
Abstract
Islet-1 (ISL-1), a LIM-homeodomain transcription factor, has been recently found to be essential for promoting postnatal pancreatic islet proliferation. However, the detailed mechanism has not yet been elucidated. In the present study, we investigated the mechanism by which ISL-1 promotes β-cell proliferation through regulation of CyclinD1 in HIT-T15 and NIT-1 cells, as well in rat islet mass. Our results provide the evidence that ISL-1 promotes adult pancreatic islet β-cell proliferation by activating CyclinD1 transcription through cooperation with Set7/9 and PDX-1 to form an ISL-1/Set7/9/PDX-1 complex. This complex functions in an ISL-1-dependent manner, with Set7/9 functioning not only as a histone methyltransferase, which increases the histone H3K4 tri-methylation of the CyclinD1 promoter region, but also an adaptor to bridge ISL-1 and PDX-1, while PDX-1 functions as a RNA pol II binding modulator. Furthermore, the formation of the ISL-1/Set7/9/PDX-1 complex is positively associated with insulin-like growth factor-1 treatment in NIT and HIT-T15 cells in vitro, while may be negatively correlated with age in vivo.
Introduction
Insulin enhancer binding protein-1 (ISL-1), a member of LIM-homeodomain family, was identified and cloned in 1990.Citation1 Previously, it has been reported to play crucial roles in heart, motor neuron and pancreas development.Citation2-6 In heart development, ISL-1 is supposed as a critical marker of the second hear field (SHF),Citation2 ISL-1 knockout will cause severe deficiency of heart in mice embryo.Citation7 In motor neuron, ISL-1 is demonstrated as a important marker in the early stage of motor neuron development,Citation8 while it is also involved in axogenesisCitation9 and motor neuron generation.Citation10 In pancreas and islet development, ISL-1 controls the development of 4 endocrine islet cell lineages and dorsal pancreas mesenchyme,Citation11 complete loss of dorsal pancreatic mesenchyme and endocrine islet cells was found in ISL-1 knock-out mice embryos.Citation6 ISL-1 is also involved in the formation of endocrine cells during the secondary transition in mouse pancreas development (E13.5 – E15.5) and controls the proliferation and survival of endocrine cells.Citation12
Recent studies further reveal that ISL-1 is also involved in postnatal physiological and pathological processes. It has been verified that ISL-1 could regulate the expression of several islet specific genes, such as proglucagon/glucagon (α cells), somatostatin (γ cells), amylin (δ cells) and insulin (β cells), although it is not the master regulator for these genes.Citation13-17 It also promotes the proliferation and the repair of injured motor neurons,Citation3,4 while overexpressing ISL-1 in endothelial cells and mesenchymal stem cells promotes the blood vessel formation.Citation18 These all suggest that ISL-1 plays an important role in adult. Our previous investigation has demonstrated that ISL-1 can promote islet β-cell proliferation in adult, however, the detailed mechanisms remain to be elucidated.
In the present study, we showed that the complex of ISL-1, Set7/9 and PDX-1 is recruited by ISL-1 to the CyclinD1 promoter. The complex exerts its functions through histone modification and RNA pol II binding in an ISL-1-dependent manner. The functions of each component were also determined. Our findings provide details of a novel mechanism by which ISL-1 regulates islet β-cell proliferation, and indicate its potential for the development of a new therapy for diabetes and insulinoma.
Results
ISL-1 binds directly to the CyclinD1 promoter in parallel with the changes in CyclinD1 expression during cell cycle progression
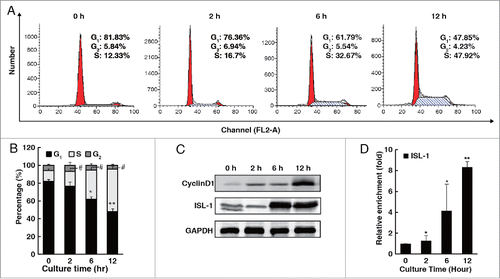
ISL-1 had been demonstrated not only to play an important tissue specific role during the early pancreas development,Citation19 but also to be required for the postnatal pancreatic islets proliferation through promoting CyclinD1 expression.Citation5 However, the detailed regulatory mechanisms of ISL-1 on CyclinD1 gene transcription requires further elucidation. To explore how ISL-1 exerts its biological functions in islet β-cell proliferation, HIT-T15 cells were synchronized (>80% of the population) in the G1 phase by serum starvation for 48 h. As expected, ISL-1 expression gradually increased following the G1/S transition of the cell cycle in parallel with the changes in CyclinD1 expression (). ChIP assays showed that the recruitment of ISL-1 to the CyclinD1 promoter increased significantly during the cell cycle progression (), which is consistent with our previous results.Citation5
Figure 1. The expression pattern of ISL-1 was parallel to that of CyclinD1 during the cell cycle. (A, B) Flow cytometry assays were performed to analyze the cell cycle distribution of HIT-T15 cells at appropriate time-points after serum starvation for 48 h. (C) HIT-T15 cells were harvested at the indicated time-points after serum starvation for 48 h and subjected to Western blotting analysis. (D) Soluble chromatin was prepared from HIT-T15 cells and then followed by immunoprecipitation with the antibody against ISL-1. The DNA extractions were amplified using the primers that cover the ISL-1 binding sites by real-time PCR. Each bar represents 3 independent experiments, mean ± SD. p values were calculated using a Student's t-test. *p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01 vs. control (0 h).
ISL-1, Set7/9 and PDX-1 form a transcriptional complex that binds directly to the CyclinD1 promoter
To investigate the co-factors of ISL-1 on the CyclinD1 promoter region, mass spectrometry was employed. Among the potential ISL-1-interactive proteins in HIT-T15 cells, we focused on the proteins that are highly expressed in β-cells specifically. Furthermore, we attempted to find the proteins that are potential to be associated with ISL-1 functionally, such as having similar binding site(s), tissue specificity, function on proliferation, etc. Owing to the high scores in this analysis and the highly expressed in β-cells correlated with cell proliferation and transcriptional regulation,Citation20,21 Set7/9 and PDX-1 were selected (Fig. S1A). Set7/9 is a pancreas-specific histone methyltransferase and interacts with Cys-His rich domain protein,Citation22 while the LIM domain of ISL-1 was just a Cys-His rich domain. PDX-1 is a pancreas-specific transcriptional factor that has a similar DNA binding site as ISL-1. These two factors play similar roles in regulating β-cell function and proliferation.
