
Abstract
Notwithstanding current multimodal treatment, including surgery, radiotherapy and chemotherapy with temozolomide (TMZ), median survival of glioblastoma (GBM) patients is about 14 months, due to the rapid emergence of cell clones resistant to treatment. Therefore, understanding the mechanisms underlying chemoresistance is mandatory to improve treatments' outcome. We generated TMZ resistant cells (TMZ-R) from a GBM cell line and from cancer stem cell-enriched cultures isolated from human GBMs. We demonstrated that TMZ resistance is partially reverted by “drug wash-out” suggesting the contribution of epigenetic mechanisms in drug resistance and supporting the possibility of TMZ rechallenge in GBM patients after prior drug exposure. The expression of histone lysine demethylase genes (KDMs) was increased in TMZ-R cells compared to parental cells, and TMZ resistance or restored sensitivity was mimicked by over-expressing or inactivating KDM5A. Methylation and expression of O6-methylguanine-DNA methyltransferase (MGMT) and drug efflux mechanisms were not altered in TMZ-R cells compared to parental TMZ sensitive cells. TMZ-R cells transiently acquired morphologic and molecular characteristics of differentiated tumor cells, features that were lost after drug wash-out. In conclusion, we demonstrated that treatment-induced TMZ resistance in GBM involves epigenetic mechanisms in a subset of slow-cycling and transiently partially differentiated cells that escape drug cytotoxicity, overcome G2 checkpoint and sustain clonal growth. We found that TMZ-R cells are sensitive to histone deacethylase inhibitors (HDACi) that synergize with TMZ. This strong synergism could be exploited to develop novel combined adjuvant therapies for this rapidly progressing and invariably lethal cancer.
Introduction
Glioblastoma (GBM) contains, within the tumor mass, small cell populations with distinct phenotypic and molecular characteristics, diverse differentiation potential and unique properties of invasiveness, proliferation, self-renewing and resistance to therapy.Citation1 These cells, identified as cancer stem cells (CSC), or tumor-initiating cells, are considered responsible for tumor recurrence and therapeutic failure.Citation2-4
The emergence of radio- and chemo-resistant cells, along with the resistance of GBM cells to apoptosis, are key factors leading to tumor recurrence; therefore, understanding the mechanisms underlying the intrinsic or acquired resistance to treatment in GBM has relevant clinical implications.Citation1 For many years GBM has been mainly treated with surgery and radiotherapy, being considered intrinsically chemoresistant. However, adjuvant treatment with the alkylating agent temozolomide (TMZ) induced a limited but significant improvement of survival, in comparison to previous protocols, particularly in patients with an epigenetically-silenced MGMT gene.Citation5,6
Epigenetics is a driving mechanism for interindividual variations of drug resistance and epigenetic modifications, because of their dynamic and reversible nature, are exploited as possible targets for innovative therapies in several tumors including GBM.Citation7 Beside MGMT methylation other epigenetic and genetic mechanisms are thought to be involved in TMZ resistance in GBM. Among these, mutation and inactivation of the Mismatch Repair mechanism,Citation8-10 miRNA modulation of signaling pathways.Citation11,12 and alteration of the extracellular matrix.Citation13 or of the drug efflux mechanisms.Citation11,14 Histone methylation and demethylation gained a particular interest in drug resistance because of the central role of these modifications in many aspects of cell physiology and pathology.Citation15-17 Lysine histone demethylases (KDMs) are a complex class of proteins, subdivided into amine oxidase (LSD1/2) and the Jumonji domain-containing protein family, which includes 28 members, structurally organized into 7 classes.Citation15 Histone demethylases are involved in many diseases, and some of them act as putative oncogenes o tumor suppressor genes and may determine the response to anticancer drugs.Citation15,18-20 In particular, KDM1A (LSD1) has been proposed as therapeutic target for GBM.Citation21
Along this line we aimed to determine whether other epigenetic factors, besides MGMT methylation, could regulate TMZ sensitivity in GBM, focusing on histone demethylase genes. In this study we demonstrate that TMZ resistance is partially reversible and that both the transient overexpression of KDM genes, in particular KDM5A, and the appearance of a transient partially differentiated phenotype characterize TMZ-R cells and contribute to the induction of resistance in GBM cultures enriched in CSCs.
Results
Generation and characterization of TMZ-resistant GBM cells
In the attempt to understand the mechanisms of drug resistance, GBM cells were grown under conditions that, mimicking an “acute response” to TMZ, rapidly kill TMZ-sensitive cells and favor the selection of cells that either are already resistant to TMZ or that rapidly develop resistance.Citation20
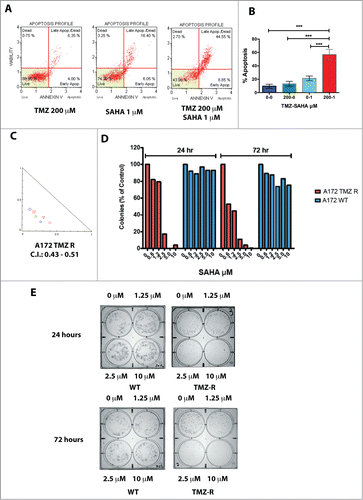
We selected TMZ-resistant (TMZ-R) cell clones from a continuous GBM cell line (A172) and 2 GBM CSC cultures (GBM 3, MGMT methylated, and GBM 5, MGMT unmethylated), by growth in medium containing 100 – 600 µM TMZ (). Within 7–10 days, only sparse cells survived in culture and, after approximately 20–30 days, small colonies of TMZ-R cells appeared. This acute treatment efficiently induced apoptosis, in a dose- and time-dependent manner, clearly evident after 72 h, as shown by annexin V staining () and caspase activation (Fig. S1A). Typically, TMZ-R cells were slow-growing, in comparison to parental (WT) cells, and accumulated in the G2/M phase (66.3% vs. 19.6% of WT cells). During TMZ treatment, the percentage of cells arrested in G2/M progressively reduced to 40.3%, indicating that TMZ-R cells partially escaped the G2 checkpoint and resumed tumor growth ().
Figure 1. Generation and characterization of TMZ-resistant GBM cells. (A) Flowchart for the generation of TMZ-resistant and wash out GBM cells. (B) Induction of apoptosis by TMZ in the A172 cell line. Apoptosis induced by TMZ cells was determined by annexin V staining. No, or negligible apoptosis was detected after 24 hrs (data not shown) whereas apoptosis was detectable at 48 and 72 hrs. This panel reports the results after 72 hrs. (C) Cell cycle analysis in A172 WT and A172 TMZ R cells. Cells were seeded in 6-well culture plates at a density of 5 × 105 cells/well and grown for 48 hrs in the presence of 200 µM TMZ or without drug (A172 WT only). The result of this analysis shows that 66.3% of the WT and 40.3 % of the TMZ R cells accumulate at G2/M upon treatment with TMZ. This indicates that TMZ R cells are a slow-cycling population that can eventually escape the G2 checkpoint. (D) Drug holiday partially restores TMZ sensitivity in GBM TMZ-R cells. GBM3 and GBM5 TMZ-resistant cells were grown in drug-free medium for at least 30 days (GBM3 and GBM5 WO) and then challenged with TMZ. Cell viability was measured by MTT assay 72 hrs. after treatment. The significance of the difference between WT, TMZR and WO cells is reported in Table S1E: Reversibility of apoptosis resistance in the TMZR GBM 3A cells after TMZ challenge, measured by annexin V staining. GBM 3 TMZR (Panel A) and GBM 3A TMZR were obtained from the same parental culture (GBM3) in 2 biologically distinct experiments of induction of drug resistance with different TMZ concentrations (400 µM for GBM 3 and 200 µM for GBM 3A). Apoptosis was measured after 72 hrs. The observed differences were significant at P < 0.05 (*) or P < 0.01 (**) (Bonferroni post-hoc).
