
Abstract
Shugoshin (SGO1) plays a pivotal role in sister chromatid cohesion during mitosis by protecting the centromeric cohesin from mitotic kinases and WAPL. Mammalian cells contain at least 6 alternatively spliced isoforms of SGO1. The relationship between the canonical SGO1A with shorter isoforms including SGO1C remains obscure. Here we show that SGO1C was unable to replace the loss of SGO1A. Instead, expression of SGO1C alone induced aberrant mitosis similar to depletion of SGO1A, promoting premature sister chromatid separation, activation of the spindle-assembly checkpoint, and mitotic arrest. In disagreement with previously published data, we found that SGO1C localized to kinetochores. However, the ability to induce aberrant mitosis did not correlate with its kinetochore localization. SGO1C mutants that abolished binding to kinetochores still triggered premature sister chromatid separation. We provide evidence that SGO1C-mediated mitotic arrest involved the sequestering of PP2A–B56 pool. Accordingly, SGO1C mutants that abolished binding to PP2A localized to kinetochores but did not induce aberrant mitosis. These studies imply that the expression of SGO1C should be tightly regulated to prevent dominant-negative effects on SGO1A and genome instability.
Introduction
Chromosome segregation is a highly regulated process in order to maintain genome integrity. Sister chromatids are held together by ring-shaped complexes called cohesin, which are important for maintaining sister-chromatid cohesion, DNA repair, and transcription regulation.Citation1-3 During S phase, cohesin is loaded onto the newly duplicated sister chromatids. This involves the binding of cohesin with sororin, which counters the removal of cohesin by PDS5 and WAPL.Citation4-6
During mitosis, cohesin is removed from chromosomes in a 2-step manner. During prophase, the CDK1, PLK1, and Aurora B collaborate to phosphorylate sororin and cohesin, triggering WAPL-dependent removal of cohesin from chromosome arms.Citation6-11 Centromeric cohesin is protected from the mitotic kinases and WAPL by a protein called Shugoshin and is only removed during anaphase. The primary signal for localizing Shugoshin to kinetochores is BUB1-dependent phosphorylation of histone H2AThr120.Citation12 Shugoshin interacts with protein phosphatase 2A (PP2A), thereby keeping cohesin and sororin in a hypophosphorylated state and maintaining centromeric cohesion.Citation11,13-16
During metaphase-anaphase transition, proper kinetochore–microtubule attachment creates tension across sister kinetochores and silences the spindle-assembly checkpoint. This allows the anaphase-promoting complex/cyclosome (APC/C) to degrade securin, thereby liberating the protease separase. Separase then cleaves centromeric cohesin to promote sister-chromatid separation. Kinetochore tension also triggers a redistribution of Shugoshin, which is also important for the cleavage of centromeric cohesin by separase.Citation17,18
Mammalian cells contain 2 members of Shugoshin, SGO1 and SGO2, functioning in centromeric cohesion during mitosis and meiosis, respectively.Citation19 There are 6 alternatively spliced isoforms of SGO1.Citation20-22 SGO1A2 is regarded as the canonical isoform and is most commonly studied. However, most loss-of-function studies of SGO1 such as RNA interference did not differentiate the roles of various SGO1 isoforms. In this study, we found that SGO1C was unable to replace the loss of SGO1A. Instead, overexpression of SGO1C induced premature separation of sister chromatids, activation of the spindle-assembly checkpoint, and mitotic arrest. We further investigated the mechanism of SGO1C-mediated mitotic arrest and found that it involved the depletion of the PP2A–B56 pool.
Results
Depletion of SGO1 induces irreversible mitotic defects
At least 6 isoforms of SGO1 are encoded in mammalian cells (Fig. S1A). SGO1A2 is regarded as the canonical isoform. A central domain found in SGO1A2 is absent in the SGO1B2 and SGO1C2 isoforms. In this study, SGO1A2 and SGO1C2 were used as representation for long (SGO1A) and short (SGO1C) forms of SGO1, respectively.
We first downregulated SGO1 and examined if SGO1A or SGO1C could rescue the effects equally. Two siRNAs (siSGO1 and siSGO1ii respectively) were designed against the N- and C-terminal regions that are common between SGO1A and SGO1C (Fig. S1A, B). Transfection of siSGO1 downregulated the mRNAs (Fig. S1C) and proteins () of both SGO1A and SGO1C.
Figure 1 (See previous page). Aberrant mitosis is induced by both depletion of SGO1 and expression of SGO1C. (A) Downregulation of both SGO1A and SGO1C with siRNA. HeLa cells were transfected with either control or SGO1 siRNA (siSGO1). After 24 h, cell-free extracts were prepared and SGO1 was enriched by immunoprecipitation with an antibody against SGO1. SGO1A and SGO1C were then detected by immunoblotting using antibodies raised in rabbit and mouse, respectively. (B) Depletion of SGO1 promotes mitotic arrest and unscheduled sister chromatid separation. Histone H2B-GFP-expressing HeLa cells were transfected with siSGO1. Individual cells were then tracked with time-lapse microscopy. Representative images of a cell undergoing protracted mitosis are shown. The arrows indicate the position of the metaphase plate, which dissolved progressively as chromatin migrated toward the poles. (C) Mitotic defects induced by siSGO1 can be rescued by SGO1A but not SGO1C. HeLa cells expressing histone H2B-GFP were transfected with control plasmid or plasmids expressing FLAG-tagged SGO1A or SGO1C (both resistant to siSGO1). A plasmid expressing mRFP was co-transfected as a marker. After 30 h, the cells were transfected with either control siRNA (siControl) or siSGO1. The cells were synchronized at S phase with thymidine block and released. Individual cells were then tracked for 24 h with time-lapse microscopy. Each horizontal bar represents one cell (n = 30). Gray: interphase; black: mitosis (from DNA condensation to anaphase); truncated bars: cell death. (D) SGO1A rescues the mitotic defects induced by siSGO1. Cells were transfected and imaged as in (C). The length of mitosis was quantified (mean±90% CI). (E) Ectopic expression of SGO1A and SGO1C in the presence of siSGO1. Cells were transfected and imaged as in (C). Lysates were prepared and the expression of SGO1 was detected with immunoblotting. Uniform loading of lysates was confirmed by immunoblotting for actin. The positions of molecular size standards (in kDa) are indicated. (F) Cell cycle defects induced with siSGO1 can be corrected with SGO1A. HeLa cells stably expressing FLAG-EGFP-SGO1A or histone H2B-GFP (control) were transfected with siSGO1. After 24 h, the cells were harvested and the DNA contents were analyzed with flow cytometry.
