
Abstract
The division cycle of unicellular yeasts is completed with the activation of a cell separation program that results in the dissolution of the septum assembled during cytokinesis between the 2 daughter cells, allowing them to become independent entities. Expression of the eng1+ and agn1+ genes, encoding the hydrolytic enzymes responsible for septum degradation, is activated at the end of each cell cycle by the transcription factor Ace2. Periodic ace2+ expression is regulated by the transcriptional complex PBF (PCB Binding Factor), composed of the forkhead-like proteins Sep1 and Fkh2 and the MADS box-like protein Mbx1. In this report, we show that Ace2-dependent genes contain several combinations of motifs for Ace2 and PBF binding in their promoters. Thus, Ace2, Fkh2 and Sep1 were found to bind in vivo to the eng1+ promoter. Ace2 binding was coincident with maximum level of eng1+ expression, whereas Fkh2 binding was maximal when mRNA levels were low, supporting the notion that they play opposing roles. In addition, we found that the expression of eng1+ and agn1+ was differentially affected by mutations in PBF components. Interestingly, agn1+ was a major target of Mbx1, since its ectopic expression resulted in the suppression of Mbx1 deletion phenotypes. Our results reveal a complex regulation system through which the transcription factors Ace2, Fkh2, Sep1 and Mbx1 in combination control the expression of the genes involved in separation at the end of the cell division cycle.
Introduction
The proliferation of all organisms depends on the process of cell division, in which a single cell duplicates and divides to produce two genetically identical daughter cells. This requires a strict temporal and spatial coordination of different processes and mechanical systems that contribute to ensuring successful cell division. Schizosaccharomyces pombe provides an excellent model to study the different control mechanisms that regulate the progression between the stages of the cell division cycle. One of these mechanisms is the transcriptional control of gene expression (for reviews, see refs.Citation1,2). The regulation of gene transcription ensures that proteins required at particular cell cycle moment are only produced when they are needed, and it is a universal mechanism used by proliferating cells for orderly cell cycle progression. In S. pombe, genome-wide gene expression analyses have identified 4 major waves of transcription through the mitotic cycle whose periodic patterns coincide with the main stages of the cell cycle.Citation3-5 The molecular mechanisms by which the expression of the different groups occurs have been partially deciphered and found to involve cis-acting DNA motifs present in the co-regulated promoters to which a trans-acting transcription factor complex specific for each group of genes binds.Citation2 The different combinations of these elements for each group results in coordinated gene expression at different cell cycle times.
For most cells, division of the nucleus (mitosis) precedes division of the cytoplasm (cytokinesis), both processes being closely interlinked in order to ensure the distribution of the genetic material among the resulting daughter cells. Cytokinesis requires the assembly and constriction of an actomyosin ring that provides the force necessary for cytoplasm partition, a process that is coordinated with the synthesis of the new membrane that separates the cells. In organisms with a cell wall, ring contraction is also coupled to the synthesis of a trilaminar structure, called the septum, between the 2 daughter cells; this is fundamental for cell integrity to be ensured during cytokinesis. In unicellular organisms, cytokinesis is followed by the controlled degradation of the central layer of the newly synthesized septum, known as the primary septum, allowing the 2 daughter cells to separate from each other physically (cell separation) and become independent entities.Citation6-8 Cytokinesis and cell separation therefore need to be highly coordinated in time and they must be spatially regulated, since they are opposite processes (septum formation and septum degradation) that must occur within minutes. To avoid cell lysis during cytokinesis, dissolution of the primary septum does not start until its synthesis has been completed, and the enzymes involved in its dissolution must be targeted to the region of the cell where the septum has been assembled.
Cell separation in S. pombe is controlled transcriptionally by the Sep1-Ace2 cascade.Citation4,9 Sep1 is a protein of the conserved forkhead familyCitation10,11 that activates the expression of the first of 2 transcriptional waves that occur at the end of the cell cycle, during the M-G1 transition, which is the moment when cytokinesis and cell separation occur.Citation3-5,12 This cluster of co-expressed genes includes genes such as plo1+, cdc15+, ppb1+, slp1+, fin1+ or sid2+, whose products are necessary for the completion of mitosis and cytokinesis. Another target of Sep1 is ace2+, which encodes a C2H2 zinc-finger transcription factor that in turn activates a second wave of gene expression, with maximum expression at the end of mitosis.Citation4,9,13 ace2+ periodicity has been shown to be conserved in multiple organisms.Citation14 Among the genes regulated by Ace2 in S. pombe are those that encode the main enzymes responsible for the dissolution of the cell division septum, such as the β-glucanase Eng1, which degrades the primary division septum;Citation15 the α-glucanase Agn1, which hydrolyses the old cell wall surrounding the septumCitation16,17 and Adg proteins of unknown function that are also necessary for cell separation.Citation9 Thus, a major function of the Sep1-Ace2 transcriptional pathway in S. pombe is to periodically trigger the expression of the genes required for completion of cytokinesis and cell separation at the end of the cell cycle. At the same time, the existence of 2 different transcription factors allows a delay in the expression of cell separation genes over those required for the completion of cytokinesis. Mutants lacking Sep1 or Ace2 have similar defects in septum degradation, producing a typical phenotype of chains of unseparated cells.Citation9,15,18 A more modest cell separation defect can be also found in mutants lacking the Eng1, Agn1 or Adg proteins.Citation9,15-17
Sep1 is part of a transcriptional complex known as PBF (Pombe cell cycle box Binding Factor), which includes at least 2 other transcription factors, namely the forkhead-like protein Fkh2 and the MADS box-like protein Mbx1.Citation12,19-22 Although forkhead proteins are generally believed to be “actipressors” (they can both activate and repress gene expression),Citation23 the 2 forkhead-like proteins of the PBF complex in S. pombe, Sep1 and Fkh2, are thought to play opposing roles in regulating mitotic transcription.Citation19,24 The activator role of Sep1 is supported by the observation that the deletion of sep1+ causes reduced transcription in mitosis, whereas its overexpression results in the induction of the target genes. Furthermore, maximum levels of Sep1 binding to mitotic gene promoters coincide with maximum transcription levels.Citation19 In contrast, Fkh2 binding is observed when gene expression is waning and the absence of this transcription factor protein elicits constitutive expression levels along the cell cycle.Citation19,20 Sep1 is required for Fkh2 activity, since it has been observed that the deletion of the sep1+ gene suppresses the lethal phenotype observed when fkh2+ is over-expressed; the two proteins have also been shown to interact with each other in cells.Citation19,24 The third factor, Mbx1, is not necessary for the periodicity of genes transcribed during M phase.Citation19 How PBF activity is regulated and integrated with the cell cycle is not yet fully understood. Recent studies have revealed that Mbx1 and Fkh2 are controlled through temporary phosphorylation by the kinases Plo1 (Polo kinase) and the cyclin-dependent kinase Cdk8, while dephosphorylation is performed by the Clp1 phosphatase.Citation24-26 In fact, Sep1 is probably a phosphoprotein controlled by Cdk1, and it is also a substrate of the Clp1 phosphatase.Citation27,28 It has also been shown that Sak1, a transcription factor of the RFX family, activates the expression of mitotic genes in collaboration with Fhk2.Citation29
Promoters of the genes thought to be under the control of PBF (including ace2+) are significantly enriched in forkhead-binding motifs.Citation3,4 These cis-acting DNA regulatory elements are conserved from humans to budding yeast. Unlike budding yeast, S. pombe forkhead-binding sites do not seem to be accompanied by consensus motifs for MADS-box-like protein binding.Citation30,31 Instead, they are often accompanied by DNA-binding sites known as PCB (Pombe cell-Cycle Box; consensus GNAACG/A).Citation12
Less is known, however, about the regulation of the genes under the control of Ace2. The promoters of the genes under the control of this transcription factor are enriched for the hexanucleotide CCAGCC.Citation3,4 This sequence has also been described as the consensus binding site for the Ace2 ortholog in S. cerevisiae and it is necessary for proper transcriptional regulation.Citation32,33
In this report, we show that Ace2 consensus motifs are required for Ace2 binding to the promoter region of their regulated genes. In addition, this recruitment occurs during periods of maximum expression and is decreased when another transcription factor, Fkh2, is detected. We also demonstrate that the PBF components Sep1, Fkh2 and Mbx1 contribute to the regulation of Ace2-dependent genes, but that there are significant differences between individual genes. Thus, Sep1 appears to be involved in activating eng1+ expression whereas Mbx1, which has not been thought to be relevant for mitotic gene control in S. pombe, is required for agn1+ expression. Our findings suggest a complex array of control mechanisms aimed at ensuring the correct timing of gene expression at the end of the cell cycle to complete cytokinesis, septation and separation.