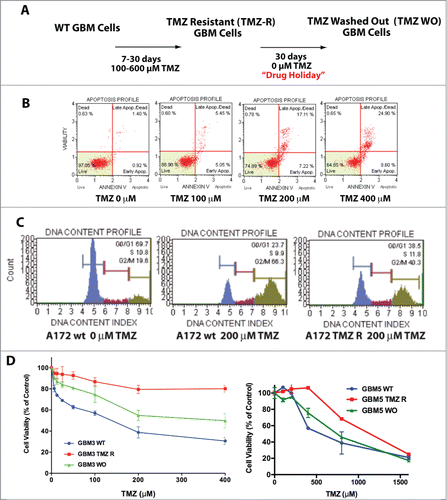
Differently from A172 WT cells, forming sparse and small colonies when grown in TMZ-containing medium, A172 resistant cells showed sustained clonal growth (Fig. S1).
To determine whether TMZ resistance is a dynamic process, we cultivated TMZ-R cells, in drug-free medium for 30 days and the resulting washed-out cells (TMZ-WO, ) were re-challenged with TMZ. Proliferative activity, evaluated in TMZ-WO sub-cultures derived from GBM 3 and GBM 5 cells (), showed that drug sensitivity was restored, at least in part, after “drug holiday.” The IC50 values at 72 hrs were 98 µM, >1000 µM and 277 µM for GBM3 WT, TMZ-R and WO cells, respectively. The IC50 of GBM5 WT cells was, as expected from the MGMT methylation status, considerably higher than that of GBM3 WT (634 µM), nevertheless treatment with TMZ further increased drug resistance to 1115 µM and after wash-out IC50 lowered to 715 µM. Differences in TMZ sensitivity between WT, TMZ-R and WO cells were highly significant, as shown in Table S1, Panel A. The partial reversibility of TMZ resistance was confirmed in a biologically distinct TMZ-R cell subpopulation obtained from GBM3 cells exposed to 200 µM TMZ (GBM3A) and assayed for TMZ-induced apoptosis (Fig. S1C). Overall these data suggest that epigenetic mechanisms contribute to the reversible drug resistance in GBM cells and, importantly, in GBM stem cell subsets.
Expression of KDM genes in GBM cells
Several studies pinpoint the role of histone demethylase genes in cancer development and in drug resistance.Citation16,18-20 To determine whether histone demethylases contribute to TMZ resistance, we selected 5 KDM genes on the basis of their potential role in GBM (KDM1A)Citation21 or in other tumors (KDM4A, KDM4B and KDM5B)Citation15,22 or in drug resistance (KDM5A).Citation20,23,24
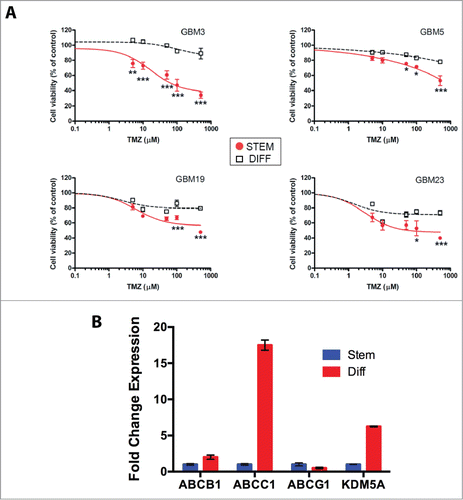
We measured the expression of these genes in 2 native CSC-enriched cultures from primary tumors (GBM3 and GBM5) and in their TMZ-R and TMZ-WO counterparts. As shown in , induction of TMZ resistance was accompanied by a marked increase in the expression of KDM1A, KDM5A and, at a lesser extent, of KDM4A in TMZ-R cells from both GBMs. KDM5B expression increased only in GBM5 TMZ-R cells, while KDM4B level was essentially unmodified in resistant cells. Importantly, the expression of these genes returned to baseline levels after drug wash-out.
Figure 2. KDM genes expression in GBM CSC cells and tumors. (A) Expression of KDM genes in 2 TMZ-resistant GBM CSC cells analyzed by qPCR in WT GBM3, GBM5 and in their TMZ-R and WO derived cultures. Fold change is relative to the expression of the WT parental cells. (B) Comparison of the mean expression levels of KDM4A, 4B, 5A and 5B in GBM and normal brain. (C) Comparison of the mean expression levels of KDM1A and KDM5A in primary GBM, recurrent GBM and normal brain. In Panels B and C the box represents the 10–90 percentile and whiskers the min-max level of expression. Significance of the mean differences was evaluated by t-test and ANOVA.
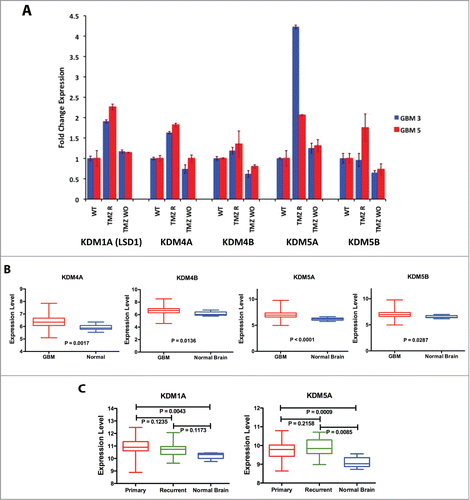
We investigated in silico the expression of KDM4A, 4B and KDM5A and 5B in a subset of 530 primary GBMs and 10 unaffected brain samples from the TCGA database (http://cancergenome.nih.gov/) utilizing the UCSC Cancer Genome Browser (https://genome-cancer.soe.ucsc.edu/).Citation25 The platform utilized for this screening (Affymetrix U133a) did not include KDM1A whose expression was analyzed, along with that of KDM5A, on the Illumina HiSeq 2000 array on a dataset of 154 primary GBM, 12 recurrent GBM and 5 normal brain samples. As shown in Figure S2A, the expression of KDM1A, 4A, 4B, 5A and 5B was widely variable in GBM samples. However, within the limits given by the small number of control non-tumor brain samples available in the TCGA database, the mean level of expression in the GBM samples was significantly higher than that of the normal brain tissue for all 5 KDM genes (). For KDM5A the mean expression difference between GBM and normal brain remained strongly significant also utilizing a different platform (). The expression of KDM1A did not significantly differ between recurrent GBM and normal brain whereas the level of KDM5A expression in recurrent samples was minimally but not significantly higher than that of primary tumors, but significantly higher than that of normal brain samples, likely supporting its implication in GBM relapse.
KDM5A is a determinant for TMZ resistance in GBM
In view of previous reports,Citation20,24 we focused our study on KDM5A. To evaluate the role of this gene in the resistance of GBM cells to TMZ, we first stably transfected A172 and GBM3 cells with KDM5A gene under the control of the CMV promoter.Citation26 In Figure S3A, is shown the increase in KDM5 enzymatic activity in A172 cells that exogenously over-express KDM5A. Transfection of A172 WT and GBM3 WT with KDM5A was accompanied by the acquisition of TMZ resistance in both cells ().