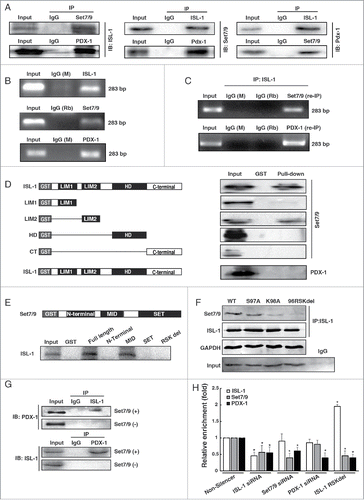
Co-IP was performed to analyze whether Set7/9, PDX-1 and ISL-1 formed a complex and bound directly to the CyclinD1 promoter. The results demonstrated that any one component of the presumptive complex co-immunoprecipitated with the other components, supporting the existence of this complex (). Furthermore, ChIP and ChIP-re-IP results confirmed that Set7/9, PDX-1 and ISL-1 occupied and co-localized on the same region of the CyclinD1 promoter (). Based on these results, we performed GST pull-down assays to verify the interactive mode among 3 factors. The results showed that Set7/9 interacted with ISL-1 via the LIM2 domain, while ISL-1 interacted with Set7/9 via its MID segment (). No interaction between PDX-1 and ISL-1 was observed, suggesting that ISL-1 may not interact with PDX-1 directly (, the buttom panel). These results are consistent with a previous report that Set7/9 interacts with a His and Cys-rich domainCitation23 and forms the protein-protein interaction via a conserved RSK amino acid sequenceCitation24,25 that was observed in the LIM2 domain of the ISL-1 protein. To further evaluate the possible interaction between Set7/9 and ISL-1, we constructed different mutants of ISL-1: S97A, K98A and RSK deletion (Fig. S1D) for Co-IP assays. The results demonstrated that all types of mutations of the presumptive Set7/9 binding sites in ISL-1, including S97A, K98A and RSK deletion, weakened the interaction between ISL-1 and Set7/9 (, right panel). A recent study demonstrated that Set7/9 interacts with PDX-1 directly in adult pancreatic β-cells.Citation26 Hence, we suspected that Set7/9 may act as an adaptor to bridge ISL-1 and PDX-1. To test this hypothesis, pcDNA3-ISL-1 and pCMV-PDX-1 plasmids were transfected into HeLa cells with or without pCMV-Set7/9. The results of Co-IP indicated that the existence of ISL-1/PDX-1 interaction could be detected only in the presence of Set7/9 (). To further evaluate whether Set7/9 serves as an adaptor bridging ISL-1 and PDX-1, ChIP was employed. ISL-1 mutant (RSK site deletion) and siRNAs targetting ISL-1, Set7/9 or PDX-1 were transfected into NIT-1 cell, respectively, and then the recruitment of ISL-1, Set7/9 or PDX-1 on the Cyclin D1 promoter was examined. The results showed that knockdown of Set7/9 or ISL-1 could decrease the enrichment of PDX-1. In contrast, PDX-1 knockdown had no influence on Set7/9 or ISL-1, it only influenced the recruitment of itself. In addition, the overexpression of ISL-1 mutant led to an obvious decline of Set7/9 and PDX-1 on the Cyclin D1 promoter (). These are consistent with Co-IP results, supporting that Set7/9 could serve as an adaptor bridging ISL-1 and PDX-1.
Figure 2. ISL-1 formed a complex with Set7/9 and PDX-1. (A) Co-IP assay was employed to detect the interactions among ISL-1, Set7/9 and PDX-1 in HIT-T15 cells. (B) Soluble chromatin was prepared from HIT-T15 cells followed by immunoprecipitation with antibodies against ISL-1, Set7/9 or PDX-1; normal IgG (Rb, rabbit; M, mouse) served as a control. (C) ChIP-re-IP assay was performed with anti-ISL-1 or rabbit IgG antibodies and then with anti-Set7/9, anti-PDX-1 or IgG antibodies for immunoprecipitation using chromatin harvested from HIT-T15 cells. (D) Full-length or truncated ISL-1 was used to construct GST-fusion proteins (Left panel) for pull-down assays with Set7/9 or PDX-1 protein (right panel). (E) Full-length or truncated Set7/9 was used to construct GST-fusion proteins for pull-down assays with ISL-1 protein. (F) The plasmids expressing ISL-1 mutants in which the Set7/9 binding sites were mutated or deleted (Left panel) were constructed and Co-IP assays were performed to detect the interaction between ISL-1 (wild-type or mutants) and Set7/9. The normal IgG was served as a negative control. ISL-1 and GAPDH served as loading and negative controls, respectively (right panel). (G) pcDNA3.1-ISL-1, pcDNA3.1-PDX-1 or pCMV-Set7/9 plasmids were transfected into HeLa cells in different combinations. Whole cell extracts were harvested after 48 h and subjected to Co-IP using anti-ISL-1 as the “IP” antibody and anti-PDX-1 as the “IB” antibody.
Taken together, our data showed that ISL-1 associates with Set7/9 and PDX-1, with Set7/9 acting as an adaptor bridging ISL-1 and PDX-1 and binding directly to the CyclinD1 promoter.
ISL-1 enhances mono- and tri-methylation levels of histone H3K4 of the CyclinD1 promoter region and RNA pol II binding via the ISL-1/PDX-1/Set7/9 complex
Previous studies showed that Set7/9 is a histone H3K4-specific methyltransferase and a novel enzymatic cofactor required for maintenance of islet gene transcription.Citation20 It is not known whether this complex activates CyclinD1 not only by the transcription regulation, but also by the epigenetic modification. ChIP assay results showed that the histone H3K4 tri-methylation level on the ISL-1-binding region of the CyclinD1 promoter was obviously increased and was consistent with the changes in ISL-1 expression during cell cycle progression (). ISL-1 overexpression significantly increased histone H3K4 mono-methylation (H3K4me1) and tri-methylation (H3K4me3) levels of the CyclinD1 promoter, while ISL-1 knockdown markedly decreased these modifications in HIT-T15 cells (). However, histone H3K9 acetylation (H3K9ac) was not altered by ISL-1-overexpression or knockdown (Fig. S1B) and the classic “repressive” modification: H3K27 tri-methylation (H3K27me3) showed an opposite trend compared to that of H3K4 methylation (Fig. S1C). Specifically, H3K4 mono- and tri-methylation exhibited more significant enrichment at the ISL-1-binding region of the CyclinD1 promoter. Therefore, the level of ISL-1 in the complex influenced the modification in the histone H3K4 methylation status of the CyclinD1 promoter.