Results
CCAGCC-binding sites are required for maximum expression mediated by Ace2
Analysis of the promoter region of the eng1+ gene revealed the presence of 2 candidate motifs that matched the predicted consensus Ace2-binding sequence (CCAGCC), located at 413 and 478 bp upstream of the ATG. To test whether these putative binding sites were important for the expression of Ace2-dependent genes, we constructed 3 versions of the eng1+ promoter region (from −600 to −1) in which either one or both copies of these sites were deleted. The three mutated promoters (referred to as eng1-Δ1, eng1-Δ2, and eng1-Δ1Δ2) were cloned upstream of the lacZ reporter gene on a plasmid and introduced into a wild-type strain. As controls, the wild-type and ace2Δ strains carrying a plasmid with the wild-type version of the eng1+ promoter (eng1-wt) were used. Analyses of βgalactosidase activity in asynchronously growing cells revealed reduced levels of enzymatic activity in cells carrying either the eng1-Δ1 or the eng1-Δ2 single-mutated promoters (around 40% and 30% of that of the wild-type, respectively) (). This effect was additive, since β-galactosidase activity was further reduced to around 10% of the wild-type activity when the 2 putative Ace2-binding sites were deleted (eng1Δ1Δ2). As expected, lacZ expression was also reduced in cells carrying the wild-type eng1+ promoter but lacking the transcriptional activator Ace2 (ace2Δ strain), although this strain still exhibited around 20% of the β-galactosidase activity found in the wild-type strain (). Thus, importantly, these results indicate that the 2 copies of the CCAGCC sequence present at the eng1+ promoter are important and necessary to achieve maximum expression of its coding region.
Figure 1. Ace2 regulates the expression of its target genes by periodically binding the CCAGCC sequence. (A) Schematic representation of the position of the 2 copies of the CCAGCC sequence in the eng1+ promoter and constructs generated. The different deletions were cloned upstream of the E. coli lacZ gene. The arrow indicates the orientation of the CCAGCC sequence. The graph shows the results of the β-galactosidase activity assay in the wild-type strain (PN1870) carrying the wild-type eng1+ promoter (WT; pMAN4), eng1-Δ1 (pMAN5), eng1-Δ2 (pMAN6), eng1-Δ1Δ2 (pMAN7) or empty vector and in the ace2Δ mutant (OL163) transformed with the wild-type eng1+ promoter (WT; pMAN4). The data are means (±SD ) of 2 independent experiments and are normalized to the wild-type strain. (B) Chromatin immunoprecipitation of Ace2-HA in a strain carrying the wild-type eng1+ promoter (WT; YMAT15) and in a strain carrying the deletion of the putative Ace2-binding sites in the eng1+ promoter (eng1-Δ1Δ2; YMAT85). Data are shown relative to the background binding in untagged control cells (OL264). The binding of Ace2 to the his3+ promoter was used as a negative control to normalize the data. The columns represent the mean of 2 independent biological repeats, indicated by dots. Each dot is the mean of at least 2 technical replicates. (C) eng1+ expression level in the wild-type strain (WT, YMAT15) and in the eng1-Δ1Δ2 (YMAT85) and ace2Δ (YMAT71) mutants determined by quantitative RT-PCR and normalized using his3+ expression. The columns represent the mean of 2 independent biological repeats, indicated by dots. Each dot is the mean of at least 2 technical replicates. (D) Expression of eng1+ and agn1+ in synchronized cells. Data were normalized using his3+ expression. The results shown are representative of the results obtained in 2 different experiments. (E) Ace2 binding to the promoters of eng1+ and eng1+ along the cell cycle. A cdc25–22 ace2-HA strain (YMAT15) was synchronized by arrest-release and samples were taken at the indicated times (minutes) after the release for mRNA purification (D) or chromatin immunoprecipitation (E) using anti-HA antibodies. The anaphase and septation indices are indicated in E. Data are shown relative to the background binding in untagged control cells (OL264). The binding of Ace2-HA to the his3+ promoter was used as a negative control to normalize the data. The results shown are representative of the results obtained in 2 different experiments.
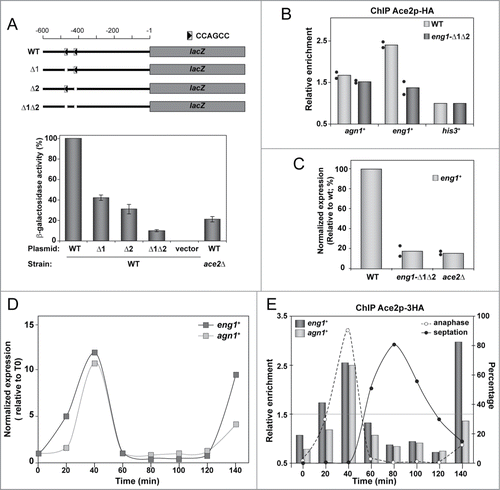
We next performed quantitative chromatin immunoprecipitation analyses (ChIP-qPCR) to determine the binding capacity of Ace2 to the CCAGCC motifs in vivo. To this end, the double-mutated version of the eng1+ promoter was integrated into its own chromosomal locus by site-directed mutagenesis (eng1-Δ1Δ2 allele) in a strain carrying Ace2 tagged with the HA epitope at its C-terminus. Both the wild-type and the eng1-Δ1Δ2 mutant strains were used to test Ace2 binding to the eng1+ promoter. As a control, we used another of the Ace2-dependent genes, agn1+. The results of ChIP-qPCR in asynchronous cultures showed that Ace2 was recruited to the wild-type promoter regions of eng1+ and agn1+, and that binding was specifically reduced in the strain carrying the eng1Δ1Δ2 allele (). This result meant that the CCAGCC motifs are also binding sites in S. pombe for Ace2 in vivo. In agreement with these data, eng1+ expression was reduced in cells lacking Ace2 or the 2 Ace2-binding sites at the eng1+ promoter (). Taken together, these results demonstrate that in fission yeast, as in S. cerevisiae, Ace2 activates the expression of its target genes directly through its binding to the CCAGCC consensus sequence motifs.
Ace2 binding to DNA is cell cycle-regulated
For a more detailed analysis of Ace2 and its function as a transcriptional activator, we synchronized cells using the cdc25–22 mutant and monitored both the expression levels of eng1+ and agn1+ and the recruitment of Ace2 to their promoters through the cell cycle. As previously described,Citation9 the expression of eng1+ and agn1+ occurred in a periodic manner, with a maximum at anaphase (). Interestingly, Ace2 was also found to bind the 2 promoters in a cyclic manner, with binding peaks coincident with the maximum mRNA levels of both genes (). Therefore, cell cycle-dependent recruitment of Ace2 to the promoters might be responsible for the periodic expression of genes under the control of this transcription factor. We also noted that Ace2 binding to the eng1+ promoter occurred earlier than to agn1+ (); this was consistent with an earlier induction of eng1+ expression seen for this gene in synchronized cultures ().
The promoters of Ace2-target genes contain additional regulatory elements
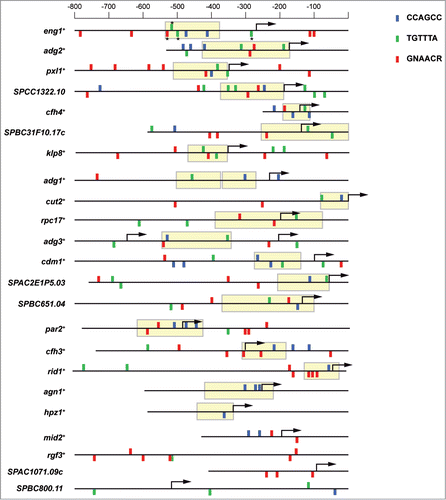
Examination of the promoters of genes under the control of Ace2 allowed us to identify other possible cis-acting regulatory elements. For this analysis, we used the upstream regions of 23 genes reported to be regulated by Ace2 by microarray experiments,Citation4,9 and the Regulatory Sequence Analysis Tools program, RSAT.Citation34 As expected, most of the promoters (19 out of 23) contained one or more copies of the Ace2 consensus-binding site. In fact, 16 promoters contained at least 2 CCAGCC motifs (). Furthermore, we found that most of these Ace2 sites were located in nucleosome-depleted regions (NDR) close to the transcription starting sites.Citation35 The localization of transcription factor binding sites in NDR regions is believed to enhance transcription factor attachment and facilitate subsequent transcription.Citation36 In S. cerevisiae, it has been shown that NDRs are important for maintaining the periodic expression of cell cycle-regulated genes.Citation37 In addition to Ace2-binding sites, the RSAT program also detected the consensus motif TGTTTA, which has been reported to be a binding site for forkhead transcription factors.Citation30 This motif was over-represented in the promoters analyzed and was frequently accompanied by PCB sites. For example, in the eng1+ promoter 2 forkhead and one PCB site were present within the NDR. In striking contrast, agn1+, the other glucanase involved in septum degradation, together with eng1+, was one of the few genes with no consensus forkhead or PCB motifs, suggesting that these two glucanases might be subject to a different type of regulation.
Figure 2. Diverse binding sites are present in the promoters of Ace2-dependent genes. Schematic representation of the promoter region of Ace2-dependent genes. Blue rectangles indicate the position of the CCAGCC sequence; green rectangles correspond to TGTTTA motifs, and red rectangles mark GNAACR sequences. The position of the rectangles indicates their orientation: direct if it is above the line and inverted if it is below. NDR regions in the promoters are indicated by yellow boxes. Genes were ordered according to the abundance of these sites within NDR. The asterisks indicate the sites mutated in this study (TGTTTA to CGGCTA and GNAACR to GNGCCR).