Figure 3. KDM5A is one of the determinants for TMZ resistance in GBM cells. (A) Cell viability measured by MTT assay in mock and KDM5A transfected A172 cells 48 hrs. after TMZ treatment (IC50 A172 WT: 243 µM; IC50 A172 KDM5A: 810 µM) . The observed differences were significant at P < 0.01 (**) or P < 0.001 (***) (2-way ANOVA and Bonferroni post-hoc). (B) Cell viability measured by MTT assay in mock and KDM5A transfected GBM3 cells 48 hrs. after TMZ treatment. IC50 for GBM3 WT and KDM5A were 183 and 641 µM, respectively. The higher IC50 value for GBM3 WT reported in this panel compared to Panel D of reflects the different incubation times in the 2 experiments (72 and 48 hrs). The observed difference were significant at P < 0.01 (**) or P < 0.001 (***) (2-way ANOVA and Bonferroni post-hoc). (C) Protection from apoptosis induced by TMZ by exogenous KDM5A. A172 cells mock or transiently transfected with KDM5A were treated with TMZ at different concentration and the level of apoptosis was measured after 24 hrs. by annexin V staining. The different sensitivity to apoptosis induced by TMZ in KDM5A and mock-transfected cells was highly significant (P < 0.0001) by linear regression analysis. (D) Silencing of the KDM5A gene sensitizes GBM cells to TMZ. Apoptosis is significantly induced by TMZ in GBM 3 and A172 TMZ-R derivatives transfected with shRNA-KDM5A and with the shGFP sequence. The cells were treated with TMZ 48 hrs. after transfection and apoptosis was measured by Annexin V staining 24 hrs after treatment. P < 0.05 (*) or P < 0.001 (***).
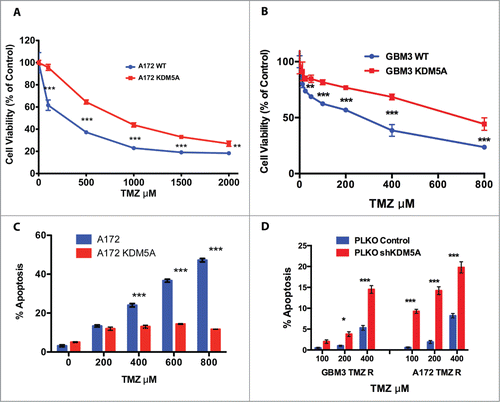
In a distinct set of experiments we evaluated TMZ-induced apoptosis in A172 WT cells transiently transfected with KDM5A. In agreement with MTT cell viability assays performed in stably transfected A172, the transient transfection of KDM5A significantly inhibited the TMZ-induced apoptosis in comparison with the cells transfected with the empty vector ().
Selective inhibitors of KDM5A activity are not yet available; therefore, to confirm the role of KDM5A in TMZ resistance, we knocked down the expression of this gene by shRNA and exposed the cells to TMZ. KDM5A is required for normal cell functionality and we observed that its stable inhibition is not compatible with cell survival. Therefore, we transiently transfected an shKDM5A construct in GBM3 TMZ-R cells and determined that KDM5A is selectively and specifically inhibited by expression of shKDM5A but not by the control shRNA (Fig. S3B). To monitor the biological effects of KDM5A inhibition we measured the apoptotic activity of TMZ in GBM3 TMZ-R and in A172 TMZ-R cells transfected with the shKDM5A construct. As shown in , knocking down KDM5A induced the recovery of TMZ pro-apoptotic effects in drug-resistant cells.
Acquired TMZ-resistance, MGMT methylation and expression and functionality of drug efflux transporters
To verify the involvement of different mechanisms in TMZ resistance concomitant to KDM5A epigenetic regulation, we utilized GBM CSCs as in vitro model to analyze the methylation status of MGMT and the functionality of drug efflux transporters before and after the induction of TMZ resistance and after drug holiday. We analyzed GBM CSCs that either are natively hypermethylated (GBM3: 90%) or unmethylated (GBM5: 5%). The mean methylation level of the MGMT promoter/enhancer in these 2 CSC cultures either after induction of TMZ resistance or after drug withdrawal, remained essentially unaltered (Table S2). Unsurprisingly MGMT expression was at the limit of detection of qPCR in hypermethylated GBM3 (Ct value > 35) but was readily detected in the unmethylated GBM5 cells (). In GBM3 the induction of drug resistance by TMZ treatment was accompanied by the transient expression of MGMT that was completely reverted by drug wash-out. However, in GBM3 cells made resistant to TMZ by transfection with KDM5A, MGMT expression remained at baseline levels (). In GBM5 cells, MGMT expression did not change after induction of TMZ resistance or after wash-out ().
Figure 4. Induction of TMZ resistance, MGMT expression and ABC-transporters functionality. (A) Expression of MGMT in WT GBM3, GBM5 and A172, utilizing the SK N AS neuroblastoma cell line as reference positive expression control. (B) Induction of TMZ resistance in GBM3 cells results in the transient expression of MGMT. After WO the expression returns to baseline levels. The exogenous expression of KDM5A does not change MGMT expression. GBM3 WT cells were utilized as reference positive control. (C) Induction of TMZ resistance does not increase MGMT expression in GBM5 cells. GBM5 WT cells were utilized as reference positive control. (D) Comparison of the mean expression levels of KDM1A and KDM5A in a 483 primary GBM dataset from TCGA. The box represents the 10–90 percentile and whiskers the min-max level of expression. Normalized mean expression values were: −0.964 for MGMT and 0.296 for KDM5A. (E) Relative mRNA expression levels of ABCB1, ABCC1 and ABCG2 in A172 WT and TMZ R cells as determined by qPCR. Fold change is relative to the expression of TMZ WT cells. (F) Cytotoxic effect of doxorubicin on A172 WT and TMZ-R cells. Cells were treated with doxorubicin (0–1 μM) for 48h and viability was determined by MTT assays. (G) Immunofluorescence images of subcellular doxorubicin distribution in A172 WT and TMZ R. Cells were treated with autofluorescent DOX (red) for 24h, nuclei and cell membranes were counterstained with DAPI (blue) and DiO (green), respectively. Overlay of images shows the predominant nuclear localization (pink) of DOX in most cells of both A172 cultures (original magnification 40X).
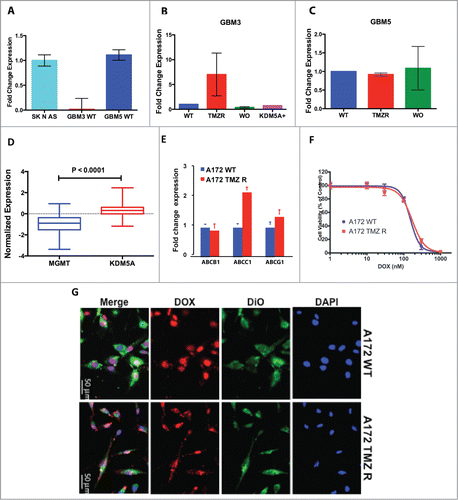
We next examined the expression of MGMT in relation to that of KDM5A in a 483-patients data set taken from the TCGA database and found evidence of weak, but significant, inverse correlation between the expression of these 2 genes (r = −0.2064: P < 0.0001) ().