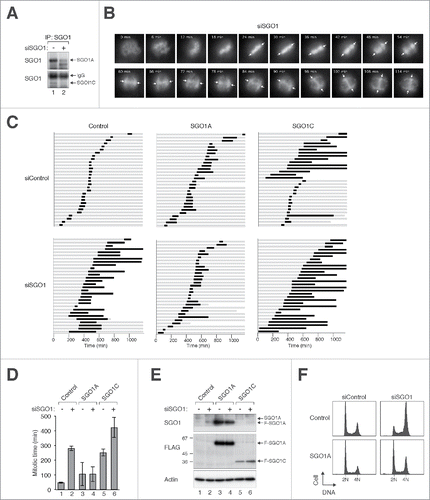
Depletion of SGO1A and SGO1C induced pronounced mitotic defects. Although siSGO1-transfected cells proceeded to metaphase normally, they were unable to undergo anaphase. Instead of the normal synchronized sister chromatid separation (Video 1), chromosomes progressively migrated toward the spindle poles in siSGO1-transfected cells (), confirming that SGO1 is important to prevent an unscheduled loss of sister chromatid cohesion. The cells were unable to recover from this mitotic state till the end of imaging period or underwent apoptosis (Video 2).
To verify that the premature sister chromatid separation in SGO1-depleted cells was not caused by degradation of securin, the activity of APC/C at individual cell level was monitored using a reporter (mRFP fused to the D-box of cyclin B1).Citation23 In contrast to the degradation of the APC/C reporter during normal anaphase (Video 3), the reporter remained stable throughout the mitotic block in siSGO1-transfected cells (Video 4). These results indicate that depletion of SGO1A and SGO1C together induced a mitotic arrest.
SGO1A is essential for mitosis
To determine if the mitotic defects triggered by siSGO1 were specific, rescue experiments were performed using recombinant SGO1A and SGO1C. We made use of the fact that the mRNA sequence targeted by siSGO1 was different between human and mouse SGO1A orthologs (Fig. S1B). Hence unlike the endogenous SGO1A, recombinant mouse SGO1A was resistant to siSGO1 (Fig. S2A). SGO1A largely overcame the mitotic arrest and cell death induced by siSGO1 (, the mitotic length is quantified in ). Expression of SGO1A was confirmed with immunoblotting (). Flow cytometry was used to verify that SGO1A could reverse the G2/M delay induced by siSGO1 (). These data indicated that overexpression of SGO1A was sufficient to compensate the effects caused by depleting both SGO1A and SGO1C.
To further verify these results, we used a second siRNA targeting a different region of SGO1 (siSGO1ii). This siRNA also targeted both SGO1A and SGO1C (Fig. S1A). Flow cytometry (Fig. S2B) and live-cell imaging (Fig. S2C) revealed that siSGO1ii induced a mitotic arrest similar to siSGO1. Although the sequences of human SGO1A targeted by siSGO1ii was similar to that of mouse SGO1A (Fig. S1B), we were able to express mouse SGO1A to a level comparable to the endogenous SGO1A before knockdown (Fig. S2A). Accordingly, co-expression of SGO1A could overcome the mitotic arrest and apoptosis caused by siSGO1ii (Fig. S2D).
Finally, we also used a siRNA targeting the central region unique to SGO1A (siSGO1A). Transfection of siSGO1A depleted SGO1A without affecting SGO1C (). As the antibodies used were raised against regions common to both SGO1A and SGO1C, this experiment also showed that both SGO1A and SGO1C were expressed endogenously and that SGO1A was more abundant than SGO1C. Using siSGO1A, we found that specific depletion of SGO1A was sufficient to induce mitotic arrest and apoptosis (), similarly to the siRNAs that targeted both SGO1A and SGO1C together (). Moreover, both cell cycle () and mitotic defects () caused by siSGO1A could be rescued with recombinant SGO1A.
Figure 2. Specific depletion of SGO1A induces mitotic defects. (A) Depletion of SGO1A. Cell-free extracts of HeLa cells transfected with siSGO1 or siSGO1A were subjected to immunoprecipitation with a mouse monoclonal antibodies against SGO1. The immunoprecipitates were immunoblotted with rabbit antibodies against SGO1. SGO1A and SGO1C could not be detected together in the total lysates (bottom panel) because of a cross-reactive band recognized by the anti-SGO1 antibodies (asterisk). The positions of molecular size standards (in kDa) are indicated. (B) Depletion of SGO1A promotes G2/M arrest. Control HeLa or HeLa stably expressing FLAG-EGFP-SGO1A were transfected with control siRNA, siSGO1, or siSGO1A. After 24 h, the cells were fixed and the cell cycle profile was analyzed with flow cytometry. (C) Mitotic defects are induced by siSGO1A. HeLa cells expressing histone H2B-mRFP alone or both histone H2B-mRFP and FLAG-EGFP-SGO1A were transfected with either control siRNA or siSGO1A. The cells were synchronized at S phase with thymidine block and released. Individual cells were then tracked for 24 h with time-lapse microscopy. Each horizontal bar represents one cell (n = 30). Gray: interphase; black: mitosis (from DNA condensation to anaphase); truncated bars: cell death. (D) SGO1A rescues the mitotic defects induced by siSGO1A. Cells were transfected and imaged as in (C). The length of mitosis was quantified (mean±90% CI).
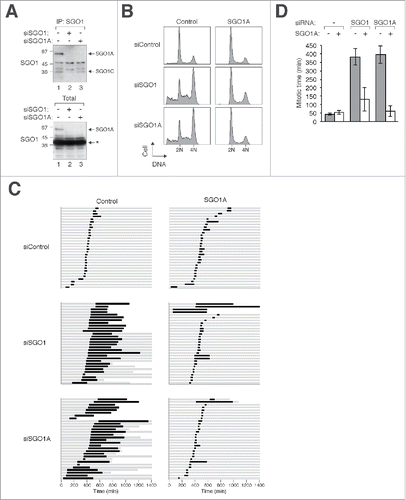
Collectively, these data suggest that the mitotic defects induced by depletion of SGO1A and SGO1C together could be ascribed to SGO1A.
SGO1C induces aberrant mitosis resembling SGO1 depletion
To perform similar rescue experiment of siSGO1 using SGO1C, silent mutations were introduced into human SGO1C to render it resistant to siSGO1 (Fig. S2E). Unlike SGO1A, SGO1C was unable to reverse the siSGO1-mediated mitotic arrest (). Remarkably, a pronounced mitotic arrest was induced by SGO1C alone.
To further verify that differential effects of SGO1A and SGO1C on mitosis, we titrated the constructs to express the 2 proteins at similar levels (). While SGO1A did not affect the cell cycle profile significantly, SGO1C was able to induce an accumulation of G2/M cells (). This was further supported by the increase of mitotic index () and the accumulation of mitotic markers including securin and phosphorylated histone H3Ser10 (). Moreover, the increase of cleaved PARP1 suggested that apoptosis was triggered after the SGO1C-mediated mitotic block.