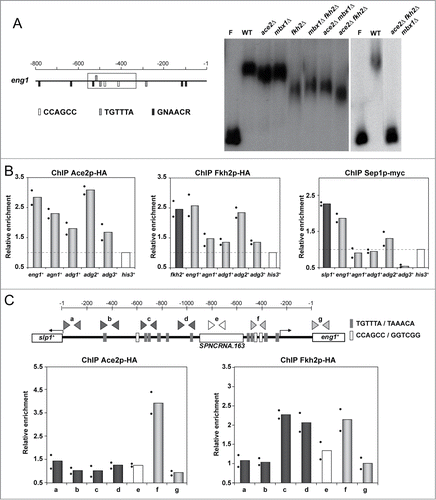
The presence of potential forkhead- and/or PCB-binding sites in many of the promoters regulated by Ace2 suggested that the PBF complex might also bind to the promoters of Ace2-dependent genes. As a first approach to investigate this possibility, we carried out electrophoretic mobility shift assays (EMSA) using a DNA fragment from the eng1+ promoter region (578 to 335) that contained 2 Ace2 sites, 2 forkhead sites, and a single PCB site as a probe. For this experiment, protein extracts from single-, double- and triple-deletion mutants lacking the regulators Ace2, Fkh2 and Mbx1 were prepared and examined for their ability to bind to labeled probe DNA in comparison with protein extracts from a wild-type strain. Incubation of the probe with a wild-type extract uncovered the presence of a retarded complex, indicating the association in vitro of a protein with the fragment of the eng1+ promoter (). Interestingly, different degrees of retardation of the probe were observed when extracts from single (ace2Δ, fkh2Δ or mbx1Δ) or double mutants (ace2Δ fkh2Δ, ace2Δmbx1Δ, or fkh2Δ mbx1Δ) were tested, supporting the idea that Ace2, along with Fkh2 and Mbx1, might contribute to the regulation of Ace2-target genes through binding to its promoter. Significantly, no mobility shift was observed upon incubation of the probe with a protein extract from the triple mutant lacking Ace2, Fkh2 and Mbx1 (ace2Δ fkh2Δ mbx1Δ), suggesting that no other proteins can bind to the eng1+ promoter region, at least in vitro. Thus, these EMSA experiments suggest that the eng1+ promoter fragment has the ability to bind Ace2, Fkh2 and Mbx1 in vitro.
Figure 3. PBF components bind to Ace2-target promoters in vitro and in vivo. (A) Gel retardation assay with the eng1+ promoter. A labeled DNA fragment corresponding to the region marked with a rectangle in the schematic representation to the left was incubated with protein extracts from the wild-type strain (PN1870, WT) and the ace2Δ (OL163), mbx1Δ (GG503), fkh2Δ (GG523), mbx1Δ fkh2Δ (GG552), ace2Δ fkh2Δ (YMAN91), ace2Δ mbx1Δ (YMAN92) and ace2Δ mbx1Δ fkh2Δ (YMAN106) mutants. The probe without protein extract (F) is also shown. (B) Chromatin immunoprecipitation of Ace2-HA, Fkh2-HA and Sep1-myc in ace2-HA (YMAT15), fkh2-HA (YMAT14) or sep1-myc (YMAT16) strains. Data are shown relative to the background binding in untagged control cells (OL264). Binding of the 3 tagged proteins to his3+ promoter was used as a negative control to normalize the data. (C) eng1+ promoter scanning. Schematic representation of the intergenic region that separates slp1+ and eng1+, in which the binding sites for Ace2 are indicated with light gray boxes and those for forkhead transcription factors with dark gray. The oligonucleotide pairs used for ChIP experiments are also indicated. Below, quantitative results of the ChIP experiments with Ace2-HA or Fkh2-HA. Data are shown relative to the background binding in untagged control cells (OL264). The binding of Ace2-HA and Fkh2-HA to the his3+ promoter was used as a negative control to normalize the data. For B and C, the columns represent the mean of 2 independent biological repeats, indicated by dots. Each dot represents the mean of at least 2 technical replicates.
The forkhead proteins Sep1 and Fkh2 bind to the promoters of Ace2-target genes in vivo
We next investigated whether the transcription factors that make up the PBF complex were recruited to the promoters of Ace2-target genes (eng1+, agn1+, adg1+, adg2+ and adg3+) in vivo. To this end, quantitative ChIP experiments using asynchronous cultures of strains containing C-terminally tagged versions of these 3 transcription factors (Sep1-myc, Fkh2-HA, and Mbx1-HA) were used. As a control, binding to slp1+ and fkh2+ promoters was also analyzed, since it is known that they are regulated by PBF.Citation19 In addition, recruitment of Ace2 was also measured for comparative purposes. As expected, Ace2 was present in the promoters of all five genes tested, although between them differences in binding were observed that correlated with the number of Ace2 binding sites present in their sequences. Thus, the promoters with at least 2 sites, such as eng1+, agn1+ and adg2+, contained the highest amount of Ace2, while adg1+ and adg3+, with only one Ace2 binding site, showed a less marked recruitment of this protein (). Importantly, the forkhead-like transcription factor Fkh2 was detected in the promoters of eng1+ (with values comparable to those of the positive control, fkh2+) and adg2+. In contrast, limited binding was observed in the promoters of agn1+, adg1+ and adg3+. Again, the reduced binding to these genes correlated with a lower number of forkhead/PCB sites present in their promoters (). For the transcription factor Sep1, we observed recruitment of this protein to the eng1+ and adg2+ promoters, but binding was lower than that detected for Fkh2 ().
We were unable to detect Mbx1 binding to any of the promoters tested (data not shown), similar to the results reported for other PBF-regulated genes.Citation24 In contrast, the Polo kinase Plo1 interacts with Mbx1 throughout the cell cycle and is detected on Mbx1-target gene promoters. Thus, we examined Plo1 association with the promoters of Ace2-dependent genes, but no binding was detected (data not shown).
Since the strongest binding detected by ChIP corresponded to the eng1+ promoter and since this gene is adjacent and divergently transcribed to slp1+, which is also regulated by PBF,Citation19 it is possible that the signal observed for eng1+ could have been due to the binding of PBF to the slp1+ promoter. To rule out this hypothesis, 7 different oligonucleotide pairs annealing at different regions of the slp1+-eng1+ intergenic region were designed and used to measure the abundance of each fragment in immunoprecipitates of a strain carrying Fkh2-HA (a to g in ). As a control, recruitment of Ace2 to the same regions was examined in an Ace2-HA strain. This analysis revealed that Ace2 ChIP-enrichment binding was restricted to the promoter region upstream of eng1+ (400–500 nt upstream of the ATG; ). No Ace2 binding to the coding region of eng1+ or to the slp1+ promoter was observed. In contrast, Fkh2 showed 2 different binding peaks upstream of eng1+ and slp1+ (c and f, ) that were separated by a region where no recruitment was observed (e, ), corresponding to a non-coding RNA region (SPNCRNA.163). This result indicated that Fkh2 binding to the eng1+ and slp1+ promoters was independent in both cases. Therefore, the ChIP analyses support the idea that the PBF transcriptional complex might be involved in the regulation of the expression of Ace2-dependent genes, at least those containing forkhead and/or PCB sites in the promoter, such as eng1+.
eng1+ and agn1+ are subject to different transcriptional regulation
The ChIP results suggested that different transcriptional regulators bound to the promoter regions of Ace2-dependent genes. To test whether they performed a function in the control of the expression of this group of genes, the effect of deleting Fkh2, Sep1, and Mbx1 on mRNA expression of Ace2-target genes was analyzed, focusing the study on eng1+ and agn1+ as representatives of genes with or without forkhead/PCB-binding sites respectively. Since it has been described that ace2+ is itself a PBF-regulated gene,Citation19,20 ace2+ transcription levels were also monitored. Quantitative mRNA measurements in asynchronously growing cells showed that eng1+ and agn1+ expression was decreased in strains lacking Sep1 or Ace2 (). This is consistent with previously published results showing that Ace2 activates the transcription of genes involved in cell separation, such as eng1+ and agn1+, and that Sep1 indirectly controls the expression of Ace2-target genes through the activation of ace2+ expression.Citation9 Strikingly, however, we found that the expression of eng1+, but not agn1+, was even lower in sep1Δ cells than in the ace2Δ mutant. This reduction in eng1+ expression in sep1Δ cells occurred when ace2+ mRNA levels were at 20% relative to the wild-type, suggesting that Sep1 might also directly influence eng1+ expression.
Figure 4. Regulation of eng1+ and agn1+ requires different factors. (A) Expression of ace2+ and the Ace2-target genes eng1+ and agn1+ measured by quantitative RTPCR in the wild-type strain (OL432) and the ace2Δ (YMAN30), sep1Δ (A131), fkh2Δ (GG523), mbx1Δ (GG503) and mbx1Δ fkh2Δ (GG552) mutants. his3+ expression was used for normalization. (B) Expression of eng1+ and agn1+ measured by quantitative RT-PCR in strains Pnmt+-ace2+ (YMAT59), mbx1Δ Pnmt1-ace2+ (YMAT61), sep1Δ Pnmt1-ace2+ (YMAT62), and fkh2Δ Pnmt1-ace2+ (YMAT60). The graph represents the quantification of the expression of each gene with respect to strain Pnmt1-ace2. act1+ expression was used for normalization. In both panels, the columns represent the mean of 2 independent biological repeats, indicated by dots. Each dot represents the mean of at least 2 technical replicates.
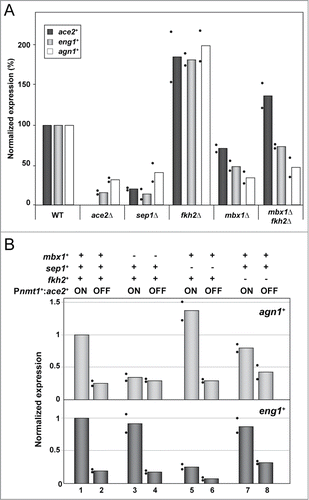
The forkhead-like protein Fkh2 has been reported to act as a repressor of the expression of ace2+ and other Sep1-dependent genes.Citation19,20 In agreement with this function, asynchronous cells lacking Fkh2 displayed up-regulated expression levels of ace2+ (). Similarly, the expression of both eng1+ and agn1+ was increased ().