Aberrant expression of the ATP-binding cassette (ABC) transporters induces active drug efflux and is thought to be central to the acquisition of multi-drug resistance in GBM cells.Citation11,14 We thus investigated whether the acquisition of TMZ resistance also involves the expression of ABC transporters comparing mRNA levels of the main transporters (ABCB1, ABCC1, ABCG2) in A172 WT and TMZ-R cells. In A172 TMZ-R cells only ABCC1 transcript was upregulated about 2-fold compared to WT cells (). Then, we ascertained whether TMZ resistance and modulation of ABC expression levels was associated with reduced sensitivity to doxorubicin (DOX), whose membrane transport is dependent on ABC function. IC50 values of DOX toxicity in A172 WT and TMZ-R cells, obtained from dose-response curves (range 0–1 µM), were 129.4 nM and 140.6 nM, respectively, showing that both cell types were equally sensitive to DOX (). We next determined the intracellular DOX localization and accumulation in A172 WT and TMZ-R cells by immunofluorescence, and we observed that most A172 WT and TMZ-R cells accumulated DOX in their nuclei, with similar doxorubicin-related fluorescent intensity (). Comparable DOX accumulation, consistent with the superimposable efficacy of the drug in both A172 cell types, indicate that under our experimental conditions, TMZ did not significantly modulate ABC transporter activity and that TMZ chemoresistant phenotype is not mediated by alteration of the expression and function of ABC transporters. Collectively, these data suggest the predominant role of KDM-mediated mechanisms of TMZ resistance in GBM cells.
HDAC inhibitors synergize with TMZ to kill TMZ-R GBM cells
It is known that HDAC inhibitors indirectly target KDM genes.Citation19,27 and reduce proliferation of CSC-enriched GBM cultures.Citation28 As shown in , A172 TMZ-R cells are resistant to apoptosis induced by TMZ even at very high drug concentrations, but retain sensitivity to the HDAC inhibitor suberanilohydroxamic acid (SAHA), an effect that is significantly increased by the combined TMZ/SAHA treatment (). A similar result was obtained with the HDAC inhibitor trichostatin A (TSA) (not shown). To determine whether SAHA synergizes with TMZ, as preliminarily suggested by the potentiation of TMZ-induced apoptosis (), we performed combination studies with the 2 drugs utilizing the Chou-Talalay algorithm.Citation29 In , is reported a representative normalized isobologram summarizing drug interactions observed in A172 TMZ-R cells treated with 200 µM TMZ and increasing concentrations of SAHA (from 0.3 to 10 µM). Combination Index (C.I.) values,Citation29 comprised between 0.43 and 0.51 for A172 TMZ R cells, were strongly suggestive of a synergic effect between SAHA and TMZ. Similar results were obtained with GBM3 cells (not shown). Interestingly, exposure to clinically relevant.Citation30 concentrations of SAHA, for 24 hrs, dramatically reduced the capacity of clonal growth of A172 TMZ-R cells, while the treatment for 72 hrs was marginally more effective (). On the contrary, SAHA exerted limited or no effects on A172 WT, indicating that this HDAC inhibitor prevalently targets TMZ-R cells ().
Figure 5. Effect of HDAC inhibitors on TMZ-R cells. (A) Resistance to apoptosis induced by TMZ in TMZ-resistant GBM cells and sensitivity to the HDAC-inhibitor SAHA. TMZ-R A172 cells were treated with 200 µM TMZ or with 1 µM SAHA or their combination (200 µM TMZ and 1 µM SAHA); apoptosis was measured by Annexin V staining after 72 hrs. (B) The combination of SAHA and TMZ exerts a significantly stronger effect compared to that of the each molecule utilized as single agent. Digital images of the wells (C) were collected and the colonies (at least 10 cells) were manually counted. (C) Colony growth of A172 WT (blue) and A172 TMZ R cells (red) treated for 24 or 72 hrs with SAHA. Identical results were obtained plating 2,500 or 5,000 cells. The result presented in this panel is that obtained with 2,500 cells/well. (D) Synergistic effects induced by TMZ (1–4 µM) and SAHA (62.5–1000 µM) combined treatment of A172 TMZ-R and GBM 3 cells evaluated by isobologram analysis according to the Chou-Talalay algorithm utilizing the CompuSyn software (www.combosyn.com). (E) Colony growth of A172 WT and A172 TMZ R treated with SAHA. A172 TMZ R (Panel A) and A172 WT (B) cells were treated for 24 or 72 hrs with serial dilutions of SAHA (From 10 to 1.25 µM), at the end of the treatment the cells were seeded in a 6 well plate at a density of 2,500 cells/well in tissue culture medium without TMZ and were grown for further 10 days in medium without drugs. At the end of the incubation period, the cells were fixed with methanol and stained with Crystal Violet.
Stemness and TMZ resistance in GBM
It is commonly accepted that sustained tumor growth and drug resistance depend on a cell subset with biological characteristics of stemness.Citation31 The induction of in vitro differentiation of GBM CSCs, by culture in serum-containing medium, results in a dramatic increased expression of astrocytic and neuronal markers (GFAP and ß-tubulin III, respectively) and in the coordinate down-modulation of the stem cell markers Olig2 and SOX2 ().Citation32-35
In the attempt to provide insights into the relation between TMZ resistance and GBM cell differentiation status, we performed cell viability assays on 4 GBM stem cell cultures, grown under stem-permissive conditions or shifted to serum-driven differentiation. In preliminary experiments we determined that the activity of TMZ is not influenced by the presence of fetal calf serum, by measuring the pro-apoptotic activity of TMZ in A172 TMZ-R cells grown either in serum-containing or in stem-permissive medium (Fig. S5A). Evaluation of cell viability by MTT assay confirmed that TMZ exerts significant cytotoxic effects in all 4 GBM CSC cultures analyzed (GBM3-5-19-23) although with different sensitivity among individual cultures (). Conversely, cells grown under differentiating conditions were significantly less sensitive to TMZ compared to their corresponding stem cultures (). Similarly, the sensitivity to the HDAC inhibitor TSA was significantly lower in differentiated cells as compared to their stem counterpart, at all the concentration tested (range 0.001-1 μM) (Fig. S5B). Next, we examined the expression of the ATP binding cassette transporters and of KDM5A in GBM3 cells grown under stem or differentiated conditions. By RT-PCR analysis we found that both ABCC1 and KDM5A were strongly over-expressed in differentiated cells ().
Figure 6. Relation between stemness and TMZ sensitivity. (A) Sensitivity to TMZ of GBM CSCs grown under stem or differentiated conditions. The P values were determined by ANOVA with the Tukey's test and are indicated for each significant point (*: P < 0.05; **: P < 0.01; *** P < 0.001 vs differentiated cells). (B) mRNA expression levels of ABCB1, ABCC1, ABCG2 and KDM5A in GBM3 differentiated cells relative to the same cells grown under stem-permissive conditions determined by quantitative PCR.
From a morphologic standpoint, GBM3 TMZ-R cells acquired a differentiated-like phenotype, showing neuronal- or astrocytic-like morphologies that were largely lost after wash-out (). To substantiate this observation and link TMZ resistance to the differentiated phenotype, we determined the expression of stemness and differentiation markers in GBM3 WT, TMZ-R and WO CSCs by immunocytofluorescence quantification. TMZ resistance significantly associated with the coordinate increased expression of the differentiation markers GFAP and ß-tubulin III and by the decrease of the stemness markers SOX2 and Olig2 (; Fig. S6A and B; Table S1B). The increased expression of KDM5A observed in TMZ-R cells does not seem responsible by itself of the acquisition of the differentiated phenotype, since the expression of GFAP and SOX2 did not change in GBM3 cells over-expressing KDM5A, when compared to WT cells ().