Figure 3 (See previous page). SGO1C induces aberrant mitotic arrest. (A) Overexpressing SGO1C but not SGO1A induces G2/M defects. Different amount of FLAG-tagged SGO1A and SGO1C was expressed in HeLa cells. A plasmid expressing histone H2B-mRFP was co-transfected as a transfection marker. Lysates were prepared and the relative abundance of SGO1A and SGO1C was analyzed with immunoblotting for FLAG. Uniform loading of lysates was confirmed by immunoblotting for actin. The cell cycle was also analyzed with flow cytometry. (B) Expression of SGO1C increases the mitotic index. HeLa cells were transfected with control or plasmids expressing EGFP-tagged SGO1A or SGO1C. A plasmid expressing histone H2B-mRFP was co-transfected for analyzing chromatin morphology. After 24 h, the mitotic index was examined (mean±SD of 3 independent experiments). (C) SGO1C promotes the appearance of mitotic and apoptotic markers. HeLa cells were transfected with vector or FLAG-SGO1C-expressing plasmids. At 40 h (lanes 1-2) or 46 h (lane 3) after transfection, the cells were harvested and analyzed with immunoblotting. The accumulation of phosphorylated histone H3Ser10 and securin (slower migrating bands) indicated that SGO1C trapped cells in mitosis. Mitotic block induced with nocodazole (NOC) for 12 h acted as a positive control. (D) SGO1C blocks cells in mitosis. HeLa cells expressing histone H2B-GFP were transfected with vector or plasmids expressing FLAG-tagged SGO1A or SGO1C. The cells were synchronized at S phase with thymidine block and released. Individual cells were then analyzed with live-cell imaging (transfected cells were identified by the co-expression of mRFP). Representative time-lapse images are shown. (E) Cells were transfected as in (D). Individual cells were then tracked for 24 h with time-lapse microscopy. Each horizontal bar represents one cell (n = 28). Gray: interphase; black: mitosis (from DNA condensation to anaphase); truncated bars: cell death. The duration of mitosis was quantified (mean±90% CI).
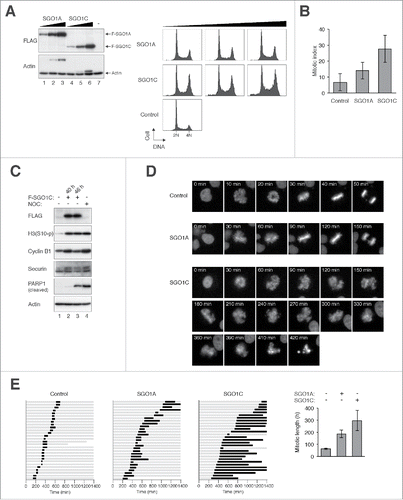
Time-lapse microscopy provided more detailed information on the mitotic defects caused by SGO1C (). Although the metaphase plate could occasionally be formed, it rapidly disintegrated into several chromosomal clusters. More often, the chromosomes dispersed before the metaphase was properly formed. Taken together, SGO1C-expressing cells underwent prolonged and aberrant mitosis.
As aberrant mitosis was induced by SGO1C alone, it was not possible to address if SGO1C could compensate the loss of function of SGO1A (). Notwithstanding, we titrated the SGO1C-expressing plasmid for it to induce a minimal effect on mitosis. Even at this expression level, SGO1C was unable to reverse the mitotic defects induced by depletion of SGO1 (with siSGO1) or SGO1A only (with siSGO1A) (Fig. S2F).
SGO1C-mediated mitosis was associated with extensive spindle defects. Chromosomes were scattered around poorly defined spindle poles (); and premature sister chromatid separation and multipolar spindle occurred at a high frequency ().
Figure 4. SGO1C induces defective mitotic spindle and activates the spindle-assembly checkpoint. (A) SGO1C disrupts proper spindle formation. HeLa cells were transfected with either vector or FLAG-SGO1C-expressing plasmids. A plasmid expressing histone H2B-mRFP was co-transfected. The cells were fixed at 40 h after transfection and stained with antibodies against CREST (centromere, red), α-tubulin (Alexa Fluor 488-conjugated) (microtubules, green), and Hoechst 33342 (DNA, blue), and examined using confocal microscopy. (B) SGO1C induces defective mitotic spindle. HeLa cells were transfected with either vector or FLAG-SGO1C-expressing plasmids. A plasmid expressing histone H2B-mRFP was co-transfected. The cells were fixed at 40 h after transfection and stained with antibodies against α-tubulin (Alexa Fluor 488-conjugated). The spindle and chromosomes were examined with fluorescence microscopy: α-tubulin (green), histone H2B-mRFP (red). The type of mitotic spindle was quantified (mean±SD of 3 independent experiments; n>50 mitotic cells each). Examples of cells undergoing normal bipolar mitosis, premature chromosome separation, and multipolar mitosis are shown at the bottom (red: DNA; green: α-tubulin). (C) The spindle-assembly checkpoint is active in the presence of SGO1C. HeLa cells were transfected with control vector or a plasmid expressing FLAG-SGO1C. At 24 h after transfection, lysates were prepared and subjected to immunoprecipitation using an antiserum against MAD2. Both the total lysates and the immunoprecipitates were then analyzed with immunoblotting. As a positive control, CDC20 was present in MAD2-immunoprecipitates from nocodazole-blocked prometaphase lysates (lane 4).
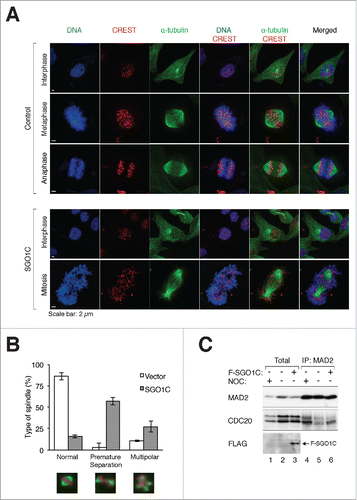
The persistence of an APC/C reporter suggested that the SGO1C-induced mitotic block was associated with inactive APC/C (data not shown). To show more directly that the spindle-assembly checkpoint was activated in the presence of SGO1C, MAD2–CDC20 complexes were analyzed with immunoprecipitation. shows that MAD2–CDC20 complexes were present in SGO1C-expressing cells (lane 6), indicating that the spindle-assembly checkpoint remained activated during SGO1C-mediated mitotic block.
Taken together, these results indicate that ectopic expression of SGO1C induces an aberrant mitosis similar to the depletion of SGO1A.
SGO1C localizes to kinetochores and induces premature sister chromatid separation
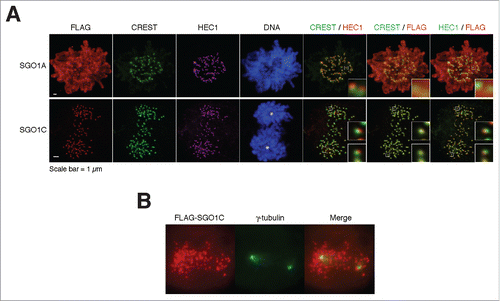
We next analyzed the effects of SGO1C on sister chromatid cohesion. Ectopically expressed SGO1C was mainly targeted to kinetochores, as it co-localized with CREST and the outer kinetochore protein HEC1 (). SGO1A localized to kinetochores and chromatin, depending on the expression level (see below). Significantly, while SGO1A-expressing cells contained paired kinetochores during early mitosis (with a pair of HEC1 franking the CREST signal), SGO1C-expressing cells generally contained separated centromeres (single HEC1).