It has been suggested that the MADS box protein Mbx1 is not required for periodic gene regulation, but that instead it plays a role in controlling the amplitude of expression, since this is reduced in mbx1Δ cells.Citation19 We noted that deletion of mbx1+ reduced the expression of the two glucanase-encoding genes. agn1+ expression was reduced in the mbx1Δ mutant to nearly the level observed in the ace2Δ and sep1Δ strains; this was surprising because the agn1+ promoter does not contain any consensus PBF binding sites (). ace2+ mRNA abundance in asynchronous cultures of the mbx1Δ strain was reduced to around 35% of the wild-type (). Nevertheless, this change in ace2+ expression could not be the only explanation for the relatively low agn1+ expression in the mbx1Δ strain. Furthermore, deletion of fkh2+ suppressed the decreased expression of ace2+ in a mbx1Δ background, but agn1+ mRNA levels were still low in the double mutant mbx1Δ fkh2Δ ().
Since the deletion of fkh2+ and mbx1+ changed the expression of ace2+,Citation19 the effect of Fkh2 and Mbx1 on Ace2-dependent genes could be indirect due to differences in the abundance of the Ace2 transcription factor in these mutant strains. Therefore, to define the possible direct role of the PBF transcriptional complex on Ace2-dependent genes, a new set of strains in which ace2+ was cloned under the control of a PBF-independent promoter was constructed. To accomplish this, the thiamine-repressible nmt1+ promoter was integrated into the genome by homologous recombination directly upstream of the ace2+ ORF in the wild-type and in mutant strains lacking Fhk2, Sep1 or Mbx1. After the induction of ace2+ transcription by growth in thiamine-free medium, the overall expression level of eng1+ and agn1+ was comparable to that of the wild-type strain (data not shown). In contrast, under non-inducing conditions (ace2+ transcription off) the Pnmt1+ace2+ strain showed low expression levels of eng1+ and agn1+, similar to those observed in the ace2Δ mutant. When the mutant strains Pnmt1+:ace2+ sep1Δ, Pnmt1+:ace2+ fkh2Δ and Pnmt1+:ace2+ mbx1Δ were grown under inducing and non-inducing conditions, different patterns of eng1+ and agn1+ expression were observed (). While agn1+ mRNA levels were markedly reduced in the absence of Mbx1 (), eng1+ was down-regulated in sep1Δ cells (). Furthermore, combining the sep1Δ mutation with the depletion of ace2+ caused an additional reduction in eng1+ expression (), indicating an additive effect of Sep1 and Ace2 in the regulation of eng1+. Cells lacking Fkh2 showed a slight decrease in the expression of both eng1+ and agn1+ (). Taken together, these results indicate that the transcriptional regulation of the Ace2-dependent genes involves different factors in addition to Ace2, and that these factors may vary between genes.
Fkh2 binds to the eng1+ promoter in a cell-cycle dependent manner
To further investigate the role of Fkh2 in the regulation of Ace2-dependent genes, we synchronized cells using the cdc25–22 mutant and followed the kinetics of Fkh2 binding to the promoters of eng1+ and agn1+ through the cell cycle. ChIP analyses revealed that Fkh2 binding to the eng1+ promoter region was periodic during the cell division cycle (), although with a different pattern to that observed for Ace2 (). Fkh2 recruitment to the eng1+ promoter increased after anaphase, when eng1+ transcription decreased (). No detectable binding was observed at anaphase, when maximum eng1+ expression occurs, consistent with Fkh2 being a transcriptional repressor. Similar binding profiles for Fkh2 have been detected in PCB-regulated promoters, where Fkh2 is also thought to play a role as a repressor.Citation24 In agreement with previous results in non-synchronized cells, no clear binding signals for Fkh2 were observed at the agn1+ promoter, supporting the notion that different molecular mechanisms operate in the transcriptional regulation of eng1+ and agn1+.
Figure 5. Fkh2 binds to the promoters of Ace2-dependent genes at different moments of the cell cycle. (A) Chromatin immunoprecipitation of Fkh2-HA in the cdc25–22 fkh2-HA strain (YMAT14) (left), and expression of eng1+ and agn1+ (right) measured by quantitative RT-PCR along the cell cycle and normalized using his3+ expression. Synchrony was induced by arrest-release and samples were taken at the indicated times (minutes) after the release for chromatin immunoprecipitation (left) using anti-HA antibodies or for mRNA purification (right). The anaphase and septation index is indicated in the left graph. Data are shown relative to the background binding in untagged control cells (OL264). Binding of Fkh2-HA to the his3+ promoter was used as a negative control to normalize the data. (B) Expression of eng1+ determined by quantitative RT-PCR in strains nda3-KM311 Pnmt1+-ace2+ (YMAT17), and nda3-KM311 fkh2Δ Pnmt1+-ace2+ (YMAT94) along the cell cycle. Cells were arrested in early mitosis by incubation at 18°C and samples were taken at the indicated times (minutes) after release for mRNA purification. Expression data were normalized using his3+ expression. (C) Chromatin immunoprecipitation of Fkh2-HA and Sep1-myc in strains carrying mutations in the 3 forkhead binding sites and an adjacent PCB site in the eng1+ promoter (Peng1–4m allele) (strains YMAT43 and YMAT70, respectively). Data are shown relative to the binding in wild-type cells (YMAT14 and YMAT69, respectively). The binding of Fkh2-HA and Sep1-myc to the his3+ promoter was used as a negative control to normalize the data. The columns represent the mean of 2 independent biological repeats, indicated by dots. Each dot represents the mean of at least 2 technical replicates. (D) Expression of eng1+ and agn1+ during the cell cycle. Synchrony was induced by arrest-release of cdc25–22 (YMAT14) or cdc25–22 Peng1–4m (YMAT43) mutants, and samples were taken at the indicated times (minutes) after release for RNA extraction. Expression was measured by quantitative RT-PCR and normalized using his3+ expression. For the time course experiments (A, B and D), the results shown are representative of the results obtained in 2 different experiments.
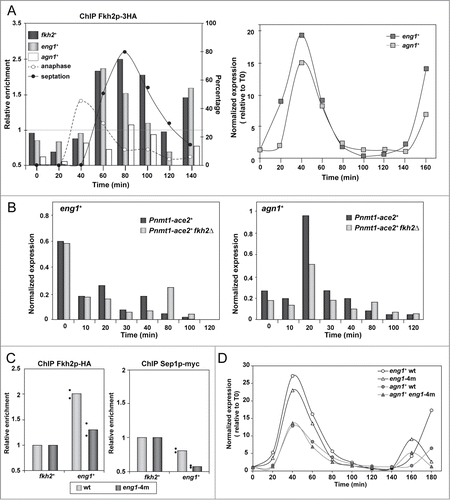
To analyze the contribution of Fkh2 to the regulation of eng1+ in more detail, we measured eng1+ expression through the cell cycle in a Pnmt1+:ace2+ fkh2Δ mutant in comparison with Pnmt1+:ace2+ cells. For this experiment, cells were arrested in early mitosis using the cold-sensitive nda3-KM311 mutant, since the cdc25–22 fkh2Δ Pnmt1+:ace2+ mutant shows synthetic sick phenotypes, which precluded its use for analysis. Under these conditions, we observed that in thiamine-free medium the expression profile for eng1+ was similar in both the Pnmt1+:ace2+ and the Pnmt1+:ace2+ fkh2Δ strains, with high mRNA levels in arrested cells (most of the cells were in metaphase), after which they decreased (). This result indicated that the observed Fkh2 recruitment to the eng1+ promoter is unlikely to be the mechanism responsible for repressing its transcription. As could be expected, agn1+ expression was also periodic in both strains, although the amplitude of the oscillation was lower in the strain lacking Fkh2. Additionally, this experiment allowed us to verify that agn1+ expression was delayed by 20 min (most of the cells were in late anaphase) as compared to that of eng1+.
We also examined the effect of point mutations in forkhead- and PCB-binding sites in the promoter of eng1+. For this, we generated strains carrying point mutations in the 3 forkhead sites and in the PCB site of the eng1+ promoter integrated at its chromosomal locus (Peng1–4m allele, indicated by asterisks in ). ChIP analyses revealed a reduction in the recruitment of Fkh2 and Sep1 to the eng1+ promoter in Peng1–4m cells as compared to the wild-type promoter (). However, these mutations did not affect Ace2 recruitment (data not shown). In spite of the reduction of Fkh2 and Sep1 recruitment, RT-qPCR analyses using RNA isolated from synchronized cultures showed similar expression profiles of eng1+ in cells carrying the mutated promoter or the wild-type promoter (). Additionally, we also examined whether Fkh2 binding to the eng1+ promoter was dependent upon Ace2 sites. Thus, Fkh2 binding was measured in a strain carrying the 2 Ace2-binding sites at the eng1+ promoter deleted (eng1-Δ1Δ2 allele, strain YMAT84). The results of ChIP analyses showed that a similar Fkh2 recruitment had occurred in both the wild-type and the mutated promoter (data not shown). Overall, these results suggest that additional regulatory elements for forkhead transcription factor binding probably exist in the eng1+ promoter, and that Ace2 and Fkh2 bind independently to different elements of the promoter.