Figure 3. ISL-1 recruited Set7/9 to modulate histone modification of the CyclinD1 promoter. (A) H3k4me3 was detected by ChIP assay in HIT-T15 cells at indicated time-points after serum starvation for 48 h. The H3K4me3 level at 0 h serum starvation served as a control. (B, C) pcDNA3.1-ISL-1 (ISL-1), pcDNA3.1-vector (control), ISL-1 siRNA or Non-silencer negative control siRNA were transfected into HIT-T15 cells. ChIP assays were employed to detect H3k4me1 or H3k4me3 after 48 h. (D) The luciferase activity of the CylinD1 promoter was measured in 293A cells transfected with the indicated plasmid combination. The cells transfected with pcDNA3 were used as a negative control. (E) NIT-1 cells was transfected with pCMV-Set7/9 (pCMV-vector served as a control) or Set7/9 siRNA (Non-silencer served as a control), and Western blotting analysis was performed using an antibody against H3k4me3. Histone H3 was used as a loading control. (F, G) NIT-1 cells were transfected with different combinations of plasmids for 48 h, and H3k4me3 or H3k4me1 of the CyclinD1 promoter were measured by ChIP assay. The cells transfected with pcDNA3 or Non-silencer were used as negative controls. *p < 0.05, **p < 0.01, vs. pcDNA3; #p < 0.05, ##p < 0.01 vs. Non-silencer, NS: non-significance.
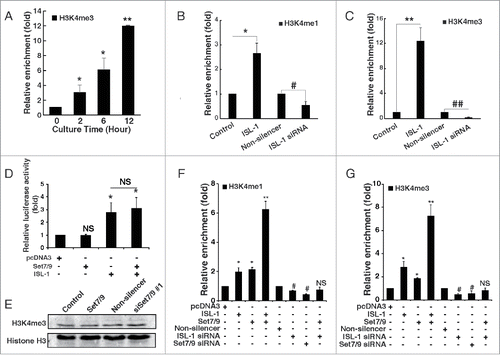
The luciferase assay was performed to investigate the synergistic effects between Set7/9 and ISL-1 on the CyclinD1 promoter. As shown in , the luc-CyclinD1 activity in HEK293A cells was significantly increased by ISL-1 transfection, whereas no obvious effects were observed when Set7/9 was transfected alone or co-transfected with ISL-1. These observations demonstrated that Set7/9 does not activate CyclinD1 transcription alone and nor does it act as a synergistic cofactor of ISL-1 at transcription level. Set7/9 is known to be a histone H3 methyltransferase. However, neither overexpression nor knockdown of Set7/9 using siRNA targeting Set7/9 (Fig. S2B) influenced the global H3K4me3 level of NIT-1 cells (), indicating that the function of Set7/9 may be independent on its expression, but is recruited by other factors to implement its function. The ChIP assay () revealed that Set7/9 and ISL-1 exhibited a synergistic effect on both H3K4me1 and H3K4me3 of the CyclinD1 promoter, with the changes in the histone modification being markedly amplified when ISL-1 and Set7/9 were co-overexpressed compared to the changes observed following expression of ISL-1 or Set7/9 alone. The positive regulatory function of Set7/9 was blocked when ISL-1 was knockeddown (), demonstrating that the function of Set7/9 is ISL-1-dependent.
It has been demonstrated that PDX-1 directly interacts with Set7/9 to enhance the expression of insulin I by improving RNA pol II binding.Citation20,26 The ChIP assay indicated that either PDX-1 alone or co-transfected with ISL-1 resulted in positive recruitment of RNA pol II to the ISL-1 binding site of the CyclinD1 promoter (). More interestingly, PDX-1 exhibited a distinct synergistic effect with ISL-1 in this recruitment (), and the positive regulatory function of PDX-1 was also blocked with ISL-1 knockdown (), demonstrating that the function of PDX-1 is also ISL-1-dependent. We also investigated whether PDX-1 in the ISL-1/Set7/9/PDX-1 complex influenced histone modification of the CyclinD1 promoter. As shown in , similar to ISL-1, PDX-1 influenced H3K4me3 modification, but no synergistic effect with ISL-1 was observed.
Figure 4. PDX-1 interacted with ISL-1 to promote the recruitment of RNA pol II. (A) NIT-1 cells were transfected with appropriate plasmids. The binding of RNA pol II to the ISL-1 site on the CyclinD1 promoter was measured by ChIP assay using an anti-RBP2 antibody. (B) ChIP assay was performed to analyze H3k4me3 of the CyclinD1 promoter region in NIT-1 cells transfected with indicated combination of plasmids. (C) Luciferase dual reporter assays were performed in 293A cells at 48 h after transfection with luc-CyclinD1-promoter and other plasmids. (D) Luciferase dual reporter assays were performed in NIT-1 cells at 48 h after transfection with luc-CyclinD1-promoter (wild-type), luc-cyclinD1-promoter in which the ISL-1 binding site TAAT was mutated to TGGT (Mutant-1) or deleted (Mutant-2) and other plasmids as shown including ISL-1 mt (the Set7/9 binding site RSK was deleted). The data represent 3 independent experiments, each performed in triplicate. *p < 0.05, **p < 0.01, vs. pcDNA3; #p < 0.05, ##p < 0.01 vs. Non-silencer or indicated column, NS, non-significance.