Figure 5. SGO1C induces premature sister chromatid separation. (A) SGO1C induces premature sister chromatid separation. Representative mitotic cells overexpressing FLAG-tagged SGO1A or SGO1C are shown. Samples were stained with antibodies against FLAG, CREST (centromeres), and HEC1 (outer kinetochore); DNA was stained with Hoechst 33342. For the SGO1C-expressing cell, the positions of the spindle poles are marked by the asterisks (based on immunostaining for α-tubulins not shown). Selected centromeres are magnified, demonstrating the presence of paired kinetochores in SGO1A-expressing cells and separated kinetochores in SGO1C-expressing cells. Note that at this expression level, FLAG-SGO1A mainly localized to the chromatin. (B) SGO1C does not localize to spindle poles. HeLa cells expressing FLAG-SGO1C were stained with antibodies against FLAG and γ-tubulin; DNA was stained with Hoechst 33342.
We also analyzed MAD2 expression at the SGO1C-expressing kinetochores. MAD2 was concentrated at kinetochores during early mitosis in normal cells. Figure S3A shows that MAD2 was also present at the prematurely segregated centromeres in SGO1C-expressing cells. We also used a cold shock method to preserve the microtubules that bind to kinetochores (K-fibers ) (Fig. S3B). These analyses indicated that K-fibers were still present in SGO1C-expressing cells, suggesting that the prematurely segregated kinetochores remained bound to the spindle. These results indicated that the spindle-assembly checkpoint was activated even though the prematurely segregated kinetochores remained bound to microtubules.
These findings differ markedly with a previous report showing that SGO1C (called sSgo1 in that study) localizes at the centrosome during interphase and spindle poles during mitosis.Citation21 Both SGO1A and SGO1C were in the nucleus during interphase (Fig S4A). As expected, SGO1A concentrated at kinetochores during mitosis. At high expression levels, SGO1A associated with both kinetochores and chromatins (Fig. S4A, B). In contrast, both FLAG- (Fig. S4A) and HA-tagged SGO1C (Fig. S4C) localized exclusively to kinetochores during mitosis. Finally, we used γ-tubulin immunostaining to confirm that SGO1C did not colocalize with spindle poles ().
These results indicate that unlike previously published data, SGO1C is found to localize to kinetochores. SGO1C promotes precocious separation of sister chromatids and activates the spindle-assembly checkpoint.
Localization of SGO1C to kinetochores is not critical for promoting mitotic arrest
To demarcate the region(s) important for SGO1C's kinetochore localization, several truncation mutants were generated (). Deletion of the C-terminal region of SGO1C to residue 157 (SGO1CCΔ157) (the position in which SGO1A starts to differ from SGO1C, see Fig. S1A) abolished binding to kinetochores (Fig. S4D). In contrast, deletion of the N-terminal region (SGO1CNΔ158) did not affect kinetochore binding. Additional C-terminal deletion mutants indicated that SGO1CCΔ252 but not SGO1CCΔ236 localized to kinetochores (Fig. S4D), suggesting that the region between 236–252 was important for kinetochore localization (summarized in ).
Figure 6. Localization of SGO1C to kinetochores is not critical for promoting aberrant mitosis. (A) Schematic diagram of SGO1C and different truncation mutants are aligned to scale. Kinetochore localization of the FLAG-tagged proteins and their ability to induce mitotic arrest are summarized. (B) The N-terminal region of SGO1C is required for inducing G2/M arrest. HeLa cells were transfected with SGO1C or truncation mutants. A plasmid expressing histone H2B-GFP was co-transfected as a marker. The cell cycle profile of transfected cells was analyzed with flow cytometry. (C) The N-terminal region of SGO1C is required for inducing mitotic defects. HeLa cells expressing histone H2B-GFP were transfected with control vector or plasmids expressing FLAG-tagged SGO1C or different truncation mutants. A plasmid expressing mRFP was co-transfected as a marker. Individual cells were then tracked for 24 h with time-lapse microscopy. Each horizontal bar represents one cell (n = 30). Gray: interphase; black: mitosis (from DNA condensation to anaphase); truncated bars: cell death. (D) Cells were transfected and imaged as in (C). The length of mitosis was quantified (mean±90% CI).
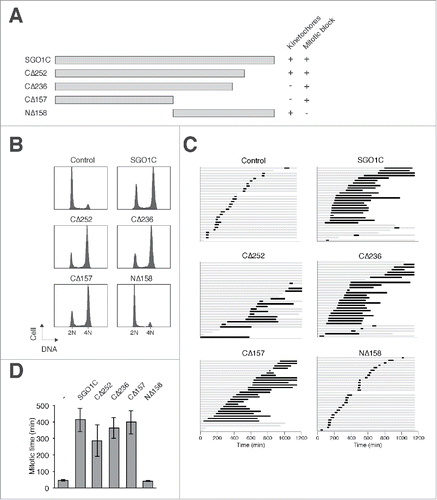
We hypothesized that kinetochore localization of SGO1C is important for its effects on mitosis, perhaps by interfering with the functions of endogenous SGO1A in a dominant-negative manner. However, although SGO1CCΔ157 and SGO1CCΔ236 were not associated with kinetochores, they nevertheless induced a G2/M arrest (). This was further confirmed using live-cell imaging, indicating that both SGO1CCΔ157 and SGO1CCΔ236 triggered mitotic arrest (). We also verified that SGO1CCΔ157 promoted premature sister chromatid separation (Fig. S4E). Collectively, these data indicate that the ability of SGO1C to induce mitotic defects does not correlate with its kinetochore localization.
Interaction with PP2A is essential for the effects of SGO1C on mitosis
Given that direct competition at kinetochores may not account for the mitotic defects induced by SGO1C, we next examined if SGO1A and SGO1C could bind different proteins. Immunoprecipitation of FLAG-tagged SGO1A or SGO1C from cell lines stably expressing these proteins was followed by mass spectrometry analysis. These analyses revealed that SGO1A and SGO1C associated with different subsets of proteins (Fig S5A). In particular, more PP2A catalytic subunits (PPP2CA and PPP2CB) as well as members of the 56 kDa regulatory subunits (called PPP2R5 or B56 or B') were found to associate with SGO1C than SGO1A.
To confirm these findings, FLAG-tagged SGO1A and SGO1C were expressed to similar levels and immunoprecipitated using antibodies against the FLAG tag. shows that more PP2A was co-immunoprecipitated with SGO1C than with SGO1A.
Figure 7. Binding to PP2A is essential for the effects of SGO1C on mitosis. (A) SGO1C binds PP2A. HeLa cells expressing histone H2B-GFP were transfected with control vector or plasmids expressing FLAG-SGO1A or FLAG-SGO1C. Lysates were prepared and subjected to immunoprecipitation using antibodies against FLAG. Both the total lysates and immunoprecipitates were then analyzed with immunoblotting. GFP analysis was included to assess protein loading and transfer. (B) 4m mutation (N60A, N61I, K62A, L68A) abolishes interaction with PP2A. HeLa cells were transfected with plasmids expressing FLAG-SGO1C (WT), FLAG-SGO1C4m, and HA-SGO1C as indicated. Lysates were prepared and subjected to immunoprecipitation using an antiserum against FLAG. Both the total lysates and immunoprecipitates were then analyzed with immunoblotting. (C) SGO14m is unable to induce mitotic arrest. HeLa cells were transfected with different amount of plasmids expressing SGO1C or SGO1C4m (1 µg, 3 µg, and 5 µg per 60-mm plates). A plasmid expressing histone H2B-GFP was co-transfected as a marker. The cell cycle profile of transfected cells was analyzed with flow cytometry. Lysates were also prepared and analyzed with immunoblotting. (D) SGO1CΔ157 containing 4m mutation does not induce mitotic arrest. HeLa cells were co-transfected with plasmids expressing histone H2B-GFP and SGO1C (WT), SGO1CCΔ157, SGO1C4m, or SGO1CCΔ157(4m). The cell cycle profile of transfected cells was analyzed with flow cytometry. Lysates were also prepared and analyzed with immunoblotting.