Mbx1 is required for agn1+ expression
The fact that Mbx1 had a specific effect on agn1+ expression was unexpected for 2 reasons: first, the agn1+ promoter had no consensus-binding sites for PBF, and second, Mbx1 has not been shown to be a critical transcription factor for gene expression until now. To obtain additional evidence that Mbx1 was controlling the expression of agn1+, we monitored agn1+ and eng1+ expression during the cell cycle progression in cdc25–22 mbx1Δ cells. Similar to the results of the asynchronous cultures, agn1+ expression was lost in the absence of Mbx1, whereas periodic eng1+ transcription was unaffected (). Therefore, agn1+ expression is dependent on Mbx1.
Figure 6. Mbx1 controls agn1+ expression. (A) Expression of eng1+ and agn1+ during the cell cycle. Synchrony was induced by arrest-release of cdc25–22 (OL264) or cdc25–22 mbx1Δ (GG549) mutants, and samples were taken at the indicated times (minutes) after release for RNA extraction. RNA blots were probed with specific probes for eng1+ and agn1+, using act1+ as a loading control. The percentage of septation at each time-point is indicated below, and was determined by counting the percentage of cells with a septum after calcofluor staining. The graph represents the quantification of the expression of each gene with respect to the wild-type (wt, value 1). (B) Overexpression of agn1+ complements the separation defects of mbx1Δ mutants. The wild-type (WT; OL432) and the agn1Δ (YSAB156), mbx1Δ (GG503) and mbx1Δ carrying Pnmt1+-agn1+ (YMAT91) mutants were grown in EMM5S medium without thiamine for 17 hours before staining the cells with aniline blue. Images show fields and details of separating cells for each strain. The graph to the right indicates the percentage of cells with a septum that have a wild-type or a V-shaped phenotype in each strain (n = 350).
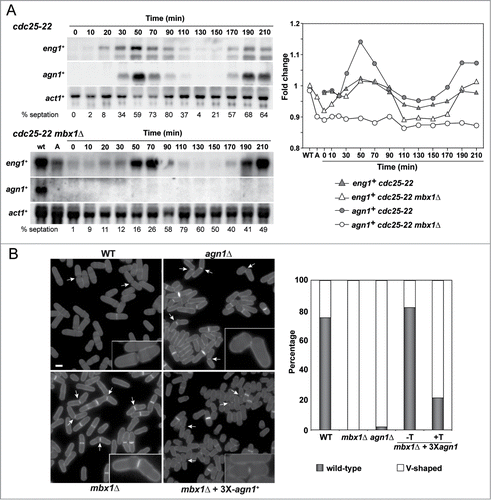
Since Agn1 is an α-1,3-glucanase required for the degradation of the cylinder of cell wall that surrounds the septum, known as the septum edging,Citation16,17 agn1Δ mutants have a typical V-shaped cell separation defect in which the 2 daughter cells remain attached by the remnants of cell wall on one side ().Citation9 It has been described that mbx1Δ mutants also have a cell separation defect,Citation19 and our results suggest that it could be attributed to the absence of Agn1. Therefore, we tested whether replacement of the agn1+ promoter by a heterologous promoter to allow agn1+ expression independent of Mbx1 could reverse the cell separation defect of mbx1Δ mutants. Quantification of the cell separation phenotype revealed that around 98% of the separating cells had a V-shaped morphology in agn1Δ mutants (). A similar percentage was observed in the mbx1Δ mutant and in mbx1Δ Pnmt1+:agn1+ cells grown under repressing conditions. Interestingly, overexpression of agn1+ largely corrected the phenotype of mbx1Δ cells, restoring a wild-type phenotype. Therefore, these results confirm the idea that transcriptional activation of agn1+ by Mbx1 is essential for normal separation of mother and daughter cells after cytokinesis.
Discussion
The regulation of cell cycle progression is of key importance for proper segregation of the genome and the generation of 2 identical cells. One way to ensure a correct order of cell cycle events is the control of specific proteins at certain phases of the cell cycle, and this can be achieved either by altering the stability of the proteins or through the regulation of gene expression. The latter mechanism ensures the coordinated expression of groups of genes at particular moments of the cell cycle, when their products are required, and it is a widespread mechanism among eukaryotic cells.
In budding yeast, the consecutive waves of gene expression during the different stages of the cell cycle are functionally linked to one another by mechanisms in which one wave of gene expression contains the transcription factor that controls the transcription of the next wave of gene expression.Citation38-41 In fission yeast, transcriptome analyses have unveiled the existence of 4 main waves of gene expression, corresponding to several stages of the cell cycle,Citation3-5 although there is a low degree of linkage between them. To date, the only direct link between 2 consecutive cell cycle waves of transcription occurs at the end of mitosis, and it is the transcriptional cascade involving Sep1 and Ace2 transcription factors.Citation1,2 In this study, we examined the regulation of genes encoding proteins required for cell separation whose expression occurs periodically at the M-G1 interval, dependent on the transcription factor Ace2. We found that the regulation of this group of genes involved other transcription factors in addition to Ace2, such as the PBF components. Furthermore, the regulation of this group of genes showed different degrees of dependence on Sep1, Fkh2 and Mbx1.
In S. cerevisiae, the paralogs Swi5 and Ace2 regulate a wave of gene expression at M-G1 by binding to the ACCAGCN sequence present at the promoters of the target genes.Citation32,33,42,43 Transcriptome analyses in S. pombe have identified a group of 23 genes that are expressed at the M-G1 transition that require the transcription factor Ace2.Citation3-5,9 Most of them contain one or more copies of the consensus sequence ACCAGCC in their promoters. eng1+ contains 2 copies of this sequence, which are necessary for the expression of the gene and for Ace2 binding, indicating that in S. pombe Ace2 has the same binding specificity as in S. cerevisiae. Several lines of evidence suggest that components of the PBF transcriptional factor are also directly involved in the regulation of this group of genes. First, in addition to Ace2-binding sites most of the promoters also contain one or more copies of the PCB (GNAACR) and/or forkhead-binding (TGTTTA) sequences in different arrangements. Interestingly, in many cases these regulatory sequences are localized to nucleosome-depleted regions (NDRs), which are normally present immediately upstream of the transcription start site (TSS). It has been proposed that the precise organization of nucleosomes in promoters regulates the interaction between transcription factors and DNA,Citation35,44,45 suggesting that they can play a role in the regulation of this group of genes. Second, according to ChIP experiments, Fkh2 can be detected at the promoter region of several of these genes (eng1+, adg2+ and adg3+), and Sep1 can also be immunoprecipitated associated with the promoter region of eng1+ and adg2+. Third, the expression of the Ace2-dependent genes was altered in sep1Δ, mbx1Δ, fkh2Δ or mbx1Δ fkh2Δ mutants, although the effect of the deletion of each factor had different effects on individual genes, suggesting a different contribution of Fkh2, Sep1 and Mbx1 to the regulation of the expression of Ace2-dependent genes. Finally, although the expression of Ace2 is itself dependent on the PBF factor,Citation4,12,46 the effect on Ace2-dependent genes might be direct, since defects in expression were observed when the ace2+ promoter was replaced with the regulated nmt1+ promoter. Together, these results indicate that the PBF directly contributes to the regulation of these genes at the M-G1 transition.
It has previously been shown that expression of the cdc15+ gene cluster requires the PBF transcription factor complex containing Sep1, Fkh2 and Mbx1,Citation12 with the 2 forkhead transcription factors playing opposing roles. The activator, Sep1, would only be bound to PCB promoters when the genes are expressed, and the repressor, Fkh2, appears to be bound when the genes are repressed.Citation19,24 However, the precise role of Fkh2 remains unclear. More recently, it has been proposed that this transcription factor would regulate the onset of mitotic transcription and the timing of mitotic entry, and that the primary function of Fkh2 could be the regulation of mitotic progression and the timing of transcription.Citation25 Our ChIP and expression analyses on synchronized cultures suggest that Ace2 and Fkh2 are also present at the promoters of Ace2-dependent genes, and that they are bound at different times of the cell cycle. Thus, Ace2 was found in the promoters of eng1+, agn1+ and adg2+ when expression was high, while Fkh2 promoter occupancy occurred when the genes were repressed. Therefore, the regulation of Ace2-dependent genes shares some similarities with the Sep1-dependent genes, although the role of Fkh2 in regulating the expression of Ace2-dependent genes is not clear either, since no increase in eng1+ expression was observed in synchronized cultures lacking Fkh2. Garg et al. (2015) have recently found by ChIP-seq that Sep1 only controls the transcription of a few genes and that a new regulator, Sak1, is apparently the main activator of mitotic gene expression.Citation29 These authors propose that Fkh2 could act as a pioneer factor to displace nucleosomes from regulatory regions in order to aid the assembly of an activating transcription complex.
It has been also shown that Mediator, a co-regulator of eukaryotic transcription that functions as a bridge between gene-specific regulators and RNA polymerase II,Citation47 is recruited to a large number of mitotic genes and regulates their transcription. This complex is present in the promoters of most Sep1-dependent genes and it has been proposed that Sep1 might be required for the recruitment of Mediator to target genes to ensure the correct regulation of periodic transcription.Citation48 Interestingly, this study also found that Mediator was recruited to the promoter of some the Ace2-dependent genes, such as chf4+, eng1+ or adg2+, but not to agn1+ or adg3+. Since genes containing a higher number of regulatory elements in their promoters (including the PCB and/or forkhead-binding sequences, ) correlate with those that recruit Mediator to the promoter, it is tempting to speculate that the presence of these regulatory elements in Ace2-dependent genes could be important for the correct regulation of periodic transcription.
