
Aging is an inexorable process in most living organisms that leads to damage accumulation and, consequently, to the loss of cellular homeostasis.Citation1 Despite the apparently irreversible nature of aging at the organism level, somatic cell reprogramming illustrates the potential reversibility of this process, overcoming age-associated barriers and resetting adult molecular marks. However, age-associated features such as genetic damage, telomere shortening or cell senescence dramatically restrict the efficiency of cell reprogramming, suggesting the importance of understanding the regulatory mechanisms involved in age-associated reprogramming barriers for developing rejuvenation approaches. Segmental progeroid syndromes, caused by defects in DNA repair systems or alterations in the nuclear lamina, represent an exacerbate phenotype of aging. The molecular characterization of these syndromes is essential for helping patients but also for supporting aging research with valuable information, as they represent important models for a better understanding of the aging biological process.
Through the derivation of induced pluripotent stem cells (iPSCs) from individuals with premature or physiological aging, we have recently found that activation of NF-κB constitutes an important barrier for somatic cell reprogramming in aging.Citation2 Our studies in this regard were first focused on the generation of iPSCs from fibroblasts of patients with Néstor-Guillermo progeria syndrome (NGPS), a severe disease caused by a homozygous mutation in the BANF1 gene (c.34G>C; p.A12T).Citation3 We found that NGPS fibroblasts showed an increased NF-κB signaling in comparison with control cells, which could contribute to their premature senescence phenotype. Interestingly, this signaling signature was dramatically repressed in NGPS-iPSCs. Consistent with this observation, experiments based on NF-κB inhibition confirmed a striking increase in the number of iPSC clones in NGPS fibroblasts, with no significant effect over cells from young donors. We also extended these results to cells from patients with other progeroid syndromes and from physiologically aged donors, thereby supporting the key relevance of NF-κB signaling in the maintenance of cellular identity.Citation2
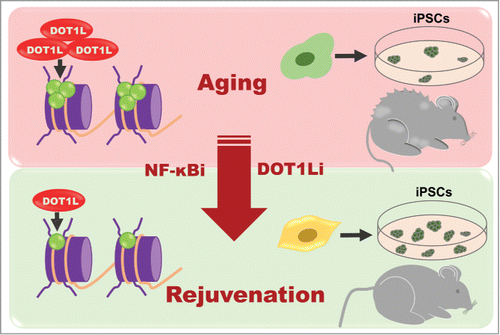
Nevertheless, the putative NF-κB targets controlling this newly identified age-associated reprogramming barrier were completely unknown. In this regard, previous studies have uncovered new chromatin-modifying proteins such as DOT1L and YY1 that function as reprogramming repressors.Citation4 Accordingly, we have provided evidence for a direct regulation of DOT1L and YY1 genes by NF-κB, and a striking over-expression of DOT1L in NGPS fibroblasts, suggesting that these proteins might be relevant effectors of NF-κB role in cellular reprogramming as well as in NF-κB-driven alterations.Citation2 Additionally, the administration of a specific DOT1L chemical inhibitor remarkably increased the reprogramming efficiency of NGPS fibroblasts.Citation2 Furthermore, the parallelism observed between rejuvenation and reprogramming enhancement through NF-κB inhibition suggested that DOT1L could be a novel target for rejuvenation-based approaches (Fig. 1). Indeed, epigenetic alterations have been proposed as a direct cause of aging.Citation1,5 In this regard, we have found that treatment of progeroid mice during lifetime with DOT1L inhibitors showed a remarkable increase in mutant mice longevity, with a notably amelioration of progeroid phenotypes.Citation2 The methylation of H3K79 by DOT1L which is a conserved epigenetic mark in many eukaryotic epigenomes, increases progressively along the aging process, suggesting that DOT1L might function as a vital clock, ticking the hours impassively.Citation6
The results discussed herein demonstrate the critical involvement of DOT1L in aging pathogenesis and provide the first in vivo evidence about the efficacy of DOT1L-inhibitory strategies to treat age-associated alterations, demonstrating the utility of studying age-associated reprogramming barriers for the development of anti-aging therapies. Moreover, the side effects caused by the prolonged treatment with different anti-inflammatory compounds which extend longevity in animal models,Citation7 might be overcome by the use of DOT1L inhibitors. Collectively, iPSCs derived from tissues of patients with progeroid syndromes may provide attractive cell-based models to study the mechanisms of aging in general, and the pathogenesis of progeria in particular. Furthermore, it seems clear that aging and cell reprogramming are well-interconnected processes and future research should be devoted to study age-associated molecular impairments, to implement cell reprogramming methodologies, and to identify new targets of rejuvenation strategies.
Figure 1. The role of DOT1L in cell reprogramming and aging. The methylation mark H3K79 increases with the inflammatory state associated with aging. Treatment with NF-κB or DOT1L inhibitors reduces the epigenetic mark H3K79 and leads to a rejuvenated state which ameliorates the reprogramming efficiency of aged cells and the age-associated features related with accelerated aging syndromes.
References
- Lopez-Otin C, et al. Cell 2013; 153(6):1194-217; PMID:23746838; http://dx.doi.org/10.1016/j.cell.2013.05.039
- Soria-Valles C, et al. Nat Cell Biol 2015; 17(8):1004-13; PMID:26214134; http://dx.doi.org/10.1038/ncb3207
- Puente XS, et al, Am J Hum Genet 2011; 88(5):650-6; PMID:21549337; http://dx.doi.org/10.1016/j.ajhg.2011.04.010
- Onder TT, et al. Nature 2012; 483(7391):598-602; PMID:22388813; http://dx.doi.org/10.1038/nature10953
- Osorio FG, et al. Aging Cell 2010; 9(6):947-57; PMID:20961378; http://dx.doi.org/10.1111/j.1474-9726.2010.00621.x
- De Vos D, et al. EMBO Rep 2011; 12(9):956-62; PMID:21760613; http://dx.doi.org/10.1038/embor.2011.131
- Osorio FG, et al. Genes Dev 2012; 26(20):2311-24; PMID:23019125; http://dx.doi.org/10.1101/gad.197954.112