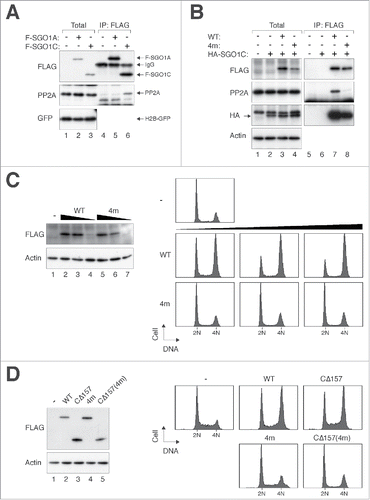
Previous crystal structure analysis and in vitro binding assays indicate that the N-terminal coiled coil region of SGO1 interacts directly with PP2A.Citation24 To examine if PP2A binding is important for SGO1C's effects on mitosis, we generated a SGO1C mutant substituting L68 with alanine (L68A), which abolished the interaction between SGO1A and PP2A in vitro.Citation24 Interestingly, SGO1CL68A retained the ability to induce mitotic arrest (Fig. S5B). However, the L68A mutation did not completely abolish the interaction between SGO1C and PP2A as expected (Fig. S5C). We therefore further mutated several residues implicated in PP2A binding (N60A, N61I, K62A, L68A).Citation24 Significantly, this mutant (called SGO1C4m herein) did not bind PP2A () and was unable to disrupt mitosis (). The localization of SGO1C4m was the same as SGO1C (Fig. S5D), indicating that kinetochore binding alone was insufficient to induce aberrant mitosis.
Finally, given that the N-terminal region of SGO1C (SGO1CCΔ157) did not localize to kinetochores but nevertheless was able to induce mitotic defects, we also generated the 4m mutations in CΔ157 (SGO1CCΔ157(4m)). shows that both SGO1C4m and SGO1CCΔ157(4m) did not disrupt mitosis, further supporting the importance of PP2A binding for SGO1C's effects on mitosis.
Collectively, these results indicate that while the interaction with PP2A–B56 is critical for the effects of SGO1C on mitosis, the localization of SGO1C to kinetochores is not an essential factor.
Discussion
SGO1 protects centromeric cohesin by directing PP2A to counteract the action of mitotic kinases. A surprising finding from this study is that there are fundamental differences between different SGO1 isoforms. As expected, depletion of both SGO1A and SGO1C () or SGO1A alone () induced a loss of sister chromatid cohesion. The rescue of the siSGO1-mediated mitotic defects by SGO1A provided further evidence that SGO1A is essential for sister chromatid cohesion (). In contrast, we were unable to show that SGO1C could replace the functions of SGO1A because expression of SGO1C already induced mitotic defects similarly to SGO1 depletion (). Even when expressed at a low level that did not affect mitosis on its own, SGO1C was unable to rescue the effects of siSGO1 (Fig. S2F).
Multiple approaches were used here to verify that ectopic expression of SGO1C blocked cells in mitosis. These include flow cytometry (), mitotic index (), histone H3Ser10 phosphorylation (), and live-cell imaging (). Mechanistically, SGO1C induced premature sister chromatid separation (), with chromosomes scattering around poorly defined spindle poles (). This is in agreement with a previous report on the overexpression of SGO1C in HEK293 cells.Citation20 We found that the prematurely segregated kinetochores contained MAD2, suggesting that the spindle-assembly checkpoint was activated (Fig. S3A). This is also supported by the presence of MAD2–CDC20 complexes () and low APC/C activity.
Wang et al. reported that SGO1C does not localize to kinetochores during any stages of the cell cycle, but instead is enriched at the centrosome in interphase and at spindle poles in mitosis.Citation21 Furthermore, the same group found that sSgo1 plays an essential role in protecting centriole cohesion, which is partly regulated by PLK1.Citation25 It is currently unclear why others Citation20 and we observed SGO1C associated with kinetochores instead. Immunostaining of γ-tubulin provided no evidence that SGO1C binds to spindle poles during mitosis ().
Given that ectopically expressed SGO1C was present at kinetochores (), one obvious hypothesis is that SGO1C competed with SGO1A for binding to kinetochores. If SGO1C lacks functions of SGO1A required for protecting centromeric cohesin, simple competition would enable SGO1C to act in a dominant-negative manner. In support of this hypothesis, the effects of SGO1C overexpression were similar to that of downregulation of SGO1A. Nevertheless, the idea that SGO1C competes with SGO1A for kinetochore binding is at odds with our findings that when SGO1A and SGO1C were tagged with 2 different epitope tags, the 2 proteins could be found at the same centromere (our unpublished data). More importantly, the ability of SGO1C to induce mitotic defects did not correlate with its kinetochore localization. SGO1C mutants that did not associate with kinetochores could still induce mitotic arrest () and premature sister chromatid separation (Fig S4E). Conversely, the PP2A binding-defective SGO1C4m (which localized to kinetochores, Fig. S5D) was unable to induce mitotic arrest (). Our data therefore suggested that kinetochore localization is not necessary for SGO1C to induce aberrant mitosis.
As SGO1A can form homodimers using the N-terminal coiled-coil domain,Citation24 another possible way that SGO1C could disrupt mitosis is by forming heterodimers with SGO1A. However, we found that although ectopically expressed SGO1A and SGO1C could form heterodimers, they are significantly weaker than the respective homodimers (our unpublished data).
Instead of the above hypotheses, our data are consistent with the model that SGO1C induces aberrant mitosis through binding to PP2A. The N-terminal coiled-coil domain required for PP2A binding is present in SGO1C.Citation24 Consistent with this, mass spectrometry indicated that both the catalytic subunits and B56 regulatory subunits were present in the SGO1C-immunoprecipitates (Fig. S5A). This is in agreement with the previous findings that among the 4 types of PP2A B subunits, only B56 was found associated with SGO1 from meiotic yeast cells Citation26 or mitotic mammalian cells.Citation14,26 Furthermore, significantly more PP2A was co-immunoprecipitated with SGO1C than with SGO1A (). Importantly, experiments using the non-PP2A binding SGO1C4m mutant showed that binding to PP2A was essential for premature sister chromatid separation (). In fact, as long as it could bind PP2A, SGO1C did not require to be associated with kinetochores to induce premature sister chromatid separation (). Together these results point to the model that SGO1C depleted the PP2A–B56 pool, thereby decreasing the amount of PP2A–B56 available for SGO1A to protect centromeric cohesin. Although SGO1C can localize to kinetochores, the lack of crucial regions including the cohesin-binding motif may preclude its protection of cohesin.