Figure 7. Stem and differentiation markers in GBM3 WT cells and in TMZ-R and WO cultures. (A) Bright field images of GBM 3 WT culture before treatment, and of TMZ R and WO cultures. (B) Expression of stem and differentiation markers evaluated by immunofluorescence in WT, TMZ R and WO conditions. Results are expressed as % mean of marker-positive cells from 3 independent experiments and bar represents standard deviations of mean. (C) Real time PCR analysis of GFAP and SOX2 expression in GBM3 WT cells and in GBM3 cells transfected with the pcDNA3/HA-FLAG-RBP2 construct.
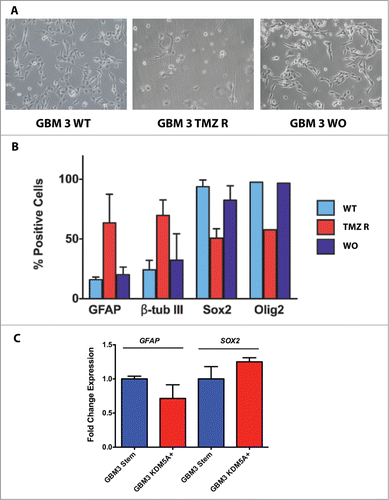
Overall these results showed that the expression of differentiation and stemness markers is deeply perturbed in cells resistant to TMZ and that, when drug sensitivity is restored after a period of “drug holiday,” the expression pattern of these markers returns to baseline levels.
Discussion
GBM is a tumor in which epigenetic factors play important roles in disease development and response to therapy.Citation1,6,36-38 The MGMT gene can repair most of the cytotoxic damage induced by TMZ,Citation39,40 and the inactivation of MGMT by hypermethylation sensitizes GBM cells to the action of TMZ, providing patients with a small, but significant, clinical advantage.Citation6 MGMT activity is not the only mechanism leading to TMZ resistance in GBM. Post-treatment mutations and reduced expression of the Mismatch Repair genes were demonstrated in vitro and in recurrent GBM, however they occur approximately in only 20% of the patients and cannot be considered a general mechanism of acquired TMZ resistance.Citation8-10 The complexity of the mechanisms of acquisition of drug resistance in GBM was highlighted in an in vivo model showing that the TMZ resistance follows both MGMT-dependent and independent pathways unrelated to the MGMT methylation status of the original tumor, a finding consistent with the development of heterogeneous mechanisms of drug resistance within the same tumor.Citation41
The present study reports that the treatment of GBM cells with TMZ rapidly induces a drug-resistant phenotype that is largely reversible, therefore strongly suggesting the intervention of epigenetic mechanisms.
Previous studies showed that “drug tolerant” cells can be generated from established lung and prostate cancer cell lines overexpressing KDM5A.Citation20,24 We have extended those findings and showed that the histone demethylase KDM5A is expressed at high levels not only in drug resistant cells derived from an established human GBM cell line but also in GBM CSCs primary cultures derived from human tumors, and that the exogenous expression and inactivation of KDM5A mimics drug resistance and sensitivity, respectively. The central role of KDM5A in the establishment of drug resistance in our GBM model early after TMZ treatment, is further suggested by the reversibility of KDM5A expression that returns to (or nearly to) baseline levels after “drug holiday,” in parallel with the reversal of the drug resistant phenotype. KDM5A is not the unique KDM gene to be modulated during the acquisition of drug resistance. Indeed, also 3 other KDM genes (KDM1A, 4A and 5B) out of the 5 tested, are transiently expressed at high levels upon induction of TMZ resistance and their expression returns nearly to baseline levels upon drug wash out. It is possible that the over expression of these other histone demethylases also contribute to the resistance to TMZ and that multiple epigenetic modifier genes participate in this mechanism. In this respect, the capacity of GBM cells to rapidly respond to drug treatment could be considered the result of an “epigenetic-driven resilient mechanism” as a consequence of the epigenetic plasticity of the cells. The in silico analysis of a large TCGA dataset showed that the expression of all the KDM genes taken into consideration in our study, and particularly that of KDM5A, is significantly higher in tumors compared to normal brain. It is tempting to speculate that the high expression of KDM5A may be related to the intrinsic high resistance to therapy of GBM.
Only few reports have longitudinally examined the variations of MGMT methylation status during the course of GBM progression.Citation42-44 The results of these studies were contradictory and their interpretation was made difficult by the different technical approaches utilized and by the heterogeneity of the tumors. In our experimental model, we have observed that in MGMT-negative GBM3 WT CSCs, the acquisition of TMZ resistance does not change MGMT methylation; nevertheless, MGMT is transiently expressed in GBM3 TMZ-R, and its expression returns to baseline levels in GBM3 WO cells. On the other hand, exogenous KDM5A overexpression is sufficient to induce TMZ resistance, without altering MGMT methylation or expression. This result suggests that MGMT methylation and expression and KDM5A expression are part of distinct pathways leading to TMZ resistance. In this respect the analysis of the TCGA data set supports this conclusion since we found that MGMT and KDM5A expression are not correlated in GBM tumors.
Our results confirm and extend those obtained in an in vivo model of GBM where methylation-independent up-regulation of MGMT was linked to the development of TMZ resistance.Citation41 In that model only 2 out of 10 TMZ-R xenografts showed elevated MGMT expression compared to controls, indicating the existence of MGMT-independent mechanisms of drug resistance Along that line, in our model we have identified KDM5A as a possible previously unknown determinant for drug resistance in GBM.
It is generally agreed that CSCs capable of multilineage differentiation exist within GBM and that this subpopulation is the only one exhibiting tumor-initiating activity and resistance to radio- and chemo-therapy;Citation1-3,31 indeed, a stem cell signature were associated with resistance to TMZ.Citation31 and hypermethylation of HOXA9 and 10 was associated to a better outcome.Citation37 Furthermore, it has been recently shown that the upregulation of BCL2, mediated by HOXA9, promoted human astrocytes immortalization and TMZ resistance.Citation45 However, a conflicting report demonstrated that TMZ preferentially depletes CSCs in GBM, leading to the concept that escaping chemotherapy could be a property of a different cell phenotype.Citation46
Our experimental model offered the opportunity to test the relationship between stemness and drug resistance. The expression of differentiation and stemness markers transiently changes in GBM cells resistant to TMZ. TMZ-R cells acquire phenotypic characteristics of differentiated cells (shift from stem cell-related to neuronal/glial marker expression and morphology) that, after drug holiday, are rapidly lost, returning to a stem-like phenotype, similar, although not identical, to that of the parental cells. Importantly, both differentiated and resistant GBM cells over-express KDM5A and it is tempting to speculate that this gene contributes to the higher chemoresistance of differentiated versus CSCs observed in our study.
How does our model reconcile with other models of drug resistance and with the clinical findings? Our working hypothesis is based on the concept that few cells resistant to chemotherapeutic drugs are already present in the tumor bulk or, more likely, acquire resistance rapidly after treatment by shifting to a slow-growing more differentiated phenotype and overexpressing MGMT, KDM5A and other epigenetic modifier genes. Eventually these cells acquire genetic alterations that render TMZ resistance irreversible.
Overall our results suggest that TMZ resistance in GBM initially is a partially reversible phenomenon that develops rapidly after treatment and is restricted to a subset of slow-growing cells capable of transient partial differentiation. Targeting these cells early, after the bulk of TMZ-sensitive cells are killed by the treatment, might provide a clinical benefit in GBM patients. In this respect, the high sensitivity of TMZ-resistant cells to HDAC inhibitors, and the synergism with TMZ, could be exploited to develop novel combined adjuvant therapies for this rapidly progressing and invariably lethal cancer.