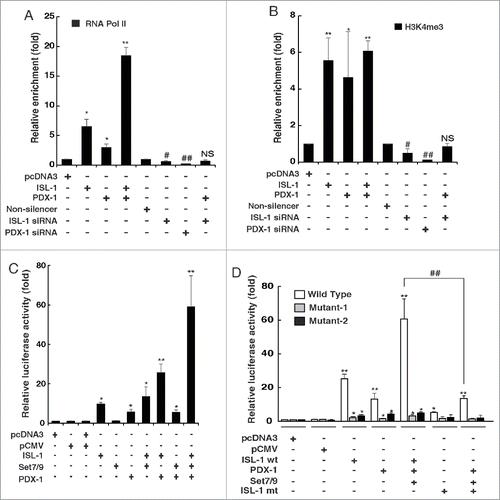
The luciferase assay demonstrated that PDX-1 activated the CyclinD1 promoter alone or synergistically with ISL-1. In contrast, Set7/9 did not regulate the CyclinD1 promoter alone, but improved the effect when co-transfected with ISL-1 and PDX-1 (). It can be speculated that this is because Set7/9 serves as an adaptor to bridge ISL-1 and PDX-1. To further investigate the importance of ISL-1, mutant luc-CyclinD1 reporters (ISL-1 binding site TAAT was mutated to TGGT or complete deletion of TAAT) were constructed (Fig. S2C). When the ISL-1-binding site on the CyclinD1 promoter was mutated, the enhanced luciferase activity mediated by ISL-1, Set7/9 or PDX-1 was dramatically reduced or absent (), as was the synergistic effect. Furthermore, luciferase assays using both luc-CyclinD1 wild-type and mutants showed that the synergistic effect was also reversed by the ISL-1 mutant in which the Set7/9 target site was deleted (), indicating that the effect of Set7/9 on the CyclinD1 promoter depends on the interaction between ISL-1 and Set7/9.
In summary, the ISL-1/Set7/9/PDX-1 complex regulates CyclinD1 at both the transcriptional level and the epigenetic level. The formation and function of this complex depends on ISL-1. Set7/9 acts as a histone methyltransferase and an adaptor, while PDX-1 improves the binding of RNA pol II.
The ISL-1/Set7/9/PDX-1 complex promotes β-cell proliferation
We have shown that ISL-1 forms an ISL-1/Set7/9/PDX-1 complex to regulate CyclinD1 at both the transcriptional and the epigenetic levels. However, whether this regulatory machenism functions on β-cell proliferation needs to be investigated. CCK-8 assay results showed that overexpression of ISL-1, Set7/9 or PDX-1 alone increased NIT-1 cell proliferation, while knockdown of these factors caused a reduction in the proliferation rate (). Moreover, Set7/9 or PDX-1 co-transfected with ISL-1 exhibited a distinct synergistic effect in promoting cell proliferation (). In contrast, knockdown of ISL-1 blocked the synergistic effect of PDX-1 or Set7/9 (, the last column from left), supporting the role of ISL-1 as an essential factor in the complex.
Figure 5. Set7/9 and PDX-1 promoted the effects of ISL-1 on β-cell proliferation. (A, B) The proliferation of NIT-1 cells transfected with the indicated plasmids in 96-well plates was analyzed with CCK-8 kits. (C, D) NIT-1 cells in 6-wells plates were transfected with the indicated plasmids. The cell confluence was measured using the Genetix clone selector after 48 h. The data represent 3 independent experiments, each performed in triplicate. *p < 0.05, **p < 0.01 vs. pcDNA3, #p < 0.05, ##p < 0.01 vs. non-silencer, NS, non-significance. +p < 0.05, ++p < 0.01 as indicated.
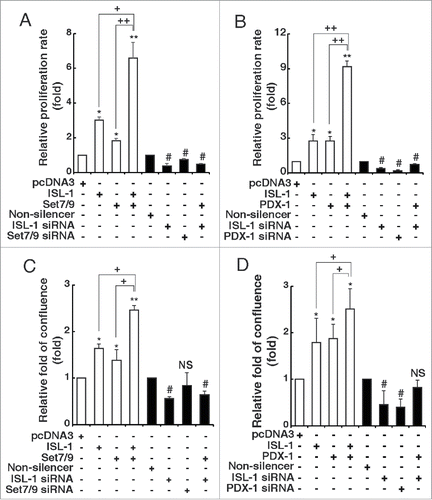
To further confirm this observation, we analyzed NIT-1 cell growth by direct measurement of the relative cell confluence rate at 48 h post-transfection using the Genetix cloneselector imager. The results exhibited a similar trend to those of the CCK-8 assay (). All these results indicate that this complex was able to promote β-cell proliferation and ISL-1 is the most critical factor in the ISL-1/Set7/9/PDX-1 complex.
Formation of the ISL-1/Set7/9/PDX-1 complex is age-related and regulated by IGF-1
Insulin-like growth factor (IGF-1) is recognized as a specified and effective growth factor functioning on adult β-cell proliferation.Citation27-29 Therefore, we investigated the effects of different doses of IGF-1 on NIT-1 cell proliferation. CCK-8 assays showed that cell proliferation was increased in a dose-dependent manner (Fig. S3), and doses of 5 ng/ml and 10 ng/ml were chosen for further investigations.
The H3K4me3 level of the ISL-1 binding site within the CyclinD1 promoter was first examined in NIT-1 cells after IGF-1 treatment. The results indicated that the H3K4me3 level was increased by IGF-1 treatment in a dose-dependent manner (), as was the expression level of ISL-1, CyclinD1 and PDX-1 (). However, the expression of Set7/9 was not influenced by IGF-1 (). This may be due to the relatively constant expression level of Set7/9, so that its function is regulated mainly through protein-protein interactions, rather than regulation of its expression.Citation25 To test this hypothesis, ChIP assays were performed to determine the level of enrichment of ISL-1, Set7/9 and PDX-1 on the CyclinD1 promoter. The results demonstrated that although Set7/9 protein level was stable, the recruitment of ISL-1, Set7/9 and PDX-1 was increased after IGF-1 treatment in an IGF-1 dose-dependent manner (). Co-IP assays were also performed to detect changes in ISL-1/Set7/9/PDX-1 formation. As shown in , the interaction of Set7/9 or PDX-1 with ISL-1 was increased in a dose-dependent manner.