Irrespective of the mechanism, an implication of this study is that the ratio between SGO1A and SGO1C may be important for maintaining the normal functions of SGO1A. Overexpression of SGO1C may lead to genome instability indirectly through inhibiting the functions of SGO1A.
While we are able to explain how ectopic expression of SGO1C causes aberrant mitosis, whether SGO1C performs any normal functions remain an interesting question. Although less abundant than SGO1A, SGO1C was certainly expressed in HeLa cells (). As the effects of siSGO1 could be rescued with SGO1A alone (), it can be argued that SGO1C may not possess a unique and indispensable function. However, as it is not possible to design siRNAs that specifically target SGO1C (Fig. S1A), it cannot be formally proven that removal of SGO1C does not induce any mitotic defects. A condition resembling SGO1C knockdown was when SGO1A was expressed in cells depleted with both SGO1A and SGO1C (Fig. S2A). No specific mitotic defect was detected in these cells, suggesting that SGO1C probably does not play an essential role in mitosis.
Materials and Methods
DNA constructs
Mouse SGO1A cDNA in pYX-Asc (IMAGE ID: 30604765) was amplified by PCR with oligonucleotides 5′-AACCATGGCTAAGGAAAGGTGTCA-3′ and 5′-CGGATCCTTATAGATTAAAATCGT-3′; the PCR product was cut with Nco I–Xba I and ligated into pUHD-P3T(PUR) Citation27 to obtain FLAG-SGO1ACΔ247 in pUHD-P3T(PUR). The Xba I–BamH I fragment from SGO1A in pYX-Asc was ligated into FLAG-SGO1ACΔ247 in pUHD-P3T(PUR) to obtain full length FLAG-SGO1A. A Nco I–Nco I fragment containing EGFP was ligated into the above plasmid to obtain FLAG-EGFP-SGO1A in pUHD-P3T(PUR). The Nhe I–BamH I fragment of FLAG-EGFP-SGO1A in pUHD-P3T(PUR) was ligated into the pIRESpuro3 (Clontech, Palo Alto, CA, USA). Human SGO1C cDNA in pDNR-LIB (IMAGE ID: 4685140) was amplified by PCR with oligonucleotides 5′-AACCATGGCCAAGGAAAGATGCC-3′ (SGO1C forward) and 5′-CGGGATCCTTACCTCAAGCAGATGT-3′ (SGO1C reverse); the PCR product was cut with Nco I–BamH I and ligated into pUHD-P3T(PUR) or pUHD-P2 Citation28 to obtain FLAG-SGO1C in pUHD-P3T(PUR) or HA-SGO1C in pUHD-P2, respectively. The Nco I–EcoR I fragment of FLAG-SGO1C in pUHD-P3T(PUR) was ligated into pUHD-P3T(PUR) to obtain FLAG-SGO1CCΔ236 in pUHD-P3T(PUR). SGO1C was amplified by PCR with oligonucleotides SGO1C forward and 5′-GGGGATCCTTATATTTGAAATGATTCTCC-3′ (for SGO1CCΔ157), SGO1C forward and 5′-AACCATGGCCAAGGAAAGATGCC-3′ (for SGO1CCΔ252), or 5′-AACCATGGCTACACCACCTGAAA-3′ and SGO1C reverse (for SGO1CNΔ158); the PCR products were cut with Nco I–BamH I and ligated into pUHD-P3T(PUR) to generate the respective constructs. The mRFP-expressing plasmid pUHD-P3R was generated by ligating the Nhe I–Nco I fragment of the PCR product of mRFP (using mRFP1 in pcDNA3.1 as a template and primers 5′-GGCTAGCATGGCCTCCTCCGAGGAC-3′ and 5′-GACCATGGAGGCGCCGGTGGAGTGGC-3′) into pUHD-P3T(PUR).
Site-directed mutagenesis was carried out by a PCR method Citation29 using the following oligonucleotides to introduce the mutations: 5′-CCATGCCAAATCATAACTAACACTTC-3′ and 5′-GAAGTGTTAGTTATGATTTGGCATGG-3′ (siSGO1-resistant SGO1C); 5′-GTTAGTTTTAGCTGCGGAAAATGAA-3′ and 5′-TTCCGCAGCTAAAACTAACATTTTG-3′ (L68A); 5′-GACGCCATCGCAATGTTAGTTTTAGCTTTG-3′ and 5′-ATTGCGATGGCGTCTTGGTAATTTTTCAGC-3′ (N60A, N61I, K62A). FLAG-SGO1CCΔ157 in pUHD-P3T(PUR) was cut with Nco I–Hind III and ligated into pGEX-KG to generate GST-SGO1CCΔ157 in pGEX-KG.
RNA interference
Stealth siRNA targeting SGO1 (CCAUGCCAAAUAAUCACCAACACUU, siSGO1) was obtained from Life Technologies (Carlsbad, CA, USA). SGO1A-specific siRNA (CCAGUCAUUUGGCAGGGAA, siSGO1A) and a second siRNA against SGO1 (GCCUGAAGGAUAUCACCAA, siSGO1ii) were obtained from Shanghai Genepharma (Shanghai, China). Cells were transfected with siRNA (10 nM) using LipofectamineTM RNAiMAX (Life Technologies).
RT-PCR
Total RNA was extracted using RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The RNA was reverse-transcribed into cDNA using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed with Power SYBR Green PCR Master Mix (Applied Biosystems) using an Applied Biosystems 7500 Real-Time PCR System. Transcripts were normalized to actin. Primers used: 5′-GAGAAGAAAACAACGAGTCTGAAGTG-3′ and 5′-TGCTCGTGGGATTCTGAATG-3′ (SGO1A); 5′-ACTTCCAGGACAAGGAGAATCATT-3′ and 5′-ACAACAGGATACAAGGAGACATTGG-3′ (SGO1C); 5′-GGGAAATCGTGCGTGACATT-3′ and 5′-GGAACCGCTCATTGCCAAT-3′ (actin).
Cell culture
The HeLa used in this study was a clone that expressed the tTA tetracycline repressor chimera.Citation28 HeLa cells stably expressing histone H2B-GFP Citation30 or APC/C reporter Citation31 were generated as described previously. HeLa cells expressing histone H2B-mRFP (mixed population) were generated by infecting cells with retroviruses expressing histone H2B-mRFP Citation32 followed by selection with hygromycin B. HeLa cells expressing FLAG-EGFP-SGO1A (or SGO1C) (isolated from single colonies) were generated by transfecting cells with FLAG-EGFP-SGO1A (or SGO1C) in pIRESpuro3 followed by selection with puromycin. HeLa cells expressing EGFP-α-tubulin (isolated from a single colony) were generated by transfecting cells with EGFP-α-tubulin in pIRESpuro3 followed by selection with puromycin. Cells were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) calf serum and 50 U/ml penicillin-streptomycin (Life Technologies). Unless stated specifically, cells were treated with the following reagents at the indicated final concentration: hygromycin B (Life Technologies; 250 µg/ml), nocodazole (Sigma-Aldrich, St. Louis, MO, USA; 0.1 µg/ml), and puromycin (Sigma-Aldrich; 15 µg/ml).
