblastoma, the most common form of primary brain cancer.Citation5 Hyper-activation of Epidermal Growth Factor Receptor (EGFR) is a critical step in the pathogenesis of glioblastomas.Citation6 However, when treated with EGFR inhibitors, EGFR expressing glioblastoma cells quickly acquire resistance by activating proto-oncogenes that substitute for EGFR.Citation4 Unfortunately, a wide spectrum of proto-oncogenes is activated during this process such that simultaneous inhibition of these gene products would not be feasible.Citation3
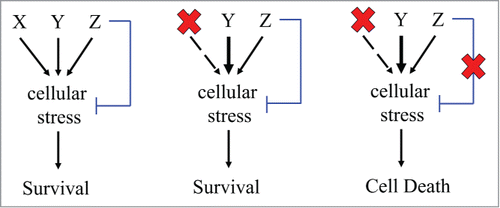
The critical observation made by the authors is that EGFR activation in glioblastoma triggers a cell state characterized by excessive accumulation of DNA damage, requiring increased utilization of DNA repair in order to ensure cell viability.Citation7 The authors showed that, once this cell state is established, it is preserved even when EGFR is inhibited and another proto-oncogene is activated. The authors isolated independent glioblastoma clones that developed distinct mechanisms of resistance to 2 clinically used EGFR inhibitors, Erlotnib and Gefitnib. Notably, all clones isolated remained universally sensitive to an inhibitor of DNA repair targeting the Polo-Like Kinase 1(PLK1). These results suggest that activating an alternate proto-oncogene to drive the existing cellular circuit would be less costly than a complete realignment of fundamental cellular processes. The work suggests potential change in therapeutic paradigm at fundamental level from one targeting unique protein to one targeting unique cell states (Fig. 1).
Because EGFR inhibitors have not been shown to alter DNA damage response and DNA repair inhibitors have not been shown to alter EGFR signaling, the authors propose that these therapeutic approaches impose parallel and orthogonal selection pressures on glioblastoma cells. Accordingly, therapeutic synergy was observed in this combined approach.
These results establish a sound foundation for combinatorial therapeutic approaches integrating inhibitors directed against oncogenes and oncogene associated cell-states. It is likely that oncogenes functionally intersect other fundamental cellular processes, such as DNA replication, transcription, or mitosis. Importantly, the aggregate of work over the past 50 years have yielded effective inhibitors to many of these processes. Characterizing the therapeutic efficacy of these inhibitors with molecular targeted agents should afford opportunities for meaningful improvement in the clinical outcome cancer patients and contribute to human history for the upcoming century.
Figure 1. Multiple oncogenes and proto-oncogenes induce similar forms of cellular stress (e.g., DNA damage). On the left: to ensure cell survival despite this stress, the oncogenes and proto-oncogenes activate cellular processes (e.g., DNA repair) to compensate for the stress (blue line). In the middle: inhibiting the onco-protein result in hyperactivation of a compensatory proto-oncogene. On the right: tumor ablation by simultaneous inhibition of the onco-protein and the stress-compensatory process.
References
- Ren R. Nat Rev Cancer 2005; 5(3):172-83; PMID:15719031; http://dx.doi.org/10.1038/nrc1567
- Weinstein IB, Joe A. Cancer Res 2008; 68(9):3077-80; discussion 3080; PMID:18451130; http://dx.doi.org/10.1158/0008-5472.CAN-07-3293
- Wheeler DL, et al. Nat Rev Clin Oncol 2010; 7(9):493-507; PMID:20551942; http://dx.doi.org/10.1038/nrclinonc.2010.97
- Shen Y, et al. Oncotarget 2015; in press
- Bartek J, Jr., et al. J Neurol Neurosurg Psychiatry 2012; 83(7):753-60; PMID:22396442; http://dx.doi.org/10.1136/jnnp-2011-300709
- Ng K, et al. J Neurooncol 2012; 107(1):1-12; PMID:22002595; http://dx.doi.org/10.1007/s11060-011-0714-2
- Nitta M, et al. PLoS One 2010; 5(5):e10767; PMID:20532243; http://dx.doi.org/10.1371/journal.pone.0010767