Methods
Cell lines, GBM CSC cultures and drugs
The human GBM cell line A172 was obtained from the Biological Bank and Cell Factory of IRCCS AOU San Martino - IST (www.iclc.it) and was grown in DMEM supplemented with 10% FBS and 2 mM L-Glutamine. The authenticity of the cells was certified by the Biological Bank utilizing 8 highly polymorphic STR loci plus amelogenin (Cell IDTM, Promega).
Cultures enriched in human GBM cancer stem cells (CSCs) were derived from primary GBM cultures grown under stem-permissive conditions in serum-free medium, containing bFGF and EGF as previously described in details.Citation32,33,47-49 Four of these cell cultures (GBM 3, 5,19 and 23) were utilized in the present study. Cell differentiation was carried out by shifting GBM CSCs in serum-containing medium (10% FBS) for at least 15 days.
TMZ resistance was induced exposing A172, GBM3 and GBM5 cells to constant concentrations of TMZ. TMZ-containing medium was changed every 48 hrs until the appearance of clones of TMZ-resistant (TMZ-R) cells. TMZ concentrations utilized for selection were: 400 µM for A172 and GBM5 cells, while 400 and 200 µM were used for GBM3 (GBM3 TMZ-R and GBM3A TMZ-R derived from the selection with 400 and 200 µM TMZ, respectively).
Temozolomide (TMZ), trichostatin A (TSA), N-hydroxy-N’-phenyl-octanediamide (SAHA, Vorinostat) and doxorubicin (DOX) were obtained from Sigma-Aldrich (Milano, Italy).
KDM5A expression vectors, shRNA and transfection into GBM cells
To overexpress KDM5A, A172 cell line was stably transfected by electroporation with the pcDNA3/HA-FLAG-RBP2 construct.Citation26 (kindly provided by W. Kaelin through Addgene, www.addgene.org). After G418 selection, we isolated clones expressing high levels of KDM5 activity. selected by screening nuclear extracts prepared from 107 cells utilizing the Nuclear Extract kit from Active Motif (Carlsbad CA, USA). KDM5 activity was determined with the JARID Demethylase Activity Assay Kit from Epigenase (Farmingdale, N.Y. USA). The same protocol was applied for transient transfection except that the cells, after 24 h recovery, were treated without G418 selection.
KDM5A expression was silenced in GBM 3 and 17 cells and in A172 cell line by transient transfection with lentiviral vectors shRNA-KDM5A pLKO1 construct (clone ID: TRCN0000329797; Sigma Aldrich).Citation20 and shRNA-GFP pLKO1 (control vector; Sigma Aldrich) by electroporation using The Neon® Transfection System and the Neon 100 µl kit (Life Technologies).
Cell survival analysis
Mitochondrial function, as an index of cell viability, was evaluated utilizing the Cell Proliferation Kit I (MTT, Roche, Monza Italy) following the manufacturer instructions. Clonogenic assay was performed as described by Franken et al.Citation50
Apoptosis and cell cycle analyses
Cell cycle analysis and apoptosis (measured by annexin V staining and caspase 1 and 3–9 activation) were determined utilizing a Muse Cell Analyzer and the dedicated kits (Millipore-Merck, Vimodrone MI, Italy) following the manufacturer instructions.
Analysis of drug interaction
Drug interaction experiments were conducted by testing the activity of TMZ and SAHA combined together at various concentrations. The Combination Index (C.I.) that allows a quantitative determination of drug interactions, was calculated according to the Chou-Talalay algorithm utilizing the CompuSyn software (www.combosyn.com).Citation29
Doxorubicin (DOX) uptake and intracellular distribution assay
A172 WT and A172 TMZ R cells were seeded in 35mm Glass Base dishes (Iwaki, Japan) and allowed to grow overnight. The cells were then exposed to 0.5 and 1μM DOX for 24hrs. Cells were counterstained with the lipophilic membrane stain Vybrant DiO Cell-Labeling Solution (Molecular Probes, Life Technologies, Monza, Italy), washed to remove non-associated drug, and fixed for 15 min at room temperature with 4% paraformaldehyde, then nuclei were counterstained with DAPI (Sigma-Aldrich), and coverslips were mounted with Mowiol (Sigma-Aldrich). Images were captured by a DM2500 microscope (Leica Microsystems, Wetzlar, Germany) equipped with a DFC350FX digital camera (Leica Microsystems).
Quantitative real-time reverse transcription-PCR
Total RNA was reverse-transcribed by QuantiTect Reverse Transcription kit (Qiagen, Milano, Italy) according to the manufacturer's instructions. All qPCR reactions were performed in triplicate using Maxima SYBR Green qPCR Master Mix (Fermentas, St Leon-Rot, Germany) and 300 nM of each primer in a final volume of 10 μL using Ep Realplex Mastercycler (Eppendorf, Milano, Italy). Reactions were assembled using an Ep Motion 5070 liquid handling station (Eppendorf, Milano, Italy). Primers for human KDM5A, KDM1A (LSD1), KDM4A, KDM4B, KDM5B, MGMT, ABCB1, ABCC1, ABCG2, SOX2 and GFAP were chosen from a public database (http://medgen.ugent.be/rtprimerdb/) or were designed by us or were obtained from published reports.Citation51 (Table S3). A standard 2-step amplification with 60°C annealing temperature was used. Relative quantification of each target gene transcript was obtained using comparative Ct method. Reference genes (ATP5b, SDHA1, and CYC1) were selected using GeNorm (PrimerDesign Ltd, Southampton, UK). For each cDNA, the duplicate Ct values were averaged and normalized (geometric mean). The copy number was expressed relative to a calibrator sample using the 2−(ΔΔCt ± SD) method.
MGMT methylation analysis
MGMT methylation analysis was conducted by pyrosequencing to precisely measure the extent of methylation at 11 CpG doublets at the 3′ end of exon 1 and within the MGMT enhancer element whose methylation is sufficient to completely abolish MGMT transcription.Citation52
DNA was modified with sodium bisulfite with the EpiTect Bisulfite kit of Qiagen (Milano, Italy) following the manufacturer instructions.
The MGMT primer sets for the pyrosequencing methylation assays (Table S3) were designed with the Pyrosequencing Assay Design software (Biotage, Uppsala, SW). The PCR reactions and pyrosequencing analysis was performed as described for other genes in previous publications.Citation37,53-55
Immunofluorescence
To determine the expression of different markers of stemness and differentiation in GBM cultures, cells were plated onto Matrigel-coated glass coverslips and then fixed with 4% paraformaldehyde for 15 min at room temperature. Cells were incubated with primary antibodies as follows: rabbit anti-Olig2 (1:500; Millipore, #AB9610), rabbit anti-Sox2 (1:500; produced by our laboratory),Citation32 rabbit anti-GFAP (1:10:000; DAKO, #Z0334), and mouse anti-β Tubulin III (1:10000; Sigma Aldrich, #T8660). Immunocomplexes were detected with secondary fluorescent antibodies as DyLight 488 goat anti-mouse IgG and DyLight 459-goat anti-rabbit IgG (Jackson ImmunoResearch, #111-485-003 and #111-505-003). Cells were counterstained with Hoechst 33342 dye (Sigma-Aldrich, #14533) to identify all nuclei. Images were acquired by automated Zeiss AxioImager M2 equipped with an Axiocam MRM (Zeiss). Results are showed as percentage of stained cells from randomly selected fields.