Figure 6. The formation of ISL-1/Set7/9/PDX-1 complex was regulated by IGF-1 in an age-related manner. (A) NIT-1 cells were treated with mouse recombinant IGF-1 protein. The H3k4me3 of the CyclinD1 promoter was verified by ChIP assay. (B) The whole cell lysates of NIT-1 cells treated with IGF-1 were used for Western blotting. (C) The enrichment of ISL-1, Set7/9 and PDX-1 at the CyclinD1 promoter was monitored by ChIP assay in NIT-1 cells treated with IGF-1. (D, E) Co-IP assays were performed to measure the ISL-1/Set7/9/PDX-1 complex in NIT-1 cells treated with IGF-1. ISL-1 and GAPDH served as a loading control and negative controls, respectively. (F) Rat islet lysates were subjected to ChIP assay using anti-ISL-1, PDX-1, Set7/9 antibodies or normal IgG. (G) Rat islet lysates were immunoprecipitated by anti-ISL-1 antibody. Western blotting was employed to detect the endogenous interaction using anti-PDX-1 or anti-Set7/9 antibodies. (H) The formation of the ISL-1/PDX-1/Set7/9 complex was detected by Co-IP assay in the islets of “young” or “old” rats. The data in (A), (C) and (F) represent 3 independent experiments, each performed in triplicate. *p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01 vs. the control, NS, non-significance.
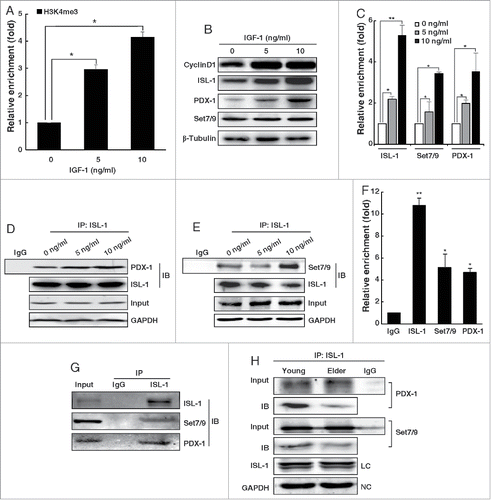
The existence and formation of the ISL-1/Set7/9/PDX-1 complex was also investigated in vivo. Rat islet masses were isolated and analyzed in ChIP assays to confirm the recruitment of ISL-1, Set7/9 and PDX-1 to the CyclinD1 promoter. The results showed that the enrichment of ISL-1, Set7/9 and PDX was approximately 11.02 ± 0.53, 4.92 ± 1.27 and 4.6 ± 0.38 times higher than that of the normal IgG control, respectively (). Co-IP assays were then performed to detect the endogenous interaction in primary rat islet cell masses. PDX-1 or Set7/9 were detected by Western blotting analysis after immunoprecipitation with ISL-1 (). ChIP and Co-IP assays both confirmed the existence of the complex. We further divided rats into “young” (aged 5 weeks) and “old” (aged 4 months) groups to study the effect of age on the formation of the complex. The Co-IP assay demonstrated that the interaction of Set7/9 and PDX-1 with ISL-1 was reduced in the “old” group compared to that in the “young” group, indicating that the formation of this complex may be negatively correlated with age (). However, this correlation requires further investigation.
Collectively, these results demonstrate that the ISL-1/Set7/9/PDX-1 complex exists not only in vitro, but also in vivo and that the formation of the complex is regulated by IGF-1.
Discussion
The study of adult pancreatic islet β-cell homeostasis is critical for the development of more effective therapies for diabetes and related diseases.Citation30 The renewal of adult islet β-cells is derived from the proliferation of existing cells, rather than from pancreatic stem cell differentiation.Citation31 CyclinD1, which functions in the G1/S phase transition of the cell cycle is an essential factor for adult β-cell proliferation.Citation32,33 In the present study, we demonstrate that ISL-1 forms a complex with Set7/9 and PDX-1 to regulate CyclinD1.
It has been reported that ISL-1 promotes both lymphoma and pancreatic islet β-cell proliferation, although a positive autocrine feed-back loop to promote its expression was observed in lymphoma but not in pancreatic islet β-cells.Citation34,35 Nevertheless, ISL-1 expression is extremely high in adult islet β-cells, indicating that the mechanism by which ISL-1 regulates β-cell proliferation is distinct and unique. As a member of a LIM-homeodomain protein family, the LIM domain of ISL-1 mediates the interactions with other factors.Citation36 In our study, ISL-1 interacts directly with Set7/9 through the LIM2 domain of ISL-1. The ISL-1 and Set7/9 heterodimer binds to the PDX-1 co-activator to provide a docking and recruitment interface with the general transcriptional machinery.
We also demonstrate that the ISL-1/Set7/9/PDX-1 complex regulates CyclinD1 expression not only at the transcriptional level, but also at the epigenetic level. The H3K4me1 and H3K4me3 levels of the CyclinD1 promoter were altered by Set7/9 in an ISL-1-dependent manner. However, direct evidence is required to confirm that the methyl-transfer is mediated by Set7/9. Set7/9 is always documented as a histone mono- and di-methyltransferase.Citation22 However, in our study, histone tri-methylation was modulated by Set7/9, possibly due to the undefined function of Set7/9 or other undefined components in this complex.Citation37 Furthermore, it has been reported that Set7/9 can function as a non-histone protein methyltransferase;Citation24 thus, raising the possibility that ISL-1 is methylated by Set7/9.
The characteristic expression of ISL-1 must be also noted. Our study demonstrates that ISL-1 is an essential factor to the formation of the ISL-1/Set7/9/PDX-1 complex that promotes β-cell proliferation. The endogenous expression of ISL-1 in β-cells is extremely high and stable, highlighting the paradox that although ISL-1 regulates CyclinD1, adult β-cell proliferation is an extremely rare event in vivo. We found that the endogenous modified ISL-1 could inhibit β-cell apoptosis and exhibit a different regulatory mechanism in this process (unpublished data). A recent study has also demonstrated that ISL-1 is essential for postnatal β-cell function in vivo, and that ablation of ISL-1 reduces glucose tolerance.Citation17 These studies indicate that ISL-1 is multi-functional and plays more substantial role in β-cells than was previously accepted. Thus, further investigation on this issue is needed.