One interesting conclusion from the observations presented here is that the regulation of Ace2-dependent genes is more complex than previously thought. Periodic expression of this group of genes requires the transcription factor Ace2,Citation9,13,49 but there are important differences in the regulation of the genes encoding the 2 main glucanases involved in cell separation, the endo-β1,3-glucananse Eng1 and the α1,3-glucananse Agn1. While the regulation of eng1+ required Ace2 and Sep1, the expression of agn1+ was dependent on Mbx1. We were unable to monitor the association of Mbx1 with the agn1+ promoter by ChIP, as previously reported,Citation19 probably because tagged versions of Mbx1 are non-functional. However, the fact that the over-expression of agn1+ complemented the separation defect of mbx1Δ mutants is a strong indication that Mbx1 regulates the expression of agn1+. In S. cerevisiae, the MADS box transcription factor Mcm1 has an important regulatory function during cell cycle expression, controlling the expression of different group of genes at the M/G1 and G2/M transitions,Citation50,51 whereas the role of Mbx1 in S. pombe is less clear. Our results indicate that agn1+ is under the control of Mbx1. These observations therefore indicate that the regulation of Ace2-dependent genes is heterogeneous, and that different components of the PBF might function differentially in each promoter. A question that arises is why cells use a different type of regulation for two genes that participate in the same biological process. One possible explanation is the observation that agn1+ overexpression is lethal for the cells, whereas the overexpression of eng1+ is not deleterious.Citation15-17 Since α-glucan is present in the septum region and surrounds the cell wall, where it plays a structural role and is essential for maintaining cell shape and viability,Citation52 it might be necessary for the cells to strictly regulate the moment when Agn1 is synthesized in order to avoid cell lysis. In contrast, Eng1 seems to be highly specific for the linear β-1,3 glucan of the primary septum,Citation53,54 not acting on other polymers of the cell wall, and therefore it might not be necessary for its expression to be tightly regulated.
Materials and Methods
Yeast strains, growth conditions and genetic manipulations
lists the yeast strains used in this work. Yeast cells were grown on YES medium or minimal medium (EMM) with the required supplements.Citation55 Yeast transformations were performed using standard procedures: the lithium acetate method or genetic crossing. For experiments using the nmt1+ promoter, cells were grown to the logarithmic phase in EMM containing 15 μM thiamine, harvested, washed 3 times with EMM, and inoculated in fresh medium without thiamine at an OD595 = 0.025. Synchronization of strains carrying the thermosensitive cdc25–22 mutation was achieved by growing the cells at the permissive temperature (25°C) to early log phase (OD595 = 0.35) and then shifting the cultures to 37°C for 4 h. Cells were released from arrest by transfer to 25°C, and samples were taken every 10 or 20 min. Synchrony was monitored by estimation of the percentage of binucleate and septated cells under the microscope.
Table 1. Yeast strains used in this study
Construction of plasmids and strains
Plasmid pMAN4, containing the eng1+ promoter was constructed by PCR amplification with oligonucleotides that generated SphI and BamHI sites at the ends and then cloning the amplified fragment into the corresponding sites of vector pSPE357, which contains the Escherichia coli lacZ gene and the ura4+ marker.Citation56 Deletion of the first (Δ1), the second (Δ2), or both (Δ1Δ2) copies of the CCAGCC sequence was achieved by recombinant PCR, generating the desired deletions as SphI-BamHI fragments, which were cloned into plasmid pSPE-357 to yield plasmids pMAN5, pMAN6 and pMAN7 respectively.
ace2+ null mutants were obtained by replacing the ace2+ coding region with the kanr cassette (which confers resistance to the antibiotic G418) or the ura4+ gen by recombinant PCR as described.Citation57 For this purpose, DNA fragments of 300–500 bp of the 5′ and 3′ flanking regions of ace2+ were PCR-amplified using specific oligonucleotide pairs. The resulting fragments were then fused by recombinant PCR to the corresponding cassette.
Strains carrying the ace2+ gene tagged with HA were constructed by a PCR-mediated strategy using the 3HA-kanMX6 module for C-terminal tagging.Citation58 Strains with ace2+ under the control of the nmt1+ promoter at its chromosomal locus were constructed using the kanMX6-P41nmt1Citation58 or the natMX6-P41nmt1-GFP (which confers resistance to the antibiotic nourseothricin) modules for inducible expression.Citation59 In all cases, the tagging and the deletion cassettes were obtained by PCR with oligonucleotides containing approximately 100 base pairs of flanking sequences homologous to the target sequence.
Strains bearing Peng1-Δ1Δ2 or Peng1–4m alleles were constructed using an in vivo site-directed mutagenesis system to create unmarked mutant alleles, in which the target locus was initially marked with the ura4+ gene, after which the marker was replaced with the mutated DNA by counterselection on medium containing 5-fluoroorotic acid (5-FOA).Citation60 For this, a DNA fragment for replacing the eng1+ promoter with the ura4+ cassette was generated by PCR amplification of plasmid pFA6a-ura4Citation58 with specific oligonucleotides, and this PCR product was used to transform strains YMAT14 and YMAT15 to the Ura+, yielding strains YMAT40 and YMAT41, respectively. A PCR product carrying the Peng1-Δ1Δ2 allele obtained by PCR using pMAN7 as template was used to transform strains YMAT40 and YMAT41, selecting 5FOAR colonies that yielded strains YMAT84 and YMAT85, respectively. A similar approach was used to insert the mutations in the 3 forkhead (GTAAACA to GTAGCG) and the PCB sites of the eng1+ promoter (Peng1–4m allele). Mutations of these 4 sites were achieved by successive rounds of PCR, and the final product was used to transform strains YMAT40 and YMAT41. Then, 5FOAR colonies were selected, yielding strains YMAT42 and YMAT43, respectively. Strains YMAT69 and YMAT70 were obtained by crossing YMAT16 and YMAT42. In all cases, proper integration of the corresponding cassettes in the S. pombe genome was confirmed by PCR. The oligonucleotide sequences used for strain constructions are available upon request.
Microscopy
Samples were observed on a Nikon Eclipse i90 microscope equipped with a Hamamatsu Orca-ER camera and controlled by MetaMorph (Molecular Devices Corporation). Visualization of septa was accomplished by staining yeast cells with aniline blue. For synchrony analysis, cells fixed in 70% ethanol were stained simultaneously with DAPI (4,6-α-diamidino-2-phenylindole) and aniline blue.
Protein extracts and β-galactosidase assay
β-galactosidase activity was determined using ONPG (o-nitrophenyl-β-D-galactopyranoside) as substrate. Yeast cells were grown to the logarithmic phase, harvested by centrifugation, washed, and suspended in lysis buffer (100 mM Tris-HCl, pH 8.0, 20% glycerol, 1 mM β-mercaptoethanol, 40 mM PMSF). After breaking the cells, lysates were cleared by centrifugation, and 50 μl of the supernatant was used in a 1-ml ONPG assay as described.Citation61 Specific enzyme activity was calculated in Miller units.
Electromobility Bandshift Assays (EMSA)
Whole cell extracts were generated from cells as described and gel retardation analysis was performed with an eng1+ promoter obtained by PCR amplification with specific oligonucleotides in the presence of α-32P dCTP, as previously described.Citation12
Northern blot analyses
Cells (109) were collected at different time intervals after release from the restrictive temperature (37°C), and total RNA was prepared as previously described.Citation15 RNAs (12.5 μg) were transferred to Hybond membranes and probed with 32P-labeled probes corresponding to eng1+ and agn1+ and act1+ obtained by PCR with specific oligonucleotides. Signals were normalized using act1+ transcript level.
Gene expression analysis by RT-qPCR
To determine the expression of genes by quantitative RT-PCR, 2.0 × 108 cells were collected by centrifugation and used for total RNA extraction using the TRIZOL method (Invitrogen), according to the manufacturer's instructions. cDNA synthesis was carried out with the SuperScript II First-Strand Synthesis System (Invitrogen), using 3 μg of RNA previously treated with DNAse I (Invitrogen). One μl of cDNA was used for the quantitative reactions in an Applied Biosystems 7300 Real-Time PCR System. The SYBR Premix Ex Taq (TaKaRa) reagent was used with primer concentrations of 0.2 μM. Serial dilutions of wild-type S. pombe genomic DNA (1/10, 1/100, 1/1000, 1/10000, 1/100000) were used to generate a standard curve for each reaction. The reaction conditions were as follows: 95°C for 45 s and 40 cycles of 95°C for 5 s and 60°C for 31 s, followed by a dissociation step at 95°C for 15 s, 60°C for 1 min and 95°C for 15 s. All PCR reactions were normalized to his3+ or act1+ transcription data. The experiments were performed at least twice using cDNA from different biological replicates with 2 technical replicates for each sample. The “Mean Normalized Expression” was calculated according to Simon.Citation62
ChIP-qPCR
Cells (109) bearing an HA or a c-myc tag from mid-log-stage cultures were collected and used for ChIP assays according to Robyr and Grunstein,Citation63 with the following modifications: cells were lysed using a FastPrep-24 bead beater at a speed setting of 4.5 for 40 s followed by 5.0 2× 40 s, at 4°C. The crude lysate was sonicated on ice using a Diagenode Bioruptor Sonicator (at settings: “high”; 30 s ON, 30 s OFF) for 30 min (15 min cumulative sonication time). Cell debris was centrifuged for 30 min at 13000 g at 4°C. A total of 15 μg of antibody specific for anti-HA (12CA5, Roche) or c-myc (9E10, Santa Cruz) was applied in a 500-μl volume of chromatin. The experiments were performed twice with 2 immunoprecipitation repeats in each experiment. Cells from the untagged control strain (OL264) were also collected, using the same experimental conditions. A 1-μl volume of ChIP DNA was used for qPCR. The reaction conditions were performed as described above.