Microscopy
Immunofluorescence microscopy was performed as previously described.Citation33 Secondary antibodies used were Alexa Fluor–594 goat anti-rabbit IgG, Alexa Fluor–633 goat anti-mouse IgG, Alexa Fluor–488 goat anti-human IgG, and Alexa Fluor–647 goat anti-human IgG (Life Technologies). For K-fiber staining, cells were grown on poly-L-lysine-coated coverslips in 35-mm plates with 2 ml of medium. After adding 500 µl of 100 mM HEPES pH 7.2 dropwise to the medium, the plates were incubated on ice for 10 min. After removing the medium, the cells were incubated with 1 ml of 100 mM PIPES; 20 mM HEPES pH 6.9, 5 mM EGTA, 2 mM MgCl2, 0.2% Triton X-100, and 3.7% paraformaldehyde at 25°C for 10 min. The cells were washed with PBS (5 min) and incubated with 1 ml of blocking solution (3% BSA, 0.2% Triton X-100 in PBS) at 25°C for 30 min. The samples were then incubated with primary antibody and processed for immunofluorescence microscopy analysis.
Live-cell imaging
Cells were imaged using a TE2000E-PFS Ti-E fluorescence microscope (Nikon, Tokyo, Japan) equipped with a SPOT BOOST EMCCD camera (Diagnostic Instrument, Sterling Heights, MI, USA) and a INU-NI-F1 temperature, humidity, and CO2 control system (Tokai Hit, Shizuoka, Japan).
Flow cytometry
Flow cytometry analysis after propidium iodide staining was performed as described previously.Citation34
Mass spectrometry
Lysates from cells stably expressing FLAG-EGFP-tagged SGO1A or SGO1C (10 mg) were prepared. The lysates were incubated with agarose (1 h at 4°C) before cleared by centrifugation (10 min in a desktop centrifuge). The supernatant was subjected to immunoprecipitation using anti-FLAG agarose (Sigma-Aldrich). The precipitated proteins were eluted using a FLAG peptide (Sigma-Aldrich). The protein solution was diluted to 1 M urea in 25 mM ammonium biocarbonate. Proteins were then reduced with 10 mM of DTT at room temperature for 3 h, before alkylated with iodoacetamide (20 mM final). After quenching the alkylation reaction with DTT (10 mM final), proteins were digested using 1:50 (w/w) of Trypsin Gold, Mass Spectrometry Grade trypsin (Promega, Madison, WI, USA) at 37°C for overnight. The samples were adjusted to 0.5% formic acid, dried in a SpeedVac concentrator (Thermo Scientific, San Jose, CA, USA), and dissolved in 0.5% formic acid in water. After desalting with µC18 reverse-phase ZipTip (Millipore, Darmstadt, Germany), the peptides were dried again before analyzed on a Thermo Scientific LTQ Velos mass spectrometer (Thermo Fisher Scientific). Peptides were analyzed using Mascot software.
Antibodies and immunological methods
Antibodies against β-actin, CDC20, cyclin B1, FLAG, FLAG (conjugated to agarose), GFP (Sigma-Aldrich), HA, phospho-histone H3Ser10, securin, γ-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), CREST (Fitzgerald Industries International, Acton, MA, USA), HEC1 (Abcam, Cambridge, UK), MAD2, cleaved PARP1(Asp214), PPP2CA (BD Biosciences, Franklin Lakes, NJ, USA), and Alexa Fluor–488 α-tubulin (Life Technologies) were obtained from the indicated suppliers. Mouse monoclonal antibody against SGO1 was obtained from Abcam. Rabbit polyclonal antibodies against SGO1 was raised against bacterially expressed GST-SGO1CCΔ157. Cell-free extracts were prepared using a NP-40 lysis buffer method as described previously.Citation35 For detection of SGO1A with immunoblotting, the lysates were dissolved in 6xSDS sample buffer directly without centrifugation. Immunoblotting and immunoprecipitation were performed as described previously.Citation35 To prepare lysates for SGO1 immunoprecipitation, lysates were prepared using NP-40 lysis buffer Citation35 supplemented with 500 mM NaCl and sonicated using a Branson sonifier 250 (30% duty cycle, 3 micro tip limit output).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
2015CC6746R-s06.avi
Download Microsoft Video (AVI) (480.7 KB)2015CC6746R-s05.avi
Download Microsoft Video (AVI) (192.2 KB)2015CC6746R-s04.avi
Download Microsoft Video (AVI) (412.4 KB)2015CC6746R-s03.avi
Download Microsoft Video (AVI) (131.6 KB)2015CC6746R-s02.pdf
Download PDF (4.6 MB)Acknowledgments
We thank Joyce Wong (BioCRF, HKUST) for help with mass spectrometry analysis, Wan Mui Chan, Kin Fan On, and Kristin Wong for technical assistance.
Funding
This work was supported in part by the Research Grants Council grants 662213 and T13-607/12R to R.Y.C.P.
Related Research Data
References
- Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet 2009; 43:525-58; PMID:19886810; http://dx.doi.org/10.1146/annurev-genet-102108-134233
- Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chromosome biology. Genes Dev 2008; 22:3089-114; PMID:19056890; http://dx.doi.org/10.1101/gad.1724308
- Onn I, Heidinger-Pauli JM, Guacci V, Unal E, Koshland DE. Sister chromatid cohesion: a simple concept with a complex reality. Annu Rev Cell Dev Biol 2008; 24:105-29; PMID:18616427; http://dx.doi.org/10.1146/annurev.cellbio.24.110707.175350
- Rankin S, Ayad NG, Kirschner MW. Sororin, a substrate of the anaphase-promoting complex, is required for sister chromatid cohesion in vertebrates. Mol Cell 2005; 18:185-200; PMID:15837422; http://dx.doi.org/10.1016/j.molcel.2005.03.017
- Lafont AL, Song J, Rankin S. Sororin cooperates with the acetyltransferase Eco2 to ensure DNA replication-dependent sister chromatid cohesion. Proc Natl Acad Sci U S A 2010; 107:20364-9; PMID:21059905; http://dx.doi.org/10.1073/pnas.1011069107
- Nishiyama T, Ladurner R, Schmitz J, Kreidl E, Schleiffer A, Bhaskara V, Bando M, Shirahige K, Hyman AA, Mechtler K, et al. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell 2010; 143:737-49; PMID:21111234; http://dx.doi.org/10.1016/j.cell.2010.10.031
- Nishiyama T, Sykora MM, Huis in 't Veld PJ, Mechtler K, Peters JM. Aurora B and Cdk1 mediate Wapl activation and release of acetylated cohesin from chromosomes by phosphorylating Sororin. Proc Natl Acad Sci U S A 2013; 110:13404-9; PMID:23901111; http://dx.doi.org/10.1073/pnas.1305020110