Our study demonstrates for the first time that PDX-1 is associated directly with CyclinD1 expression. PDX-1 has been accepted as a crucial factor for pancreatic development and the maintenance of β-cell function.Citation38 It has recently been demonstrated both in vitro and in vivo, that PDX-1 is involved in β-cell proliferation.Citation39 However, the functions of PDX-1 in β-cell proliferation and differentiation are exerted through several different pathways. Therefore, the mechanism of the synergistic effect of PDX-1 and ISL-1 on cell proliferation requires further clarification.
Taken together, our findings reveal a novel regulatory mechanism by which adult pancreatic islet β-cell proliferation is controlled. ISL-1 functions as a crucial adult β-cell proliferation regulator, the function of which is mediated predominantly through the formation of a complex with Set7/9 and PDX-1 to regulate CyclinD1. In the complex, Set7/9 functions as a histone methyltransferase and an adaptor linking ISL-1 and PDX-1, while PDX-1 promotes the binding of RNA pol II to the ISL-1 binding site of the CyclinD1 promoter. Ultimately, the function of this complex depends on ISL-1, further confirming the importance of ISL-1 in adult pancreatic islet β-cells.
Materials and Methods
Cell culture
The hamster pancreatic islet cell line HIT-T15 (ATCC: CRL-1777) and the mouse insulinoma cell line NIT-1 (ATCC: CRL-1777) were maintained in RPMI 1640 containing 10% fetal bovine serum (FBS). HIT-T15 cells were used in most experiments except those involving siRNA targeting set7/9 and pdx-1, because the hamster genome has not been completely sequenced.
Cell proliferation and cell cycle assays
The cell proliferation assay was carried out using CCK-8 and EdU assays as described previously.Citation5 Cell cycle analysis was performed as described previously.Citation5
Luciferase assays
Plasmid transfection and luciferase activity detection were performed as described previously.Citation40 All experiments were performed in triplicate and 3 independently repeated experiments were performed.
Chromatin immunoprecipitation (ChIP) and ChIP-re-IP assays
ChIP and ChIP-re-IP experiments were performed as described by Shang et al.Citation41 using the following primers covering a 283 bp region of the rat and hamster CyclinD1 promoter: F: 5′-AGCTTCGGTGTCTGGTTC-3′, R: 5′-ATTCCAGCAACGCTCAAGATG-3′, or the primers covering a 258 bp region of the mouse CyclinD1 promoter: F: 5′-CGGCTCACAAGTTTATC-3′, R: 5′- AGCCTATCGTGTCTCAAC. The following antibodies were used: trimethyl-histone H3 (Lys4) (#17–678, Millipore, Billerica, MA, USA); Set7/9 (A301-747A, Bethyl, Montgomery, TX, USA); monomethyl-histone H3 (Lys4) (ab8895), PDX-1 (ab47267), ISL-1 (ab109517) and RPB2 (ab10338) (all from Abcam, Cambridge, UK).
Quantitative real-time PCR
Total RNA was extracted using Trizol Reagent (Invitrogen, Grand island, NY, USA) based on the manufacturer's instructions. Amplifications were performed in the ABI 7300 Real-Time PCR System using the following primers: ISL-1: F: 5′-CTGCTTTTCAGCAACTGGTCA-3′, R: 5′-TAGGACTGGCTACCATGCTGT-3′; CyclinD1: F: 5′-GCGTACCCTGACACCCCTCTC-3′, R: 5′- CTCCTCTTCGCCTGATCC-3′; GAPDH: F: 5′-CGACCACTTTGTCAAGCTCA-3′, R: 5′-AGGGGTCTACATGGCAACTG-3′.
Immunoprecipitation and Western blotting analysis
Cell lysates were prepared using RIPA lysis buffer (P0013E, Beyotime, China) containing protease inhibitor cocktail (469313200, Roche, Basel, Switzerland) following the manufacturer's instructions. Immunoprecipitation and Western blotting analysis were carried out as described previously.Citation42
The following antibodies were used: ISL-1 for Co-IP (H00003670-M05, Abnova, Taipei, China); ISL-1 for Western blotting (ab109517, Abcam); PDX-1 (ab47267, Abcam); Set7/9 (A301-747A, Bethyl); Set7/9 (#2813). GAPDH (#2118) and β-tubulin (#2146) (both from Cell Signaling Technology, Danvers, MA, USA).
GST pull-down assay
GST fused with full-length ISL-1 or various truncated ISL-1 proteins were prepared as described previously.Citation43 Biotin-labeled wild-type Set7/9 or PDX-1 were synthesized using the TNT Quick Coupled Transcription/Translation System (L5020, Promega, Madison, WI, USA).
IGF-1 treatment
NIT-1 cell proliferation was stimulated by insulin-like growth factor −1 (IGF-1) treatment. IGF-1 (I8779, Sigma, St. Louis, MO, USA) was dissolved in PBS containing 0.1% BSA and then was added to the cell cultures at the indicated dose and time.
Animal experiments
“Young” (aged 5 weeks, 80–100 g) and “old” (aged 4 months, 350–400 g) male SD rats were purchase from the Department of Laboratory Animal Science of Peking University and subject to euthanasia by 2% nembutal sodium. Collagenase V (5 ml, 2.5 μg/ml, Sigma) was injected into pancreas via the bile duct. Pancreatic tissue was isolated immediately, placed into 1× HBSS and digested with collagenase V (15 ml) at 37°C for 10 min with 330 rpm shaking. The suspension was then centrifuged (1,000 rpm, 5 min), and the pellet was harvested and filtered (100 µm mesh nylon filter). Dithizone (40 µl) was added into the filtered suspension and incubated for 10 min at 37°C. The dithizone stained islet masses were picked and cultured in RPMI 1640 containing 15% FBS. All animal experiments were performed in accordance with the ethical principles and guidelines for scientific experiments on animals of the Swiss Academy of Medical Sciences (1995). All protocols were approved by the Animal Care and Use Committee of Peking University (LA 2010–066).