For ChIP experiments along the cell cycle, cells were synchronized in cdc25–22 block-release experiments, and collected every 20 min after release from the restrictive temperature (37°C). Samples for each time-point were divided into 2 aliquots, one (109 cells) processed for ChIP and the other (2 × 108 cells) processed for mRNA expression analyses. Cells from the untagged control strain were also collected using the same experimental setting.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank Andrés Clemente and Sergio Moreno for helpful comments and discussions and Nick Skinner for language revision.
Funding
This research was supported by grants from the Comisión Interministerial de Ciencia y Tecnología (BFU2010–15884).
References
- Bahler J. A transcriptional pathway for cell separation in fission yeast. Cell Cycle 2005; 4:39-41; PMID:15611619; http://dx.doi.org/10.4161/cc.4.1.1336
- McInerny CJ. Cell cycle regulated gene expression in yeasts. Adv Genet 2011; 73:51-85; PMID:21310294; http://dx.doi.org/10.1016/B978-0-12-380860-8.00002-1
- Oliva A, Rosebrock A, Ferrezuelo F, Pyne S, Chen H, Skiena S, Futcher B, Leatherwood J. The cell cycle-regulated genes of Schizosaccharomyces pombe. PLoS Biol 2005; 3:e225; PMID:15966770; http://dx.doi.org/10.1371/journal.pbio.0030225
- Rustici G, Mata J, Kivinen K, Lió P, Penkett CJ, Burns G, Hayles J, Brazma A, Nurse P, Bähler J. Periodic gene expression program of the fission yeast cell cycle. Nat Genet 2004; 36:809-17; PMID:15195092; http://dx.doi.org/10.1038/ng1377
- Peng X, Karuturi RK, Miller LD, Lin K, Jia Y, Kondu P, Wang L, Wong LS, Liu ET, Balasubramanian MK, et al. Identification of cell cycle-regulated genes in fission yeast. Mol Biol Cell 2005; 16:1026-42; PMID:15616197; http://dx.doi.org/10.1091/mbc.E04-04-0299
- Sipiczki M. Splitting of the fission yeast septum. FEMS Yeast Res 2007; 7:761-70; PMID:17596184; http://dx.doi.org/10.1111/j.1567-1364.2007.00266.x
- Roncero C, Sánchez Y. Cell separation and the maintenance of cell integrity during cytokinesis in yeast: the assembly of a septum. Yeast 2010; 27:521-30; PMID:20641019; http://dx.doi.org/10.1002/yea.1779
- Weiss EL. Mitotic exit and separation of mother and daughter cells. Genetics 2012; 192:1165-202; PMID:23212898; http://dx.doi.org/10.1534/genetics.112.145516
- Alonso-Núñez ML, An H, Martín-Cuadrado AB, Mehta S, Petit C, Sipiczki M, del Rey F, Gould KL, Vázquez de Aldana CR. Ace2p controls the expression of genes required for cell separation in Schizosaccharomyces pombe. Mol Biol Cell 2005; 16:2003-17; PMID:15689498; http://dx.doi.org/10.1091/mbc.E04-06-0442
- Ribár B, Bánrévi A, Sipiczki M. sep1+ encodes a transcription-factor homologue of the HNF-3/forkhead DNA-binding-domain family in Schizosaccharomyces pombe. Gene 1997; 202:1-5; PMID:9427538; http://dx.doi.org/10.1016/S0378-1119(97)00390-9
- Ribár B, Grallert A, Oláh E, Szállási Z. Deletion of the sep1+ forkhead transcription factor homologue is not lethal but causes hyphal growth in Schizosaccharomyces pombe. Biochem Biophys Res Commun 1999; 263:465-74; PMID:10491317; http://dx.doi.org/10.1006/bbrc.1999.1333
- Anderson M, Ng SS, Marchesi V, MacIver FH, Stevens FE, Riddell T, Glover DM, Hagan IM, McInerny CJ. plo1+ regulates gene transcription at the M-G1 interval during the fission yeast mitotic cell cycle. EMBO J 2002; 21:5745-55; PMID:12411492; http://dx.doi.org/10.1093/emboj/cdf564
- Petit C, Mehta S, Roberts RH, Gould KL. Ace2p contributes to fission yeast septin ring assembly by regulating mid2+ expression. J Cell Sci 2005; 118:5731-42; PMID:16317047; http://dx.doi.org/10.1242/jcs.02687
- Fernandez MA, Rueda C, Peddada SD. Identification of a core set of signature cell cycle genes whose relative order of time to peak expression is conserved across species. Nucleic Acids Res 2012; 40:2823-32; PMID:22135306; http://dx.doi.org/10.1093/nar/gkr1077
- Martín-Cuadrado AB, Dueñas E, Sipiczki M, Vázquez de Aldana CR, del Rey F. The endo-b-1,3-glucanase Eng1p is required for dissolution of the primary septum during cell separation in Schizosaccharomyces pombe. J Cell Sci 2003; 116:1689-98; PMID:12665550; http://dx.doi.org/10.1242/jcs.00377
- Dekker N, Speijer D, Grün CH, van den Berg M, de Haan A, Hochstenbach F. Role of the a-glucanase Agn1p in fission-yeast cell separation. Mol Biol Cell 2004; 15:3903-14; PMID:15194814; http://dx.doi.org/10.1091/mbc.E04-04-0319
- García I, Jiménez D, Martín V, Durán A, Sánchez Y. The a-glucanase Agn1p is required for cell separation in Schizosaccharomyces pombe. Biol Cell 2005; 97:569-76; PMID:15850449; http://dx.doi.org/10.1042/BC20040096
- Sipiczki M, Grallert B, Miklos I. Mycelial and syncytial growth in Schizosaccharomyces pombe induced by novel septation mutations. J Cell Sci 1993; 104:485-93; PMID:8505375
- Buck V, Ng SS, Ruiz-Garcia AB, Papadopoulou K, Bhatti S, Samuel JM, Anderson M, Millar JB, McInerny CJ. Fkh2p and Sep1p regulate mitotic gene transcription in fission yeast. J Cell Sci 2004; 117:5623-32; PMID:15509866; http://dx.doi.org/10.1242/jcs.01473
- Bulmer R, Pic-Taylor A, Whitehall SK, Martin KA, Millar JB, Quinn J, Morgan BA. The forkhead transcription factor Fkh2 regulates the cell division cycle of Schizosaccharomyces pombe. Eukaryot Cell 2004; 3:944-54; PMID:15302827; http://dx.doi.org/10.1128/EC.3.4.944-954.2004
- Szilagyi Z, Batta G, Enczi K, Sipiczki M. Characterisation of two novel fork-head gene homologues of Schizosaccharomyces pombe: their involvement in cell cycle and sexual differentiation. Gene 2005; 348:101-9; PMID:15777722; http://dx.doi.org/10.1016/j.gene.2004.12.043
- Zilahi E, Salimova E, Simanis V, Sipiczki M. The S. pombe sep1 gene encodes a nuclear protein that is required for periodic expression of the cdc15 gene. FEBS Lett 2000; 481:105-8; PMID:10996305; http://dx.doi.org/10.1016/S0014-5793(00)01990-6
- Voth WP, Yu Y, Takahata S, Kretschmann KL, Lieb JD, Parker RL, Milash B, Stillman DJ. Forkhead proteins control the outcome of transcription factor binding by antiactivation. EMBO J 2007; 26:4324-34; PMID:17898805; http://dx.doi.org/10.1038/sj.emboj.7601859
- Papadopoulou K, Ng SS, Ohkura H, Geymonat M, Sedgwick SG, McInerny CJ. Regulation of gene expression during M-G1-phase in fission yeast through Plo1p and forkhead transcription factors. J Cell Sci 2008; 121:38-47; PMID:18057023; http://dx.doi.org/10.1242/jcs.019489
- Szilagyi Z, Banyai G, Lopez MD, McInerny CJ, Gustafsson CM. Cyclin-dependent kinase 8 regulates mitotic commitment in fission yeast. Mol Biol Cell 2012; 32:2099-109; PMID:22451489; http://dx.doi.org/10.1128/MCB.06316-11
- Papadopoulou K, Chen JS, Mead E, Feoktistova A, Petit C, Agarwal M, Jamal M, Malik A, Spanos A, Sedgwick SG, et al. Regulation of cell cycle-specific gene expression in fission yeast by the Cdc14p-like phosphatase Clp1p. J Cell Sci 2010; 123:4374-81; PMID:21098641; http://dx.doi.org/10.1242/jcs.073056
- Grallert A, Grallert B, Ribar B, Sipiczki M. Coordination of initiation of nuclear division and initiation of cell division in Schizosaccharomyces pombe: genetic interactions of mutations. J Bacteriol 1998; 180:892-900; PMID:9473044
- Chen JS, Broadus MR, McLean JR, Feoktistova A, Ren L, Gould KL. Comprehensive proteomics analysis reveals new substrates and regulators of the fission yeast clp1/cdc14 phosphatase. Mol Cell Proteomics 2013; 12:1074-86; PMID:23297348; http://dx.doi.org/10.1074/mcp.M112.025924