Statistical analysis
Statistical significance was determined by analysis of variance (ANOVA) or the Student's t-test and post-hoc analyses were conducted using either Tukey's test or Bonferroni correction. The relation between the expression of genes was computed by Pearson and non-parametric Spearman correlation. The accepted values of significance were 0.05. All statistical analyses were performed using Graph Pad Prism software (GraphPad Software, San Diego CA, USA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1090063_Supplemental_Material.zip
Download Zip (17.7 MB)Acknowledgments
We thank Dr. Claudio Brigati from our Institute for sowing the seeds of this work. MR dedicates this work to the memory of Prof. Marcello Siniscalco: mentor, colleague and friend.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
Funding
This study was supported by the “Fondazione Compagnia di San Paolo – Torino,” by the Italian Ministry of Health, Core Grant “Ricerca Corrente” to the IRCCS AOU San Martino-IST, by funds “5 × 1000” and by the Italian Association for Cancer Research (AIRC) [Grant IG13563]. BB is the recipient of the “Young Investigators” grant GR-2008-1143408 from the Italian Ministry of Health.
Related Research Data
References
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev 2012; 26:756-84; PMID:22508724; http://dx.doi.org/10.1101/gad.187922.112
- Dirks PB. Brain tumor stem cells: the cancer stem cell hypothesis writ large. Mol Oncol 2010; 4:420-30; PMID:20801091; http://dx.doi.org/10.1016/j.molonc.2010.08.001
- Yamada R, Nakano I. Glioma stem cells: their role in chemoresistance. World Neurosurg 2012; 77:237-40; PMID:22501017; http://dx.doi.org/10.1016/j.wneu.2012.01.004
- Florio T, Barbieri F. The status of the art of human malignant glioma management: the promising role of targeting tumor-initiating cells. Drug Discov Today 2012; 17:1103-10; PMID:22704957; http://dx.doi.org/10.1016/j.drudis.2012.06.001
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987-96; PMID:15758009; http://dx.doi.org/10.1056/NEJMoa043330
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352:997-1003; PMID:15758010; http://dx.doi.org/10.1056/NEJMoa043331
- Ivanov M, Kacevska M, Ingelman-Sundberg M. Epigenomics and interindividual differences in drug response. Clin Pharmacol Ther 2012; 92:727-36; PMID:23093317; http://dx.doi.org/10.1038/clpt.2012.152
- Cahill DP, Codd PJ, Batchelor TT, Curry WT, Louis DN. MSH6 inactivation and emergent temozolomide resistance in human glioblastomas. Clin Neurosurg 2008; 55:165-71; PMID:19248684
- Shinsato Y, Furukawa T, Yunoue S, Yonezawa H, Minami K, Nishizawa Y, Ikeda R, Kawahara K, Yamamoto M, Hirano H, et al. Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget 2013; 4:2261-70; PMID:24259277; http://dx.doi.org/10.18632/oncotarget.1302
- Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, Louis DN. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 2009; 15:4622-9; PMID:19584161; http://dx.doi.org/10.1158/1078-0432.CCR-08-3012
- Munoz JL, Rodriguez-Cruz V, Ramkissoon SH, Ligon KL, Greco SJ, Rameshwar P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget 2015; 6:1190-201; PMID:25595896; http://dx.doi.org/10.18632/oncotarget.2778
- Wang Z, Yang J, Xu G, Wang W, Liu C, Yang H, Yu Z, Lei Q, Xiao L, Xiong J, et al. Targeting miR-381-NEFL axis sensitizes glioblastoma cells to temozolomide by regulating stemness factors and multidrug resistance factors. Oncotarget 2015; 6:3147-64; PMID:25605243; http://dx.doi.org/10.18632/oncotarget.3061
- Hiddingh L, Tannous BA, Teng J, Tops B, Jeuken J, Hulleman E, Boots-Sprenger SH, Vandertop WP, Noske DP, Kaspers GJ, et al. EFEMP1 induces gamma-secretase/Notch-mediated temozolomide resistance in glioblastoma. Oncotarget 2014; 5:363-74; PMID:24495907; http://dx.doi.org/10.18632/oncotarget.1620
- Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer 2010; 10:147-56; PMID:20075923; http://dx.doi.org/10.1038/nrc2789
- Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev 2008; 22:1115-40; PMID:18451103; http://dx.doi.org/10.1101/gad.1652908
- Agger K, Christensen J, Cloos PA, Helin K. The emerging functions of histone demethylases. Curr Opin Genet Dev 2008; 18:159-68; PMID:18281209; http://dx.doi.org/10.1016/j.gde.2007.12.003
- Bezecny P. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol 2014; 31:985; PMID:24838514; http://dx.doi.org/10.1007/s12032-014-0985-5
- Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature 2013; 502:480-8; PMID:24153301; http://dx.doi.org/10.1038/nature12751
- Huang Y, Vasilatos SN, Boric L, Shaw PG, Davidson NE. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res Treat 2012; 131:777-89; PMID:21452019; http://dx.doi.org/10.1007/s10549-011-1480-8
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141:69-80; PMID:20371346; http://dx.doi.org/10.1016/j.cell.2010.02.027
- Sareddy GR, Nair BC, Krishnan SK, Gonugunta VK, Zhang QG, Suzuki T, Miyata N, Brenner AJ, Brann DW, Vadlamudi RK. KDM1 is a novel therapeutic target for the treatment of gliomas. Oncotarget 2013; 4:18-28; PMID:23248157; http://dx.doi.org/10.18632/oncotarget.725
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013; 23:811-25; PMID:23764003; http://dx.doi.org/10.1016/j.ccr.2013.05.003
- Hou J, Wu J, Dombkowski A, Zhang K, Holowatyj A, Boerner JL, Yang ZQ. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am J Transl Res 2012; 4:247-56; PMID:22937203
- Yan H, Chen X, Zhang Q, Qin J, Li H, Liu C, Calhoun-Davis T, Coletta LD, Klostergaard J, Fokt I, et al. Drug-tolerant cancer cells show reduced tumor-initiating capacity: depletion of CD44 cells and evidence for epigenetic mechanisms. PLoS One 2011; 6:e24397; PMID:21935404; http://dx.doi.org/10.1371/journal.pone.0024397
- Goldman M, Craft B, Swatloski T, Ellrott K, Cline M, Diekhans M, Ma S, Wilks C, Stuart J, Haussler D, et al. The UCSC cancer genomics browser: update 2013. Nucleic Acids Res 2013; 41:D949-54; PMID:23109555; http://dx.doi.org/10.1093/nar/gks1008
- Klose RJ, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P, Gilliland DG, Zhang Y, Kaelin WG, Jr. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 2007; 128:889-900; PMID:17320163; http://dx.doi.org/10.1016/j.cell.2007.02.013
- Rotili D, Mai A. Targeting Histone Demethylases: a new avenue for the fight against cancer. Genes Cancer 2011; 2:663-79; PMID:21941621; http://dx.doi.org/10.1177/1947601911417976
- Alvarez AA, Field M, Bushnev S, Longo MS, Sugaya K. The effects of histone deacetylase inhibitors on Glioblastoma-Derived stem cells. J Mol Neurosci 2014; 55(1):7-20
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70:440-6; PMID:20068163; http://dx.doi.org/10.1158/0008-5472.CAN-09-1947
- Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007; 12:1247-52; PMID:17962618; http://dx.doi.org/10.1634/theoncologist.12-10-1247
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 2008; 26:3015-24; PMID:18565887; http://dx.doi.org/10.1200/JCO.2007.15.7164
- Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, Ravetti GL, Zona GL, Daga A, Corte G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 2009; 27:40-8; PMID:18948646; http://dx.doi.org/10.1634/stemcells.2008-0493
- Griffero F, Daga A, Marubbi D, Capra MC, Melotti A, Pattarozzi A, Gatti M, Bajetto A, Porcile C, Barbieri F, et al. Different response of human glioma tumor-initiating cells to epidermal growth factor receptor kinase inhibitors. J Biol Chem 2009; 284:7138-48; PMID:19147502; http://dx.doi.org/10.1074/jbc.M807111200
- Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, Griffero F, Marubbi D, Spaziante R, Bellora F, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol 2009; 182:3530-9; http://dx.doi.org/10.4049/jimmunol.0802845
- Gritti M, Wurth R, Angelini M, Barbieri F, Peretti M, Pizzi E, Pattarozzi A, Carra E, Sirito R, Daga A, et al. Metformin repositioning as antitumoral agent: selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014; 5:11252-68; PMID:25361004; http://dx.doi.org/10.18632/oncotarget.2617
- Costa BM, Smith JS, Chen Y, Chen J, Phillips HS, Aldape KD, Zardo G, Nigro J, James CD, Fridlyand J, et al. Reversing HOXA9 oncogene activation by PI3K inhibition: epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res 2010; 70:453-62; PMID:20068170; http://dx.doi.org/10.1158/0008-5472.CAN-09-2189
- Di Vinci A, Casciano I, Marasco E, Banelli B, Ravetti GL, Borzi L, Brigati C, Forlani A, Dorcaratto A, Allemanni G, et al. Quantitative methylation analysis of HOXA3, 7, 9, and 10 genes in glioma: association with tumor WHO grade and clinical outcome. J Cancer Res Clin Oncol 2012; 138:35-47; PMID:21947269; http://dx.doi.org/10.1007/s00432-011-1070-5
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010; 17:510-22; PMID:20399149; http://dx.doi.org/10.1016/j.ccr.2010.03.017
- Johannessen TC, Bjerkvig R. Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev Anticancer Ther 2012; 12:635-42; PMID:22594898; http://dx.doi.org/10.1586/era.12.37
- Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res 2000; 6:2585-97; PMID:10914698
- Kitange GJ, Mladek AC, Carlson BL, Schroeder MA, Pokorny JL, Cen L, Decker PA, Wu W, Lomberk GA, Gupta SK, et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res 2012; 18:4070-9; PMID:22675172; http://dx.doi.org/10.1158/1078-0432.CCR-12-0560
- Brandes AA, Franceschi E, Tosoni A, Bartolini S, Bacci A, Agati R, Ghimenton C, Turazzi S, Talacchi A, Skrap M, et al. O(6)-methylguanine DNA-methyltransferase methylation status can change between first surgery for newly diagnosed glioblastoma and second surgery for recurrence: clinical implications. Neuro Oncol 2010; 12:283-8; PMID:20167816; http://dx.doi.org/10.1093/neuonc/nop050
- Oberstadt MC, Bien-Moller S, Weitmann K, Herzog S, Hentschel K, Rimmbach C, Vogelgesang S, Balz E, Fink M, Michael H, et al. Epigenetic modulation of the drug resistance genes MGMT, ABCB1 and ABCG2 in glioblastoma multiforme. BMC Cancer 2013; 13:617; PMID:24380367; http://dx.doi.org/10.1186/1471-2407-13-617
- Park CK, Kim JE, Kim JY, Song SW, Kim JW, Choi SH, Kim TM, Lee SH, Kim IH, Park SH. The changes in MGMT promoter methylation status in initial and recurrent Glioblastomas. Transl Oncol 2012; 5:393-7; PMID:23066447; http://dx.doi.org/10.1593/tlo.12253
- Pojo M, Goncalves CS, Xavier-Magalhaes A, Oliveira AI, Goncalves T, Correia S, Rodrigues AJ, Costa S, Pinto L, Pinto AA, et al. A transcriptomic signature mediated by HOXA9 promotes human glioblastoma initiation, aggressiveness and resistance to temozolomide. Oncotarget 2015; 6:7657-74; PMID:25762636; http://dx.doi.org/10.18632/oncotarget.3150
- Beier D, Rohrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, Proescholdt M, Brawanski A, Bogdahn U, Trampe-Kieslich A, et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res 2008; 68:5706-15; PMID:18632623; http://dx.doi.org/10.1158/0008-5472.CAN-07-6878
- Carra E, Barbieri F, Marubbi D, Pattarozzi A, Favoni RE, Florio T, Daga A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle 2013; 12:491-500; PMID:23324350; http://dx.doi.org/10.4161/cc.23372
- Wurth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, Sirito R, Massollo M, Marini C, Zona G, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013; 12:145-56; PMID:23255107; http://dx.doi.org/10.4161/cc.23050
- Gatti M, Pattarozzi A, Bajetto A, Wurth R, Daga A, Fiaschi P, Zona G, Florio T, Barbieri F. Inhibition of CXCL12/CXCR4 autocrine/paracrine loop reduces viability of human glioblastoma stem-like cells affecting self-renewal activity. Toxicology 2013; 314:209-20; http://dx.doi.org/10.1016/j.tox.2013.10.003
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nature Protoc 2006; 1:2315-9; PMID:17406473; http://dx.doi.org/10.1038/nprot.2006.339
- Riganti C, Salaroglio IC, Caldera V, Campia I, Kopecka J, Mellai M, Annovazzi L, Bosia A, Ghigo D, Schiffer D. Temozolomide downregulates P-glycoprotein expression in glioblastoma stem cells by interfering with the Wnt3a/glycogen synthase-3 kinase/β-catenin pathway. Neuro Oncol 2013; 15:1502-17; PMID:23897632; http://dx.doi.org/10.1093/neuonc/not104
- Nakagawachi T, Soejima H, Urano T, Zhao W, Higashimoto K, Satoh Y, Matsukura S, Kudo S, Kitajima Y, Harada H, et al. Silencing effect of CpG island hypermethylation and histone modifications on O6-methylguanine-DNA methyltransferase (MGMT) gene expression in human cancer. Oncogene 2003; 22:8835-44; PMID:14647440
- Banelli B, Bonassi S, Casciano I, Mazzocco K, Di Vinci A, Scaruffi P, Brigati C, Allemanni G, Borzi L, Tonini GP, et al. Outcome prediction and risk assessment by quantitative pyrosequencing methylation analysis of the SFN gene in advanced stage, high-risk, neuroblastic tumor patients. Int J Cancer 2010; 126:656-68; PMID:19626586; http://dx.doi.org/10.1002/ijc.24768
- Banelli B, Brigati C, Di Vinci A, Casciano I, Forlani A, Borzi L, Allemanni G, Romani M. A pyrosequencing assay for the quantitative methylation analysis of the PCDHB gene cluster, the major factor in neuroblastoma methylator phenotype. Lab Invest 2011; 92:458-65; PMID:22157715; http://dx.doi.org/10.1038/labinvest.2011.169
- Banelli B, Merlo DF, Allemanni G, Forlani A, Romani M. Clinical potentials of methylator phenotype in stage 4 high-risk neuroblastoma: an open challenge. PLoS One 2013; 8:e63253; PMID:23717404; http://dx.doi.org/10.1371/journal.pone.0063253