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Comparisons between groups were analyzed by Student's t-test using SPSS version 17.0. Differences were considered to be statistically significant at p < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1069926_supplemental_files.zip
Download Zip (545.9 KB)Acknowledgments
We thank Prof. Shang YF and Prof. Zhu WG, Peking University School of Basic Medical Sciences, for the gifts of luc-CyclinD1 and Set7/9 construct, respectively.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81170713, 81472022, 81370236, 81371889, 81071675), the Natural Science Foundation of Beijing (No. 5122021), the Leading Academic Discipline Project of Beijing Education Bureau, and the 111 Project of China (B07001).
Related Research Data
References
- Karlsson O, Thor S, Norberg T, Ohlsson H, Edlund T. Insulin gene enhancer binding protein Isl-1 is a member of a novel class of proteins containing both a homeo- and a Cys-His domain. Nature 1990; 344:879-82; PMID:1691825; http://dx.doi.org/10.1038/344879a0
- Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 2009; 460:113-7; PMID:19571884; http://dx.doi.org/10.1038/nature08191
- Reimer MM, Sorensen I, Kuscha V, Frank RE, Liu C, Becker CG, Becker T. Motor neuron regeneration in adult zebrafish. J Neurosci 2008; 28:8510-6; PMID:18716209; http://dx.doi.org/10.1523/JNEUROSCI.1189-08.2008
- Shi Y, Zhao S, Li J, Mao B. Islet-1 is required for ventral neuron survival in Xenopus. Biochem Biophys Res Commun 2009; 388:506-10; PMID:19666005; http://dx.doi.org/10.1016/j.bbrc.2009.08.017
- Guo T, Wang W, Zhang H, Liu Y, Chen P, Ma K, Zhou C. ISL1 promotes pancreatic islet cell proliferation. PloS one 2011; 6:e22387; PMID:21829621; http://dx.doi.org/10.1371/journal.pone.0022387
- Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature 1997; 385:257-60; PMID:9000074; http://dx.doi.org/10.1038/385257a0
- Lin L, Cui L, Zhou W, Dufort D, Zhang X, Cai CL, Bu L, Yang L, Martin J, Kemler R, et al. Beta-catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proc Natl Acad Sci U S A 2007; 104:9313-8; PMID:17519333; http://dx.doi.org/10.1073/pnas.0700923104
- Ericson J, Thor S, Edlund T, Jessell TM, Yamada T. Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science 1992; 256:1555-60; PMID:1350865; http://dx.doi.org/10.1126/science.1350865
- Hol EM, Schwaiger FW, Werner A, Schmitt A, Raivich G, Kreutzberg GW. Regulation of the LIM-type homeobox gene islet-1 during neuronal regeneration. Neuroscience 1999; 88:917-25; PMID:10363827; http://dx.doi.org/10.1016/S0306-4522(98)00263-2
- Pfaff SL, Mendelsohn M, Stewart CL, Edlund T, Jessell TM. Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell 1996; 84:309-20; PMID:8565076; http://dx.doi.org/10.1016/S0092-8674(00)80985-X
- Lyttle BM, Li J, Krishnamurthy M, Fellows F, Wheeler MB, Goodyer CG, Wang R. Transcription factor expression in the developing human fetal endocrine pancreas. Diabetologia 2008; 51:1169-80; PMID:18491072; http://dx.doi.org/10.1007/s00125-008-1006-z
- Du A, Hunter CS, Murray J, Noble D, Cai CL, Evans SM, Stein R, May CL. Islet-1 is required for the maturation, proliferation, and survival of the endocrine pancreas. Diabetes 2009; 58:2059-69; PMID:19502415; http://dx.doi.org/10.2337/db08-0987
- Hashimoto T, Nakamura T, Maegawa H, Nishio Y, Egawa K, Kashiwagi A. Regulation of ATP-sensitive potassium channel subunit Kir6.2 expression in rat intestinal insulin-producing progenitor cells. J Biol Chem 2005; 280:1893-900; PMID:15528203; http://dx.doi.org/10.1074/jbc.M410759200
- Wang M, Drucker DJ. Activation of amylin gene transcription by LIM domain homeobox gene isl-1. Mol Endocrinol 1996; 10:243-51; PMID:8833653
- Wang M, Drucker DJ. The LIM domain homeobox gene isl-1 is a positive regulator of islet cell-specific proglucagon gene transcription. J Biol Chem 1995; 270:12646-52; PMID:7759514; http://dx.doi.org/10.1074/jbc.270.21.12646
- Leonard J, Serup P, Gonzalez G, Edlund T, Montminy M. The LIM family transcription factor Isl-1 requires cAMP response element binding protein to promote somatostatin expression in pancreatic islet cells. Proc Natl Acad Sci U S A 1992; 89:6247-51; PMID:1352885; http://dx.doi.org/10.1073/pnas.89.14.6247
- Ediger BN, Du A, Liu J, Hunter CS, Walp ER, Schug J, Kaestner KH, Stein R, Stoffers DA, May CL. Islet-1 Is essential for pancreatic beta-cell function. Diabetes 2014; 63:4206-17; PMID:25028525; http://dx.doi.org/10.2337/db14-0096
- Barzelay A, Ben-Shoshan J, Entin-Meer M, Maysel-Auslender S, Afek A, Barshack I, Keren G, George J. A potential role for islet-1 in post-natal angiogenesis and vasculogenesis. Thromb Haemost 2010; 103:188-97; PMID:20062933; http://dx.doi.org/10.1160/TH09-07-0433
- Zhuang S, Zhang Q, Zhuang T, Evans SM, Liang X, Sun Y. Expression of Isl1 during mouse development. Gene Expr Patterns 2013; 13:407-12; PMID:23906961; http://dx.doi.org/10.1016/j.gep.2013.07.001
- Deering TG, Ogihara T, Trace AP, Maier B, Mirmira RG. Methyltransferase Set7/9 maintains transcription and euchromatin structure at islet-enriched genes. Diabetes 2009; 58:185-93; PMID:18984737; http://dx.doi.org/10.2337/db08-1150