- Garg A, Futcher B, Leatherwood J. A new transcription factor for mitosis: in Schizosaccharomyces pombe, the RFX transcription factor Sak1 works with forkhead factors to regulate mitotic expression. Nucleic Acids Res 2015; PMID:25908789; http://dx.doi.org/10.1093/nar/gkv274
- Pic A, Lim FL, Ross SJ, Veal EA, Johnson AL, Sultan MR, West AG, Johnston LH, Sharrocks AD, Morgan BA. The forkhead protein Fkh2 is a component of the yeast cell cycle transcription factor SFF. EMBO J 2000; 19:3750-61; PMID:10899128; http://dx.doi.org/10.1093/emboj/19.14.3750
- Pierrou S, Hellqvist M, Samuelsson L, Enerback S, Carlsson P. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J 1994; 13:5002-12; PMID:7957066
- Dohrmann PR, Butler G, Tamai K, Dorland S, Greene JR, Thiele DJ, Stillman DJ. Parallel pathways of gene regulation: homologous regulators SWI5 and ACE2 differentially control transcription of HO and chitinase. Genes Dev 1992; 6:93-104; PMID:1730413; http://dx.doi.org/10.1101/gad.6.1.93
- Dohrmann PR, Voth WP, Stillman DJ. Role of negative regulation in promoter specificity of the homologous transcriptional activators Ace2p and Swi5p. Mol Cell Biol 1996; 16:1746-58; PMID:8657150
- Thomas-Chollier M, Defrance M, Medina-Rivera A, Sand O, Herrmann C, Thieffry D, van Helden J. RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res 2011; 39:W86-91; PMID:21715389; http://dx.doi.org/10.1093/nar/gkr377
- Soriano I, Quintales L, Antequera F. Clustered regulatory elements at nucleosome-depleted regions punctuate a constant nucleosomal landscape in Schizosaccharomyces pombe. BMC genomics 2013; 14:813; PMID:24256300; http://dx.doi.org/10.1186/1471-2164-14-813
- Whitehouse I, Tsukiyama T. Opening windows to the genome. Cell 2009; 137:400-2; PMID:19410536; http://dx.doi.org/10.1016/j.cell.2009.04.026
- Bai L, Charvin G, Siggia ED, Cross FR. Nucleosome-depleted regions in cell-cycle-regulated promoters ensure reliable gene expression in every cell cycle. Dev Cell 2010; 18:544-55; PMID:20412770; http://dx.doi.org/10.1016/j.devcel.2010.02.007
- Breeden LL. Periodic transcription: a cycle within a cycle. Curr Biol 2003; 13:R31-8; PMID:12526763; http://dx.doi.org/10.1016/S0960-9822(02)01386-6
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 2002; 298:799-804; PMID:12399584; http://dx.doi.org/10.1126/science.1075090
- Simon I, Barnett J, Hannett N, Harbison CT, Rinaldi NJ, Volkert TL, Wyrick JJ, Zeitlinger J, Gifford DK, Jaakkola TS, et al. Serial regulation of transcriptional regulators in the yeast cell cycle. Cell 2001; 106:697-708; PMID:11572776; http://dx.doi.org/10.1016/S0092-8674(01)00494-9
- Tyers M. Cell cycle goes global. Curr Opin Cell Biol 2004; 16:602-13; PMID:15530770; http://dx.doi.org/10.1016/j.ceb.2004.09.013
- Doolin MT, Johnson AL, Johnston LH, Butler G. Overlapping and distinct roles of the duplicated yeast transcription factors Ace2p and Swi5p. Mol Microbiol 2001; 40:422-32; PMID:11309124; http://dx.doi.org/10.1046/j.1365-2958.2001.02388.x
- Laabs TL, Markwardt DD, Slattery MG, Newcomb LL, Stillman DJ, Heideman W. ACE2 is required for daughter cell-specific G1 delay in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 2003; 100:10275-80; PMID:12937340; http://dx.doi.org/10.1073/pnas.1833999100
- Tsankov A, Yanagisawa Y, Rhind N, Regev A, Rando OJ. Evolutionary divergence of intrinsic and trans-regulated nucleosome positioning sequences reveals plastic rules for chromatin organization. Genome Res 2011; 21:1851-62; PMID:21914852; http://dx.doi.org/10.1101/gr.122267.111
- Lantermann AB, Straub T, Stralfors A, Yuan GC, Ekwall K, Korber P. Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol 2010; 17:251-7; PMID:20118936; http://dx.doi.org/10.1038/nsmb.1741
- Fankhauser C, Reymond A, Cerutti L, Utzig S, Hofmann K, Simanis V. The S. pombe cdc15 gene is a key element in the reorganization of F-actin at mitosis. Cell 1995; 82:435-44; PMID:7634333; http://dx.doi.org/10.1016/0092-8674(95)90432-8
- Conaway RC, Conaway JW. Function and regulation of the Mediator complex. Curr Opin Genet Dev 2011; 21:225-30; PMID:21330129; http://dx.doi.org/10.1016/j.gde.2011.01.013
- Banyai G, Lopez MD, Szilagyi Z, Gustafsson CM. Mediator can regulate mitotic entry and direct periodic transcription in fission yeast. Mol Cell Biol 2014; 34:4008-18; PMID:25154415; http://dx.doi.org/10.1128/MCB.00819-14
- Dekker N, de Haan A, Hochstenbach F. Transcription regulation of the a-glucanase gene agn1+ by cell separation transcription factor Ace2p in fission yeast. FEBS Lett 2006; 580:3099-106; PMID:16678171; http://dx.doi.org/10.1016/j.febslet.2006.04.061
- Althoefer H, Schleiffer A, Wassmann K, Nordheim A, Ammerer G. Mcm1 is required to coordinate G2-specific transcription in Saccharomyces cerevisiae. Mol Cell Biol 1995; 15:5917-28; PMID:7565744
- Mai B, Miles S, Breeden LL. Characterization of the ECB binding complex responsible for the M/G(1)-specific transcription of CLN3 and SWI4. Mol Cell Biol 2002; 22:430-41; PMID:11756540; http://dx.doi.org/10.1128/MCB.22.2.430-441.2002
- Grun C, Hochstenbach F, Humbel B, Verkleij AJ, Sietsma J, Klis FM, Kamerling JP, Vliegenthart JF. The structure of cell wall a-glucan from fission yeast. Glycobiology 2005; 15:245-57; PMID:15470229; http://dx.doi.org/10.1093/glycob/cwi002
- Martín-Cuadrado AB, Encinar del Dedo J, de Medina-Redondo M, Fontaine T, del Rey F, Latgé JP, Vázquez de Aldana CR. The Schizosaccharomyces pombe endo-1,3-b-glucanase Eng1 contains a novel carbohydrate binding module required for septum localization. Mol Microbiol 2008; 69:188-200; PMID:18466295; http://dx.doi.org/10.1111/j.1365-2958.2008.06275.x
- Martín-Cuadrado AB, Fontaine T, Esteban PF, Encinar del Dedo J, de Medina-Redondo M, del Rey F, Latgé JP, Vázquez de Aldana CR. Characterization of the endo-β-1,3-glucanase activity of S. cerevisiae Eng2 and other members of the GH81 family. Fungal Genet Biol 2008; 45:542-53; PMID:17933563; http://dx.doi.org/10.1016/j.fgb.2007.09.001
- Moreno S, Klar A, Nurse P. Molecular genetics analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol 1991; 194:795-823; PMID:2005825; http://dx.doi.org/10.1016/0076-6879(91)94059-L
- Lafuente MJ, Petit T, Gancedo C. A series of vectors to construct lacZ fusions for the study of gene expression in Schizosaccharomyces pombe. FEBS Lett 1997; 420:39-42; PMID:9450546; http://dx.doi.org/10.1016/S0014-5793(97)01486-5
- Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in Saccharomyces cerevisiae. Yeast 1996; 12:259-65; PMID:8904338; http://dx.doi.org/10.1002/(SICI)1097-0061(19960315)12:3%3c259::AID-YEA901%3e3.0.CO;2-C
- Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 1998; 14:943-51; PMID:9717240; http://dx.doi.org/10.1002/(SICI)1097-0061(199807)14:10%3c943::AID-YEA292%3e3.0.CO;2-Y
- Van Driessche B, Tafforeau L, Hentges P, Carr AM, Vandenhaute J. Additional vectors for PCR-based gene tagging in Saccharomyces cerevisiae and Schizosaccharomyces pombe using nourseothricin resistance. Yeast 2005; 22:1061-8; PMID:16200506; http://dx.doi.org/10.1002/yea.1293
- Storici F, Resnick MA. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol 2006; 409:329-45; PMID:16793410; http://dx.doi.org/10.1016/S0076-6879(05)09019-1
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, N. Y.: Cold Spring Harbor Laboratory; 1972.
- Simon P. Q-Gene: processing quantitative real-time RT-PCR data. Bioinformatics 2003; 19:1439-40; PMID:12874059; http://dx.doi.org/10.1093/bioinformatics/btg157
- Robyr D, Grunstein M. Genomewide histone acetylation microarrays. Methods 2003; 31:83-9; PMID:12893177; http://dx.doi.org/10.1016/S1046-2023(03)00091-4