- Hauf S, Roitinger E, Koch B, Dittrich CM, Mechtler K, Peters JM. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol 2005; 3:e69; PMID:15737063; http://dx.doi.org/10.1371/journal.pbio.0030069
- Dreier MR, Bekier MEn, Taylor WR. Regulation of sororin by Cdk1-mediated phosphorylation. J Cell Sci 2011; 124:2976-87; PMID:21878504; http://dx.doi.org/10.1242/jcs.085431
- Zhang N, Panigrahi AK, Mao Q, Pati D. Interaction of Sororin protein with polo-like kinase 1 mediates resolution of chromosomal arm cohesion. J Biol Chem 2011; 286:41826-37; PMID:21987589; http://dx.doi.org/10.1074/jbc.M111.305888
- Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, Peters JM. Wapl controls the dynamic association of cohesin with chromatin. Cell 2006; 127:955-67; PMID:17113138; http://dx.doi.org/10.1016/j.cell.2006.09.040
- Kawashima SA, Yamagishi Y, Honda T, Ishiguro K, Watanabe Y. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science 2010; 327:172-7; PMID:19965387; http://dx.doi.org/10.1126/science.1180189
- Tang Z, Shu H, Qi W, Mahmood NA, Mumby MC, Yu H. PP2A is required for centromeric localization of Sgo1 and proper chromosome segregation. Dev Cell 2006; 10:575-85; PMID:16580887; http://dx.doi.org/10.1016/j.devcel.2006.03.010
- Kitajima TS, Sakuno T, Ishiguro K, Iemura S, Natsume T, Kawashima SA, Watanabe Y. Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature 2006; 441:46-52; PMID:16541025; http://dx.doi.org/10.1038/nature04663
- Gandhi R, Gillespie PJ, Hirano T. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr Biol 2006; 16:2406-17; PMID:17112726; http://dx.doi.org/10.1016/j.cub.2006.10.061
- Liu H, Rankin S, Yu H. Phosphorylation-enabled binding of SGO1-PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat Cell Biol 2013; 15:40-9; PMID:23242214; http://dx.doi.org/10.1038/ncb2637
- Liu H, Jia L, Yu H. Phospho-H2A and cohesin specify distinct tension-regulated Sgo1 pools at kinetochores and inner centromeres. Curr Biol 2013; 23:1927-33; PMID:24055156; http://dx.doi.org/10.1016/j.cub.2013.07.078
- Lee J, Kitajima TS, Tanno Y, Yoshida K, Morita T, Miyano T, Miyake M, Watanabe Y. Unified mode of centromeric protection by shugoshin in mammalian oocytes and somatic cells. Nat Cell Biol 2008; 10:42-52; PMID:18084284; http://dx.doi.org/10.1038/ncb1667
- Kitajima TS, Kawashima SA, Watanabe Y. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature 2004; 427:510-7; PMID:14730319; http://dx.doi.org/10.1038/nature02312
- Suzuki H, Akiyama N, Tsuji M, Ohashi T, Saito S, Eto Y. Human Shugoshin mediates kinetochore-driven formation of kinetochore microtubules. Cell Cycle 2006; 5:1094-101; PMID:16687935; http://dx.doi.org/10.4161/cc.5.10.2747
- Wang X, Yang Y, Dai W. Differential subcellular localizations of two human Sgo1 isoforms: implications in regulation of sister chromatid cohesion and microtubule dynamics. Cell Cycle 2006; 5:635-40; PMID:16582621
- McGuinness BE, Hirota T, Kudo NR, Peters JM, Nasmyth K. Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells. PLoS Biol 2005; 3:e86; PMID:15737064; http://dx.doi.org/10.1371/journal.pbio.0030086
- Ma HT, Poon RY. Orderly inactivation of the key checkpoint protein mitotic arrest deficient 2 (MAD2) during mitotic progression. J Biol Chem 2011; 286:13052-9; PMID:21335556; http://dx.doi.org/10.1074/jbc.M110.201897
- Xu Z, Cetin B, Anger M, Cho US, Helmhart W, Nasmyth K, Xu W. Structure and function of the PP2A-shugoshin interaction. Mol Cell 2009; 35:426-41; PMID:19716788; http://dx.doi.org/10.1016/j.molcel.2009.06.031
- Wang X, Yang Y, Duan Q, Jiang N, Huang Y, Darzynkiewicz Z, Dai W. sSgo1, a major splice variant of Sgo1, functions in centriole cohesion where it is regulated by Plk1. Dev Cell 2008; 14:331-41; PMID:18331714; http://dx.doi.org/10.1016/j.devcel.2007.12.007
- Riedel CG, Katis VL, Katou Y, Mori S, Itoh T, Helmhart W, Galova M, Petronczki M, Gregan J, Cetin B, et al. Protein phosphatase 2A protects centromeric sister chromatid cohesion during meiosis I. Nature 2006; 441:53-61; PMID:16541024; http://dx.doi.org/10.1038/nature04664
- Ma HT, Tsang YH, Marxer M, Poon RY. Cyclin A2-cyclin-dependent kinase 2 cooperates with the PLK1-SCFbeta-TrCP1-EMI1-anaphase-promoting complex/cyclosome axis to promote genome reduplication in the absence of mitosis. Mol Cell Biol 2009; 29:6500-14; PMID:19822658; http://dx.doi.org/10.1128/MCB.00669-09
- Yam CH, Siu WY, Lau A, Poon RY. Degradation of cyclin A does not require its phosphorylation by CDC2 and cyclin-dependent kinase 2. J Biol Chem 2000; 275:3158-67; PMID:10652300; http://dx.doi.org/10.1074/jbc.275.5.3158
- Horton RM, Pease LR. Recombination and mutagenesis of DNA sequences using PCR. In: McPherson MJ, ed(s). Directed mutagenesis: a practical approach. Oxford:IRL Press at OUP, 1991:217-47.
- Chan YW, Ma HT, Wong W, Ho CC, On KF, Poon RY. CDK1 inhibitors antagonize the immediate apoptosis triggered by spindle disruption but promote apoptosis following the subsequent rereplication and abnormal mitosis. Cell Cycle 2008; 7:1449-61; PMID:18418077; http://dx.doi.org/10.4161/cc.7.10.5880
- Chow JP, Poon RY, Ma HT. Inhibitory phosphorylation of cyclin-dependent kinase 1 as a compensatory mechanism for mitosis exit. Mol Cell Biol 2011; 31:1478-91; PMID:21262764; http://dx.doi.org/10.1128/MCB.00891-10
- Marxer M, Foucar CE, Man WY, Chen Y, Ma HT, Poon RY. Tetraploidization increases sensitivity to Aurora B kinase inhibition. Cell Cycle 2012; 11:2567-77; PMID:22722494; http://dx.doi.org/10.4161/cc.20947
- Han X, Poon RY. Critical differences between isoforms of securin reveal mechanisms of separase regulation. Mol Cell Biol 2013; 33:3400-15; PMID:23798554; http://dx.doi.org/10.1128/MCB.00057-13
- Siu WY, Arooz T, Poon RY. Differential responses of proliferating versus quiescent cells to adriamycin. Exp Cell Res 1999; 250:131-41; PMID:10388527; http://dx.doi.org/10.1006/excr.1999.4551
- Poon RY, Toyoshima H, Hunter T. Redistribution of the CDK inhibitor p27 between different cyclin.CDK complexes in the mouse fibroblast cell cycle and in cells arrested with lovastatin or ultraviolet irradiation. Mol Biol Cell 1995; 6:1197-213; PMID:8534916; http://dx.doi.org/10.1091/mbc.6.9.1197