- Feanny MA, Fagan SP, Ballian N, Liu SH, Li Z, Wang X, Fisher W, Brunicardi FC, Belaguli NS. PDX-1 expression is associated with islet proliferation in vitro and in vivo. J Surg Res 2008; 144:8-16; PMID:17583748; http://dx.doi.org/10.1016/j.jss.2007.04.018
- Zhang X, Bruice TC. Enzymatic mechanism and product specificity of SET-domain protein lysine methyltransferases. Proc Natl Acad Sci U S A 2008; 105:5728-32; PMID:18391193; http://dx.doi.org/10.1073/pnas.0801788105
- Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev 2008; 22:2980-93; PMID:18981476; http://dx.doi.org/10.1101/gad.1689708
- Pradhan S, Chin HG, Esteve PO, Jacobsen SE. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics 2009; 4:383-7; PMID:19684477; http://dx.doi.org/10.4161/epi.4.6.9450
- Liu X, Wang D, Zhao Y, Tu B, Zheng Z, Wang L, Wang H, Gu W, Roeder RG, Zhu WG. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc Natl Acad Sci U S A 2011; 108:1925-30; PMID:21245319; http://dx.doi.org/10.1073/pnas.1019619108
- Ogihara T, Vanderford NL, Maier B, Stein RW, Mirmira RG. Expression and function of Set7/9 in pancreatic islets. Islets 2009; 1:269-72; PMID:21099283; http://dx.doi.org/10.4161/isl.1.3.9779
- Huang Y, Chang Y. Regulation of pancreatic islet beta-cell mass by growth factor and hormone signaling. Prog Mol Biol Transl Sci 2014; 121:321-49; PMID:24373242; http://dx.doi.org/10.1016/B978-0-12-800101-1.00010-7
- Hill DJ, Hogg J. Growth factor control of pancreatic B cell hyperplasia. Baillieres Clin Endocrinol Metab 1991; 5:689-98; PMID:1755812; http://dx.doi.org/10.1016/S0950-351X(10)80010-2
- Nielsen JH, Galsgaard ED, Moldrup A, Friedrichsen BN, Billestrup N, Hansen JA, Lee YC, Carlsson C. Regulation of beta-cell mass by hormones and growth factors. Diabetes 2001; 50:S25-S9; PMID:11272193; http://dx.doi.org/10.2337/diabetes.50.2007.S25
- Liu C, Wu H. From Beta Cell Replacement to Beta Cell Regeneration: Implications for Antidiabetic Therapy. J Diabetes Sci Technol 2014; 8:1221-6; PMID:25355714; http://dx.doi.org/10.1177/1932296814540611
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429:41-6; PMID:15129273; http://dx.doi.org/10.1038/nature02520
- Lee YC, Nielsen JH. Regulation of beta cell replication. Mol Cell Endocrinol 2009; 297:18-27; PMID:18824066; http://dx.doi.org/10.1016/j.mce.2008.08.033
- Heit JJ, Karnik SK, Kim SK. Intrinsic regulators of pancreatic beta-cell proliferation. Ann Rev Cell Dev Biol 2006; 22:311-38; PMID:NOT_FOUND; http://dx.doi.org/10.1146/annurev.cellbio.22.010305.104425
- Zhang Q, Yang Z, Wang W, Guo T, Jia Z, Ma K, Zhou C. A positive feedback regulation of ISL-1 in DLBCL but not in pancreatic beta-cells. Biochem Biophys Res Commun 2014; 449:295-300; PMID:24845569; http://dx.doi.org/10.1016/j.bbrc.2014.05.021
- Zhang Q, Yang Z, Jia Z, Liu C, Guo C, Lu H, Chen P, Ma K, Wang W, Zhou C. ISL-1 is overexpressed in non-Hodgkin lymphoma and promotes lymphoma cell proliferation by forming a p-STAT3/p-c-Jun/ISL-1 complex. Mol Cancer 2014; 13:181; PMID:25070240; http://dx.doi.org/10.1186/1476-4598-13-181
- Dawid IB, Breen JJ, Toyama R. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet 1998; 14:156-62; PMID:9594664; http://dx.doi.org/10.1016/S0168-9525(98)01424-3
- Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009; 138:660-72; PMID:19703393; http://dx.doi.org/10.1016/j.cell.2009.05.050
- Kaneto H, Miyatsuka T, Kawamori D, Yamamoto K, Kato K, Shiraiwa T, Katakami N, Yamasaki Y, Matsuhisa M, Matsuoka TA. PDX-1 and MafA play a crucial role in pancreatic beta-cell differentiation and maintenance of mature beta-cell function. Endocr J 2008; 55:235-52; PMID:17938503; http://dx.doi.org/10.1507/endocrj.K07E-041
- Hayes HL, Moss LG, Schisler JC, Haldeman JM, Zhang Z, Rosenberg PB, Newgard CB, Hohmeier HE. Pdx-1 activates islet alpha- and beta-cell proliferation via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Mol Cell Biol 2013; 33:4017-29; PMID:23938296; http://dx.doi.org/10.1128/MCB.00469-13
- Wang J, Jia Z, Zhang C, Sun M, Wang W, Chen P, Ma K, Zhang Y, Li X, Zhou C. miR-499 protects cardiomyocytes from H 2O 2-induced apoptosis via its effects on Pdcd4 and Pacs2. RNA Biol 2014; 11:339-50; PMID:24646523; http://dx.doi.org/10.4161/rna.28300
- Zhang Y, Zhang H, Liang J, Yu W, Shang Y. SIP, a novel ankyrin repeat containing protein, sequesters steroid receptor coactivators in the cytoplasm. EMBO J 2007; 26:2645-57; PMID:17476305; http://dx.doi.org/10.1038/sj.emboj.7601710
- Liu Z, Li T, Liu Y, Jia Z, Li Y, Zhang C, Chen P, Ma K, Affara N, Zhou C. WNT signaling promotes Nkx2.5 expression and early cardiomyogenesis via downregulation of Hdac1. Biochim Biophys Acta 2009; 1793:300-11; PMID:18851995; http://dx.doi.org/10.1016/j.bbamcr.2008.08.013
- Zhang H, Wang WP, Guo T, Yang JC, Chen P, Ma KT, Guan YF, Zhou CY. The LIM-homeodomain protein ISL1 activates insulin gene promoter directly through synergy with BETA2. J Mol Biol 2009; 392:566-77; PMID:19619559; http://dx.doi.org/10.1016/j.jmb.2009.07.036